Bivalent Inhibitor with Selectivity for Trimeric MMP-9 Amplifies Neutrophil Chemotaxis and Enables Functional Studies on MMP-9 Proteoforms

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Proteins, Reagents and Buffers
2.2. Gelatin and Peptide Degradation Assays
2.3. Isolation, Degranulation and Chemotaxis of Human Neutrophils
2.4. Compound Toxicity and Stability Testing
2.5. Mouse Air Pouch Model
2.6. Cytospin Preparation and Manual Cell Counting
2.7. Flow Cytometry
2.8. Mouse Endotoxemia Model
2.9. Protein Extraction and Analysis
2.10. Statistics
3. Results
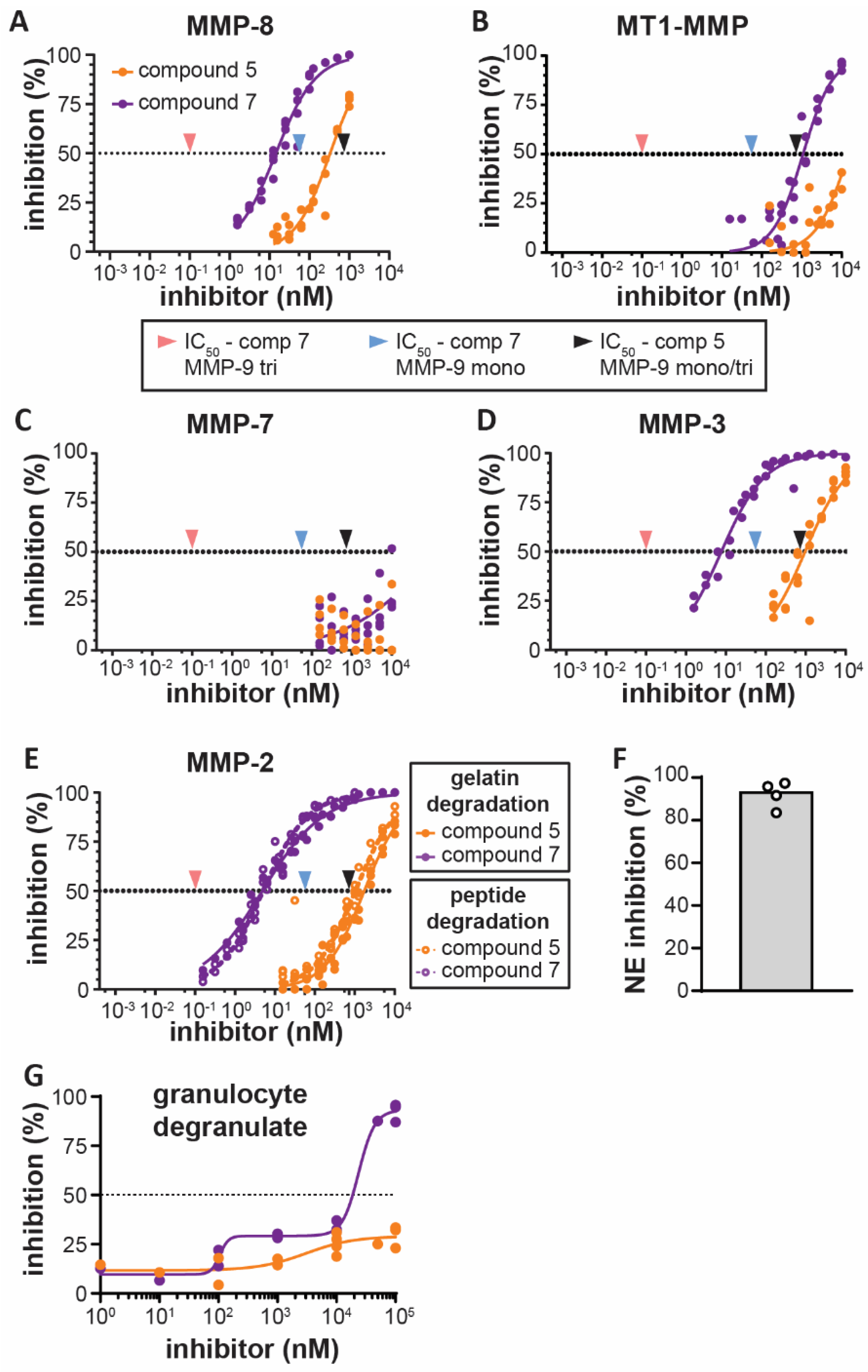
3.1. Bivalent Carboxylate Inhibitor Efficiently Inhibits Homotrimeric MMP-9
3.2. Bivalent Inhibitor Is Most Potent Against Trimeric MMP-9, Out of Several Leukocyte-Derived Proteases
3.3. Bivalent Inhibitor Most Efficiently Inhibits Gelatinolysis in Secretions of Stimulated Human Granulocytes
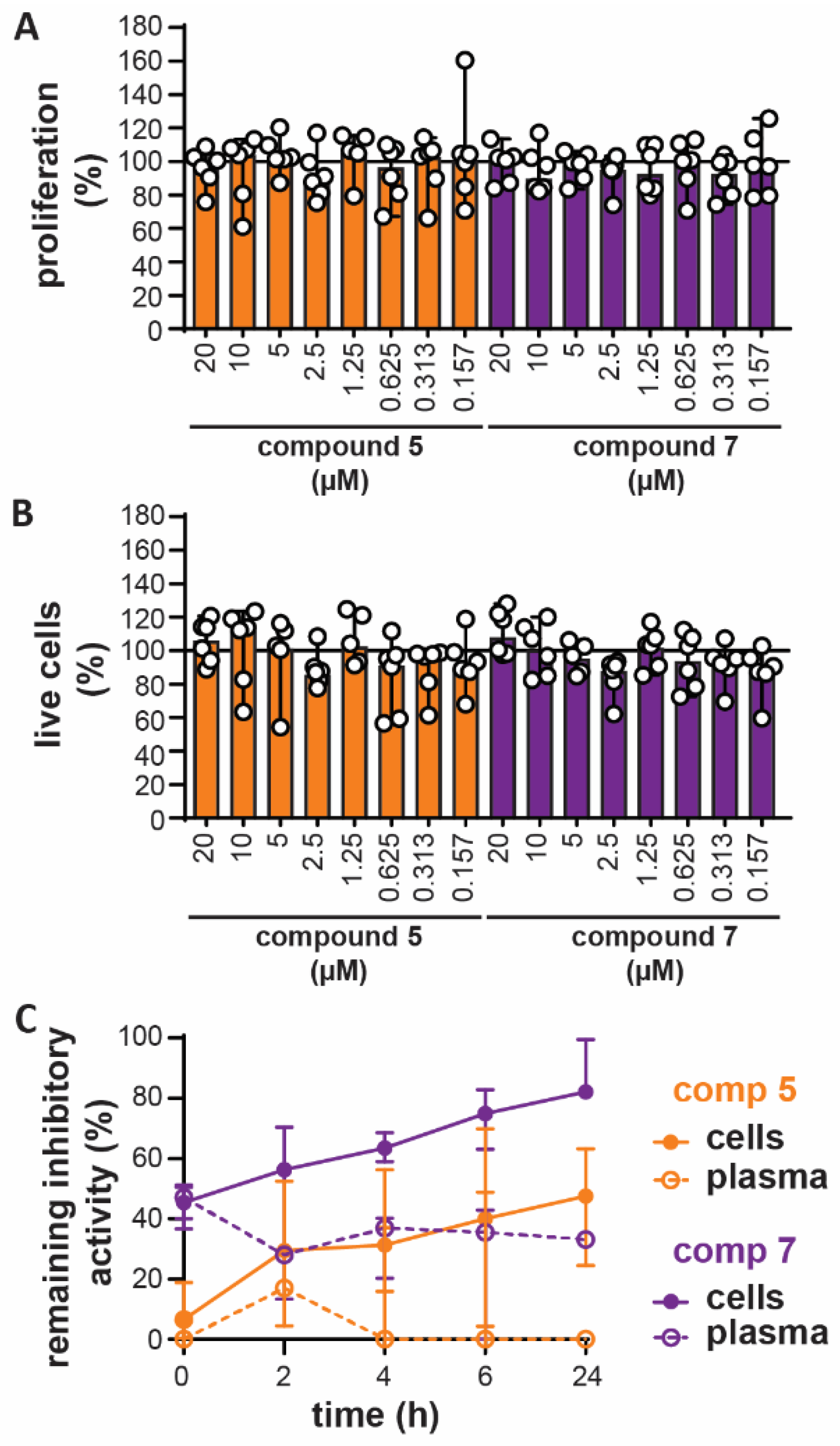
3.4. In Vitro Toxicity and Ex Vivo Stability
3.5. Bivalent Inhibitor Amplifies In Vivo Chemotaxis of Neutrophils Towards LPS injection
3.6. Bivalent Inhibitor Does Not Differentially Affect Disease Scores in a Mouse Endotoxemia Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.P.; Nicholls, A.J.; Wong, C.H.Y. Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef]
- Silva, M.T. When two is better than one: Macrophages and neutrophils work in concert in innate immunity as complementary and cooperative partners of a myeloid phagocyte system. J. Leukoc. Biol. 2010, 87, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Pederzoli-Ribeil, M.; Gabillet, J.; Witko-Sarsat, V. Proteases from Inflammatory Cells: Regulation of Inflammatory Response. In Proteases and Their Receptors in Inflammation; Springer Science and Business Media LLC: Berlin, Germany, 2011; pp. 73–100. [Google Scholar]
- Proost, P.; Struyf, S.; Van Damme, J.; Fiten, P.; Ugarte-Berzal, E.; Opdenakker, G. Chemokine isoforms and processing in inflammation and immunity. J. Autoimmun. 2017, 85, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef]
- Sela-Passwell, N.; Rosenblum, G.; Shoham, T.; Sagi, I. Structural and functional bases for allosteric control of MMP activities: Can it pave the path for selective inhibition? Biochim. Biophys. Acta 2010, 1803, 29–38. [Google Scholar] [CrossRef]
- Hu, J.; Van den Steen, P.E.; Sang, Q.X.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [PubMed]
- D’Haese, A.; Wuyts, A.; Dillen, C.; Dubois, B.; Billiau, A.; Heremans, H.; Van Damme, J.; Arnold, B.; Opdenakker, G. In vivo neutrophil recruitment by granulocyte chemotactic protein-2 is assisted by gelatinase B/MMP-9 in the mouse. J. Interferon Cytokine Res. 2000, 20, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wu, C.; Korpos, E.; Zhang, X.; Agrawal, S.M.; Wang, Y.; Faber, C.; Schafers, M.; Korner, H.; Opdenakker, G.; et al. Focal MMP-2 and MMP-9 activity at the blood-brain barrier promotes chemokine-induced leukocyte migration. Cell Rep. 2015, 10, 1040–1054. [Google Scholar] [CrossRef] [PubMed]
- Pruijt, J.F.; Fibbe, W.E.; Laterveer, L.; Pieters, R.A.; Lindley, I.J.; Paemen, L.; Masure, S.; Willemze, R.; Opdenakker, G. Prevention of interleukin-8-induced mobilization of hematopoietic progenitor cells in rhesus monkeys by inhibitory antibodies against the metalloproteinase gelatinase B (MMP-9). Proc. Natl. Acad. Sci. USA 1999, 96, 10863–10868. [Google Scholar] [CrossRef]
- Vandooren, J.; Swinnen, W.; Ugarte-Berzal, E.; Boon, L.; Dorst, D.; Martens, E.; Opdenakker, G. Endotoxemia shifts neutrophils with TIMP-free gelatinase B/MMP-9 from bone marrow to the periphery and induces systematic upregulation of TIMP-1. Haematologica 2017, 102, 1671–1682. [Google Scholar] [CrossRef]
- Dubois, B.; Starckx, S.; Pagenstecher, A.; Oord, J.; Arnold, B.; Opdenakker, G. Gelatinase B deficiency protects against endotoxin shock. Eur. J. Immunol. 2002, 32, 2163–2171. [Google Scholar] [CrossRef]
- Hu, J.; Van den Steen, P.E.; Dillen, C.; Opdenakker, G. Targeting neutrophil collagenase/matrix metalloproteinase-8 and gelatinase B/matrix metalloproteinase-9 with a peptidomimetic inhibitor protects against endotoxin shock. Biochem. Pharmacol. 2005, 70, 535–544. [Google Scholar] [CrossRef]
- Steinberg, J.; Halter, J.; Schiller, H.J.; Dasilva, M.; Landas, S.; Gatto, L.A.; Maisi, P.; Sorsa, T.; Rajamaki, M.; Lee, H.M.; et al. Metalloproteinase inhibition reduces lung injury and improves survival after cecal ligation and puncture in rats. J. Surg. Res. 2003, 111, 185–195. [Google Scholar] [CrossRef]
- Rahman, M.; Zhang, S.; Chew, M.; Syk, I.; Jeppsson, B.; Thorlacius, H. Platelet shedding of CD40L is regulated by matrix metalloproteinase-9 in abdominal sepsis. J. Thromb. Haemost. 2013, 11, 1385–1398. [Google Scholar] [CrossRef]
- Vandooren, J.; Born, B.; Solomonov, I.; Zajac, E.; Saldova, R.; Senske, M.; Ugarte-Berzal, E.; Martens, E.; Van den Steen, P.E.; Van Damme, J.; et al. Circular trimers of gelatinase B/matrix metalloproteinase-9 constitute a distinct population of functional enzyme molecules differentially regulated by tissue inhibitor of metalloproteinases-1. Biochem. J. 2015, 465, 259–270. [Google Scholar] [CrossRef]
- Serifova, X.; Ugarte-Berzal, E.; Opdenakker, G.; Vandooren, J. Homotrimeric MMP-9 is an active hitchhiker on alpha-2-macroglobulin partially escaping protease inhibition and internalization through LRP-1. Cell. Mol. Life. Sci. 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gilliam, A.; Maitra, R.; Damaj, M.I.; Tajuba, J.M.; Seltzman, H.H.; Thomas, B.F. Synthesis and biological evaluation of bivalent ligands for the cannabinoid 1 receptor. J. Med. Chem. 2010, 53, 7048–7060. [Google Scholar] [CrossRef] [PubMed]
- Decker, M. Homobivalent quinazolinimines as novel nanomolar inhibitors of cholinesterases with dirigible selectivity toward butyrylcholinesterase. J. Med. Chem. 2006, 49, 5411–5413. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Radomski, M.W.; Medina, C.; Gilmer, J.F. MMP inhibition by barbiturate homodimers. Bioorg. Med. Chem. Lett. 2013, 23, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Nuti, E.; Rosalia, L.; Cuffaro, D.; Camodeca, C.; Giacomelli, C.; Da Pozzo, E.; Tuccinardi, T.; Costa, B.; Antoni, C.; Vera, L.; et al. Bifunctional inhibitors as a new tool to reduce cancer cell invasion by impairing MMP-9 homodimerization. ACS Med. Chem. Lett. 2017, 8, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Rossello, A.; Nuti, E.; Catalani, M.P.; Carelli, P.; Orlandini, E.; Rapposelli, S.; Tuccinardi, T.; Atkinson, S.J.; Murphy, G.; Balsamo, A. A new development of matrix metalloproteinase inhibitors: Twin hydroxamic acids as potent inhibitors of MMPs. Bioorg. Med. Chem. Lett. 2005, 15, 2311–2314. [Google Scholar] [CrossRef]
- Van den Steen, P.E.; Van Aelst, I.; Hvidberg, V.; Piccard, H.; Fiten, P.; Jacobsen, C.; Moestrup, S.K.; Fry, S.; Royle, L.; Wormald, M.R.; et al. The hemopexin and O-glycosylated domains tune gelatinase B/MMP-9 bioavailability via inhibition and binding to cargo receptors. J. Biol. Chem. 2006, 281, 18626–18637. [Google Scholar] [CrossRef]
- Vandooren, J.; Geurts, N.; Martens, E.; Van den Steen, P.E.; De Jonghe, S.; Herdewijn, P.; Opdenakker, G. Gelatin degradation assay reveals MMP-9 inhibitors and function of O-glycosylated domain. World J. Biol. Chem. 2011, 2, 14–24. [Google Scholar] [CrossRef]
- De Buck, M.; Berghmans, N.; Pörtner, N.; Vanbrabant, L.; Cockx, M.; Struyf, S.; Opdenakker, G.; Proost, P.; Van Damme, J.; Gouwy, M. Serum amyloid A1alpha induces paracrine IL-8/CXCL8 via TLR2 and directly synergizes with this chemokine via CXCR2 and formyl peptide receptor 2 to recruit neutrophils. J. Leukoc. Biol. 2015, 98, 1049–1060. [Google Scholar] [CrossRef]
- Folkman, J.; Merler, E.; Abernathy, C.; Williams, G. Isolation of a tumor factor responsible for angiogenesis. J. Exp. Med. 1971, 133, 275–288. [Google Scholar] [CrossRef]
- Shrum, B.; Anantha, R.V.; Xu, S.X.; Donnelly, M.; Haeryfar, S.M.; McCormick, J.K.; Mele, T. A robust scoring system to evaluate sepsis severity in an animal model. BMC Res. Notes 2014, 7, 233. [Google Scholar] [CrossRef] [PubMed]
- Antoni, C.; Vera, L.; Devel, L.; Catalani, M.P.; Czarny, B.; Cassar-Lajeunesse, E.; Nuti, E.; Rossello, A.; Dive, V.; Stura, E.A. Crystallization of bi-functional ligand protein complexes. J. Struct. Biol. 2013, 182, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Rorvig, S.; Ostergaard, O.; Heegaard, N.H.; Borregaard, N. Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: Correlation with transcriptome profiling of neutrophil precursors. J. Leukoc. Biol. 2013, 94, 711–721. [Google Scholar] [CrossRef]
- Adrover, J.M.; Aroca-Crevillen, A.; Crainiciuc, G.; Ostos, F.; Rojas-Vega, Y.; Rubio-Ponce, A.; Cilloniz, C.; Bonzon-Kulichenko, E.; Calvo, E.; Rico, D.; et al. Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat. Immunol. 2020, 21, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Jin, S.L.; Milla, M.E.; Bickett, D.M.; Burkhart, W.; Carter, H.L.; Chen, W.J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef]
- Meissner, F.; Scheltema, R.A.; Mollenkopf, H.J.; Mann, M. Direct proteomic quantification of the secretome of activated immune cells. Science 2013, 340, 475–478. [Google Scholar] [CrossRef]
- Huang, W.C.; Sala-Newby, G.B.; Susana, A.; Johnson, J.L.; Newby, A.C. Classical macrophage activation up-regulates several matrix metalloproteinases through mitogen activated protein kinases and nuclear factor-kappaB. PLoS ONE 2012, 7, e42507. [Google Scholar]
- Li, Q.; Park, P.W.; Wilson, C.L.; Parks, W.C. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell 2002, 111, 635–646. [Google Scholar] [CrossRef]
- Okada, Y.; Takeuchi, N.; Tomita, K.; Nakanishi, I.; Nagase, H. Immunolocalization of matrix metalloproteinase 3 (stromelysin) in rheumatoid synovioblasts (B cells): Correlation with rheumatoid arthritis. Ann. Rheum. Dis. 1989, 48, 645–653. [Google Scholar] [CrossRef]
- Nerusu, K.C.; Warner, R.L.; Bhagavathula, N.; McClintock, S.D.; Johnson, K.J.; Varani, J. Matrix metalloproteinase-3 (stromelysin-1) in acute inflammatory tissue injury. Exp. Mol. Pathol. 2007, 83, 169–176. [Google Scholar] [CrossRef]
- Song, J.; Wu, C.; Zhang, X.; Sorokin, L.M. In vivo processing of CXCL5 (LIX) by matrix metalloproteinase (MMP)-2 and MMP-9 promotes early neutrophil recruitment in IL-1beta-induced peritonitis. J. Immunol. 2013, 190, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Van den Steen, P.E.; Proost, P.; Wuyts, A.; Van Damme, J.; Opdenakker, G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood 2000, 96, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Lalu, M.M.; Gao, C.Q.; Schulz, R. Matrix metalloproteinase inhibitors attenuate endotoxemia induced cardiac dysfunction: A potential role for MMP-9. Mol. Cell. Biochem. 2003, 251, 61–66. [Google Scholar] [CrossRef]
- Shapira, L.; Soskolne, W.A.; Houri, Y.; Barak, V.; Halabi, A.; Stabholz, A. Protection against endotoxic shock and lipopolysaccharide-induced local inflammation by tetracycline: Correlation with inhibition of cytokine secretion. Infect. Immun. 1996, 64, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Solorzano, C.C.; Ksontini, R.; Pruitt, J.H.; Auffenberg, T.; Tannahill, C.; Galardy, R.E.; Schultz, G.P.; MacKay, S.L.; Copeland, E.M.; Moldawer, L.L. A matrix metalloproteinase inhibitor prevents processing of tumor necrosis factor alpha (TNF alpha) and abrogates endotoxin-induced lethality. Shock 1997, 7, 427–431. [Google Scholar] [CrossRef]
- Pagenstecher, A.; Stalder, A.K.; Kincaid, C.L.; Volk, B.; Campbell, I.L. Regulation of matrix metalloproteinases and their inhibitor genes in lipopolysaccharide-induced endotoxemia in mice. Am. J. Pathol. 2000, 157, 197–210. [Google Scholar] [CrossRef]
- Opdenakker, G.; Van den Steen, P.E.; Dubois, B.; Nelissen, I.; Van Coillie, E.; Masure, S.; Proost, P.; Van Damme, J. Gelatinase B functions as regulator and effector in leukocyte biology. J. Leukoc. Biol. 2001, 69, 851–859. [Google Scholar]
- Tester, A.M.; Cox, J.H.; Connor, A.R.; Starr, A.E.; Dean, R.A.; Puente, X.S.; Lopez-Otin, C.; Overall, C.M. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS ONE 2007, 2, e312. [Google Scholar] [CrossRef]
- Fingleton, B. Matrix metalloproteinases as regulators of inflammatory processes. Biochim. Biophys. Acta. Mol. Cell. Res. 2017, 1864, 2036–2042. [Google Scholar] [CrossRef]
- Iyer, R.P.; de Castro Bras, L.E.; Patterson, N.L.; Bhowmick, M.; Flynn, E.R.; Asher, M.; Cannon, P.L.; Deleon-Pennell, K.Y.; Fields, G.B.; Lindsey, M.L. Early matrix metalloproteinase-9 inhibition post-myocardial infarction worsens cardiac dysfunction by delaying inflammation resolution. J. Mol. Cell. Cardiol. 2016, 100, 109–117. [Google Scholar] [CrossRef]
- Ducharme, A.; Frantz, S.; Aikawa, M.; Rabkin, E.; Lindsey, M.; Rohde, L.E.; Schoen, F.J.; Kelly, R.A.; Werb, Z.; Libby, P.; et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Investig. 2000, 106, 55–62. [Google Scholar] [CrossRef]
- Zajac, E.; Schweighofer, B.; Kupriyanova, T.A.; Juncker-Jensen, A.; Minder, P.; Quigley, J.P.; Deryugina, E.I. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood 2013, 122, 4054–4067. [Google Scholar] [CrossRef]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MMP-9 Preparation/Substrate | MMP-9 Form | Monovalent Inhibitor (Compound 5) | Bivalent Inhibitor (Compound 7) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Model | n | IC25 | IC50 | IC75 | Model | n | IC25 | IC50 | IC75 | ||
| (R2) | (CI) (nM) | (CI) (nM) | (CI) (nM) | (R2) | (CI) (nM) | (CI) (nM) | (CI) (nM) | ||||
| Recombinant 1/gelatin degradation | mix 4 | dose response | 7 | 359 | 1407 | 5509 | biphasic | 9 | 28 | 182 | 627 |
| (0.98) | (325–396) | (1307–1517) | (4879–6260) | (0.97) | (19–38) | (158–207) | (539–740) | ||||
| mono | dose response | 7 | 392 | 1516 | 5859 | dose response | 10 | 48 | 191 | 754 | |
| (0.96) | (339–449) | (1369–1683) | (4951–7024) | (0.92) | (39–58) | (166–220) | (612–946) | ||||
| tri | dose response | 7 | 526 | 2253 | 9660 | biphasic | 10 | 0.03 | 0.18 | 18 | |
| (0.97) | (466–589) | (2058–2477) | (8228–NA) | (0.93) | (0.019–0.027) | (0.14–0.24) | (5–42) | ||||
| Recombinant 1/peptide degradation 2 | mix 4 | dose response | 4 | 228 | 726 | 2309 | dose response | 6 | 1.4 | 15 | 169 |
| (0.99) | (209–248) | (682–773) | (2097–2553) | (0.94) | (1.0–1.9) | (12–19) | (125–230) | ||||
| mono | dose response | 4 | 232 | 718 | 2228 | dose response | 4 | 10.5 | 56 | 296 | |
| (0.99) | (220–244) | (693–745) | (2109–2357) | (0.96) | (7.7–14.2) | (45–69) | (225–397) | ||||
| tri | dose response | 4 | 237 | 724 | 2211 | dose response | 4 | 0.0017 | 0.10 | 6.0 | |
| (0.99) | (223–252) | (693–757) | (2070–2366) | (0.82) | (0.0006–0.0046) | (0.05–0.18) | (3.1–12.5) | ||||
| neutrophil-derived 3/gelatin degradation | mono | dose response | 2 | 300 | 1154 | 4431 | dose response | 3 | 10 | 43 | 181 |
| (0.97) | (259–345) | (1026–1312) | (NA–NA) | (0.96) | (8–13) | (35–52) | (139–240) | ||||
| tri | dose response | 2 | 479 | 4223 | 17212 | biphasic | 4 | 0.008 | 0.066 | 0.6 | |
| (0.95) | (398–577) | (NA-NA) | (NA–NA) | (0.93) | (NA–0.013) | (0.05–0.09) | (0.4–1.1) | ||||
| Proteases | Assay Substrate | Monovalent Inhibitor (Compound 5) | Bivalent Inhibitor (Compound 7) | ||||
|---|---|---|---|---|---|---|---|
| Model | n | IC50 | Model | n | IC50 | ||
| (R2) | (CI) (nM) | (R2) | (CI) (nM) | ||||
| MMP-9 monomers | Peptide 1 | dose response | 4 | 718 | dose response | 4 | 56 |
| (0.99) | (693–745) | (0.96) | (45–69) | ||||
| MMP-9 trimers | Peptide 1 | dose response | 4 | 724 | dose response | 4 | 0.10 |
| (0.99) | (693–757) | (0.82) | (0.05–0.18) | ||||
| neutrophil elastase | Peptide 2 and gelatin | NA | 3 | no inhibition † | NA | 3 | no inhibition † |
| TACE/ADAM17 | Peptide 3 | NA | 2 | no inhibition † | NA | 2 | no inhibition † |
| MMP-8/neutrophil collagenase | Peptide 1 | dose response | 5 | 331 | dose response | 5 | 14.5 |
| (0.94) | (288–385) | (0.96) | (12.5–16.7) | ||||
| MMP-14/MT1-MMP | Peptide 1 | dose response | 4 | 14795 | dose response | 4 | 1076 |
| (0.91) | (12048–18437) | (0.92) | (885–1311) | ||||
| MMP-7 | Peptide 1 | dose response | 4 | > 10000 | dose response | 4 | >10000 |
| (NA) | (NA–NA) | (0.29) | (NA–NA) | ||||
| MMP-3 | Peptide 1 | dose response | 4 | 965 | dose response | 4 | 7.7 |
| (0.86) | (753–1225) | (0.96) | (6.4–9.1) | ||||
| MMP-2 | gelatin | dose response | 5 | 1724 | dose response | 5 | 4.8 |
| (0.97) | (1529–1953) | (0.99) | (4.3–5.4) | ||||
| Peptide 1 | dose response | 6 | 1029 | dose response | 6 | 5.0 | |
| (0.91) | (869–1238) | (0.97) | (4.4–5.6) | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuti, E.; Rossello, A.; Cuffaro, D.; Camodeca, C.; Van Bael, J.; van der Maat, D.; Martens, E.; Fiten, P.; Pereira, R.V.S.; Ugarte-Berzal, E.; et al. Bivalent Inhibitor with Selectivity for Trimeric MMP-9 Amplifies Neutrophil Chemotaxis and Enables Functional Studies on MMP-9 Proteoforms. Cells 2020, 9, 1634. https://doi.org/10.3390/cells9071634
Nuti E, Rossello A, Cuffaro D, Camodeca C, Van Bael J, van der Maat D, Martens E, Fiten P, Pereira RVS, Ugarte-Berzal E, et al. Bivalent Inhibitor with Selectivity for Trimeric MMP-9 Amplifies Neutrophil Chemotaxis and Enables Functional Studies on MMP-9 Proteoforms. Cells. 2020; 9(7):1634. https://doi.org/10.3390/cells9071634
Chicago/Turabian StyleNuti, Elisa, Armando Rossello, Doretta Cuffaro, Caterina Camodeca, Jens Van Bael, Dries van der Maat, Erik Martens, Pierre Fiten, Rafaela Vaz Sousa Pereira, Estefania Ugarte-Berzal, and et al. 2020. "Bivalent Inhibitor with Selectivity for Trimeric MMP-9 Amplifies Neutrophil Chemotaxis and Enables Functional Studies on MMP-9 Proteoforms" Cells 9, no. 7: 1634. https://doi.org/10.3390/cells9071634
APA StyleNuti, E., Rossello, A., Cuffaro, D., Camodeca, C., Van Bael, J., van der Maat, D., Martens, E., Fiten, P., Pereira, R. V. S., Ugarte-Berzal, E., Gouwy, M., Opdenakker, G., & Vandooren, J. (2020). Bivalent Inhibitor with Selectivity for Trimeric MMP-9 Amplifies Neutrophil Chemotaxis and Enables Functional Studies on MMP-9 Proteoforms. Cells, 9(7), 1634. https://doi.org/10.3390/cells9071634