The Anti-Cancer Drug Dabrafenib Is a Potent Activator of the Human Pregnane X Receptor

 , , , ,
, , , ,  and
and 
Abstract
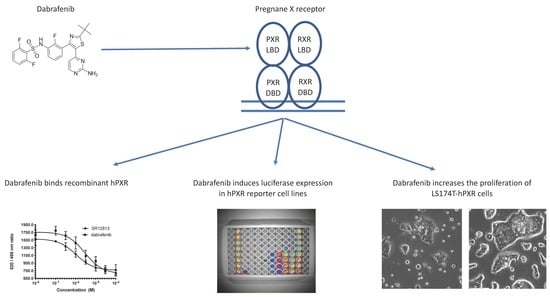
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. PXR and CAR Reporter Cell Lines
2.3. Transactivation Assays
2.4. LS174T-hPXR Proliferation Assay (P-SCREEN)
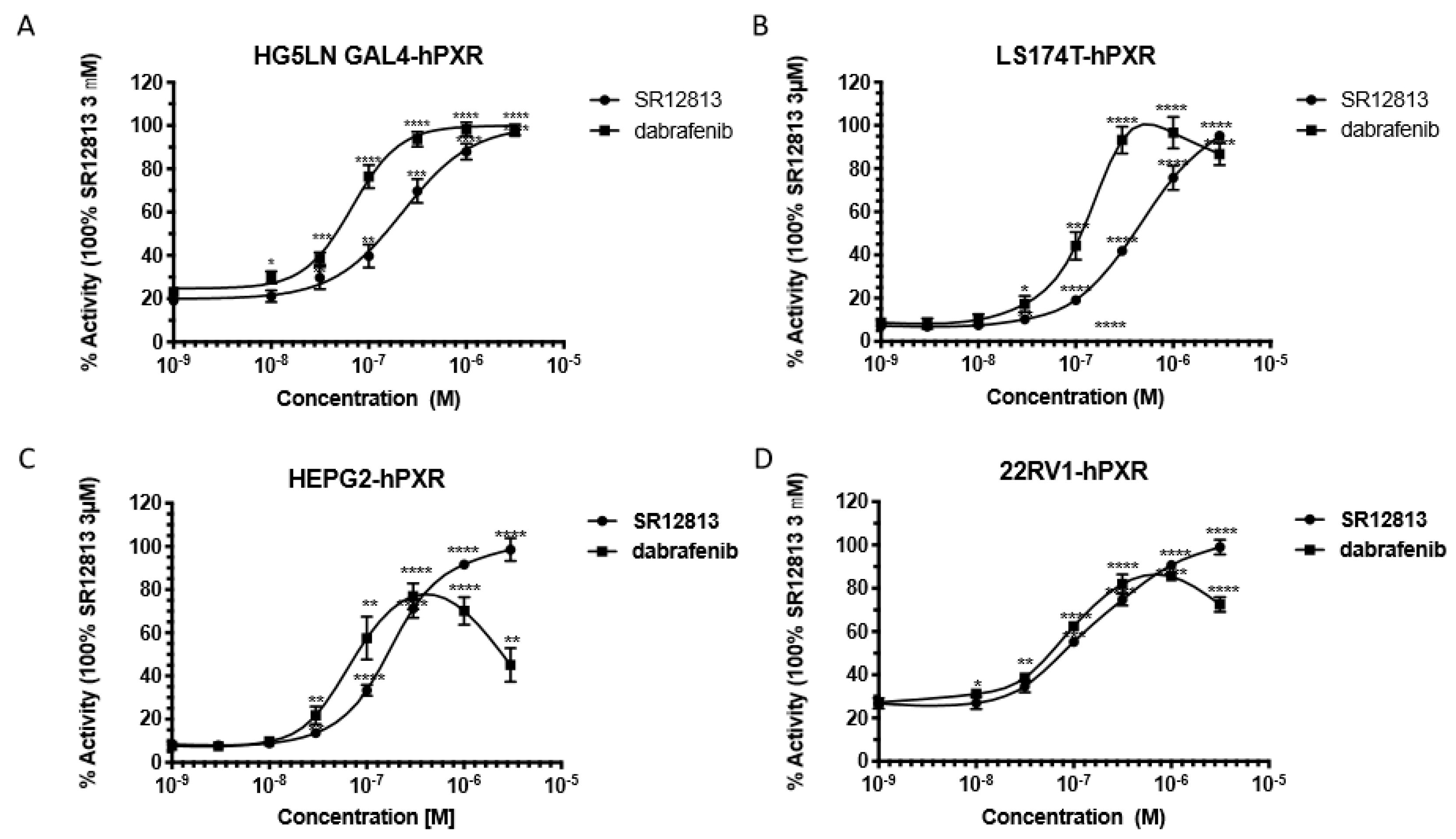
2.5. Lanthascreen TR-FRET PXR Competitive Binding Assay
2.6. Isolation and Primary Culture of Human Hepatocytes
2.7. Real Time-Quantitative Polymerase Chain Reaction (RT-qPCR)
3. Results
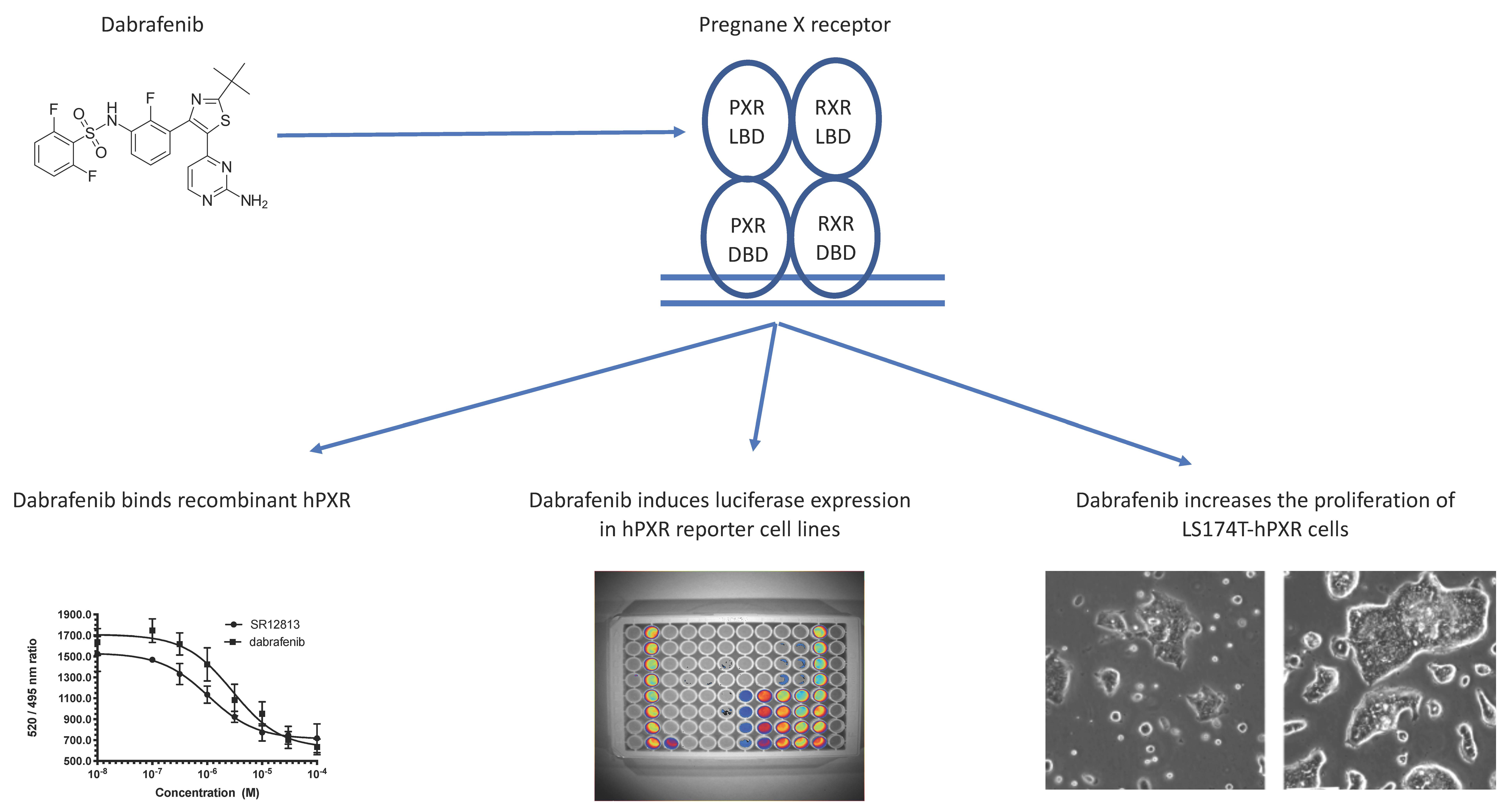
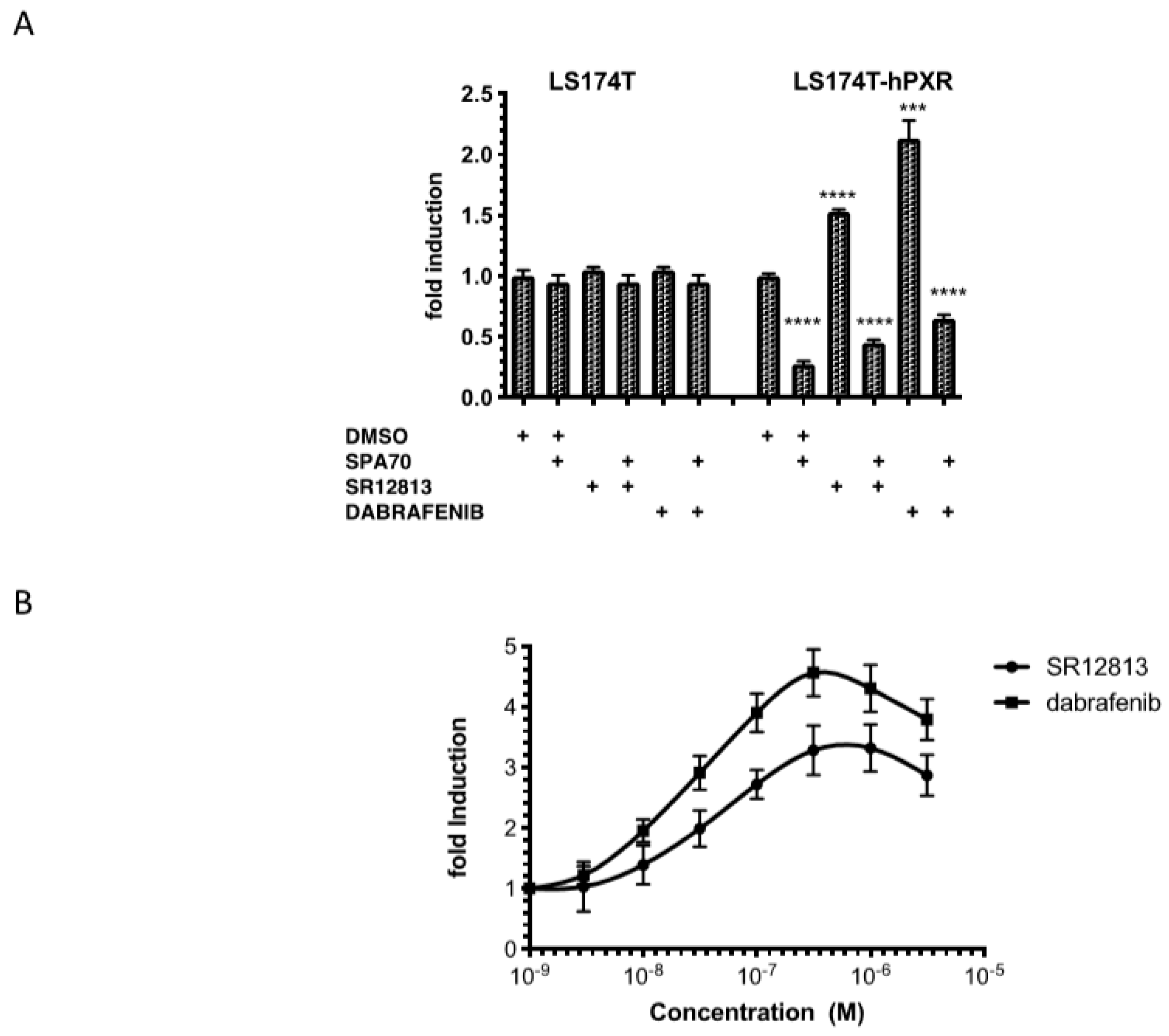
3.1. Transactivation of PXR by Dabrafenib in Human Cancer Cell Lines
3.2. Effects of Dabrafenib on the Expression of hPXR Target Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yu, P.; Du, Y.; Yang, L.; Fan, S.; Wu, J.; Zheng, S. Significance of multidrug resistance gene-related proteins in the postoperative chemotherapy of gastric cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 7945–7950. [Google Scholar]
- Wang, T.; Wei, J.; Wang, N.; Ma, J.L.; Hui, P.P. The glucosylceramide synthase inhibitor PDMP sensitizes pancreatic cancer cells to MEK/ERK inhibitor AZD-6244. Biochem. Biophys. Res. Commun. 2015, 456, 821–826. [Google Scholar] [CrossRef]
- Harmsen, S.; Meijerman, I.; Beijnen, J.H.; Schellens, J.H.M. Nuclear receptor mediated induction of cytochrome P450 3A4 by anticancer drugs: A key role for the pregnane X receptor. Cancer Chemother. Pharmacol. 2009, 64, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Hashimoto, M.; Honkakoski, P.; Negishi, M. Regulation of gene expression by CAR: An update. Arch. Toxicol. 2015, 89, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Mnif, W.; Pascussi, J.M.; Pillon, A.; Escande, A.; Bartegi, A.; Nicolas, J.C.; Cavaillès, V.; Duchesne, M.J.; Balaguer, P. Estrogens and antiestrogens activate hPXR. Toxicol. Lett. 2007, 170, 19–29. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Y.; Wang, M.T.; Zeng, S.; Nie, D. Human pregnane X receptor and resistance to chemotherapy in prostate cancer. Cancer Res. 2007, 67, 10361–10367. [Google Scholar] [CrossRef]
- Gupta, D.; Venkatesh, M.; Wang, H.; Kim, S.; Sinz, M.; Goldberg, G.L.; Whitney, K.; Longley, C.; Mani, S. Expanding the roles for pregnane X receptor in cancer: Proliferation and drug resistance in ovarian cancer. Clin. Cancer Res. 2008, 14, 5332–5340. [Google Scholar] [CrossRef]
- Harmsen, S.; Meijerman, I.; Febus, C.L.; Maas-Bakker, R.F.; Beijnen, J.H.; Schellens, J.H.M. PXR-mediated induction of P-glycoprotein by anticancer drugs in a human colon adenocarcinoma-derived cell line. Cancer Chemother. Pharmacol. 2010, 66, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Chen, K.; He, J.; Pan, F.; Li, J.; Chen, J.; Chen, W.; Liang, H. Association of pregnane X receptor with multidrug resistance-related protein 3 and its role in human colon cancer chemoresistance. J. Gastrointest. Surg. 2009, 13, 1831–1838. [Google Scholar] [CrossRef]
- Masuyama, H.; Nakamura, K.; Nobumoto, E.; Hiramatsu, Y. Inhibition of pregnane X receptor pathway contributes to the cell growth inhibition and apoptosis of anticancer agents in ovarian cancer cells. Int. J. Oncol. 2016, 49, 1211–1220. [Google Scholar] [CrossRef]
- Moon, J.Y.; Gwak, H.S. Role of the nuclear pregnane X X receptor in drug metabolism and the clinical response. Recept. Clin. Investig. 2015, 2, e996. [Google Scholar]
- Nallani, S.C.; Goodwin, B.; Maglich, J.M.; Buckley, D.J.; Buckley, A.R.; Desai, P.B. Induction of cytochrome P450 3A by paclitaxel in mice: Pivotal role of the nuclear xenobiotic receptor, pregnane X receptor. Drug Metab. Dispos. Biol. 2003, 31, 681–684. [Google Scholar] [CrossRef]
- Yoshinari, K. Role of Nuclear Receptors PXR and CAR in Xenobiotic-Induced Hepatocyte Proliferation and Chemical Carcinogenesis. Biol. Pharm. Bull. 2019, 42, 1243–1252. [Google Scholar] [CrossRef]
- Koutsounas, I.; Theocharis, S.; Patsouris, E.; Giaginis, C. Pregnane X receptor (PXR) at the crossroads of human metabolism and disease. Curr. Drug Metab. 2013, 14, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Yamazaki, Y.; Negishi, M. Pregnane X Receptor Represses HNF4α Gene to Induce Insulin-Like Growth Factor-Binding Protein IGFBP1 that Alters Morphology of and Migrates HepG2 Cells. Mol. Pharmacol. 2015, 88, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Venkatesh, M.; Li, H.; Goetz, R.; Mukherjee, S.; Biswas, A.; Zhu, L.; Kaubisch, A.; Wang, L.; Pullman, J.; et al. Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice. J. Clin. Investig. 2011, 121, 3220–3232. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, M.; Zhai, Y.; Xie, W. The antiapoptotic role of pregnane X receptor in human colon cancer cells. Mol. Endocrinol. 2008, 22, 868–880. [Google Scholar] [CrossRef]
- Puszkiel, A.; Noé, G.; Bellesoeur, A.; Kramkimel, N.; Paludetto, M.N.; Thomas-Schoemann, A.; Vidal, M.; Goldwasser, F.; Chatelut, E.; Blanchet, B. Clinical Pharmacokinetics and Pharmacodynamics of Dabrafenib. Clin. Pharmacokinet. 2019, 58, 451–467. [Google Scholar] [CrossRef]
- Dhillon, S. Dabrafenib plus Trametinib: A Review in Advanced Melanoma with a BRAF (V600) Mutation. Target. Oncol. 2016, 11, 417–428. [Google Scholar] [CrossRef]
- Lawrence, S.K.; Nguyen, D.; Bowen, C.; Richards-Peterson, L.; Skordos, K.W. The metabolic drug-drug interaction profile of Dabrafenib: In vitro investigations and quantitative extrapolation of the P450-mediated DDI risk. Drug. Metab. Dispos. Biol. 2014, 42, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, M.; Boulahtouf, A.; Delfosse, V.; Thouennon, E.; Bourguet, W.; Balaguer, P. Reporter cell lines for the characterization of the interactions between human nuclear rceptors and endocrine disruptors. Front. Endocrinol. 2015, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Delfosse, V.; Dendele, B.; Huet, T.; Grimaldi, M.; Boulahtouf, A.; Gerbal-Chaloin, S.; Beucher, B.; Roecklin, D.; Muller, C.; Rahmani, R.; et al. Synergistic activation of human pregnane X receptor by binary cocktails of pharmaceutical and environmental compounds. Nat. Commun. 2015, 6, 8089. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.M.; Sonnenschein, C.; Chung, K.L.; Fernandez, M.F.; Olea, N.; Olea Serrano, F. The E-SCREEN assay as a tool to identify estrogens; an update on estrogenic environmental pollutants. Environ. Health Perspect. 1995, 103, 113–122. [Google Scholar] [PubMed]
- Lin, W.; Wang, Y.M.; Chai, S.C.; Lv, L.; Zheng, J.; Wu, J.; Zhang, Q.; Wang, Y.D.; Griffin, P.R.; Chen, T. SPA70 is a potent antagonist of human pregnane X receptor. Nat. Commun. 2017, 8, 741. [Google Scholar] [CrossRef]
- Pichard, L.; Raulet, E.; Fabre, G.; Ferrini, J.B.; Ourlin, J.C.; Maurel, P. Human hepatocyte culture. Methods Mol. Biol. 2006, 320, 283–293. [Google Scholar] [PubMed]
- Bershas, D.A.; Ouellet, D.; Mamaril-Fishman, D.B.; Nebot, N.; Carson, S.W.; Blackman, S.C.; Morrison, R.A.; Adams, J.L.; Jurusik, K.E.; Knecht, D.M.; et al. Metabolism and disposition of oral dabrafenib in cancer patients: Proposed participation of aryl nitrogen in carbon-carbon bond cleavage via decarboxylation following enzymatic oxidation. Drug Metab. Dispos. Biol. 2013, 41, 2215–2224. [Google Scholar] [CrossRef]
- Wistuba, W.; Gnewuch, C.; Liebisch, G.; Schmitz, G.; Langmann, T. Lithocholic acid induction of the FGF19 promoter in intestinal cells is mediated by PXR. World J. Gastroenterol. 2007, 13, 4230–4235. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Creusot, N.; Gassiot, M.; Alaterre, E.; Chiavarina, B.; Grimaldi, M.; Boulahtouf, A.; Toporova, L.; Gerbal-Chaloin, S.; Daujat-Chavanieu, M.; Matheux, A.; et al. The Anti-Cancer Drug Dabrafenib Is a Potent Activator of the Human Pregnane X Receptor. Cells 2020, 9, 1641. https://doi.org/10.3390/cells9071641
Creusot N, Gassiot M, Alaterre E, Chiavarina B, Grimaldi M, Boulahtouf A, Toporova L, Gerbal-Chaloin S, Daujat-Chavanieu M, Matheux A, et al. The Anti-Cancer Drug Dabrafenib Is a Potent Activator of the Human Pregnane X Receptor. Cells. 2020; 9(7):1641. https://doi.org/10.3390/cells9071641
Chicago/Turabian StyleCreusot, Nicolas, Matthieu Gassiot, Elina Alaterre, Barbara Chiavarina, Marina Grimaldi, Abdelhay Boulahtouf, Lucia Toporova, Sabine Gerbal-Chaloin, Martine Daujat-Chavanieu, Alice Matheux, and et al. 2020. "The Anti-Cancer Drug Dabrafenib Is a Potent Activator of the Human Pregnane X Receptor" Cells 9, no. 7: 1641. https://doi.org/10.3390/cells9071641
APA StyleCreusot, N., Gassiot, M., Alaterre, E., Chiavarina, B., Grimaldi, M., Boulahtouf, A., Toporova, L., Gerbal-Chaloin, S., Daujat-Chavanieu, M., Matheux, A., Rahmani, R., Gongora, C., Evrard, A., Pourquier, P., & Balaguer, P. (2020). The Anti-Cancer Drug Dabrafenib Is a Potent Activator of the Human Pregnane X Receptor. Cells, 9(7), 1641. https://doi.org/10.3390/cells9071641