Cell-Type Targeted NF-kappaB Inhibition for the Treatment of Inflammatory Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Relevance of NF-kappaB in Chronic Inflammatory Diseases
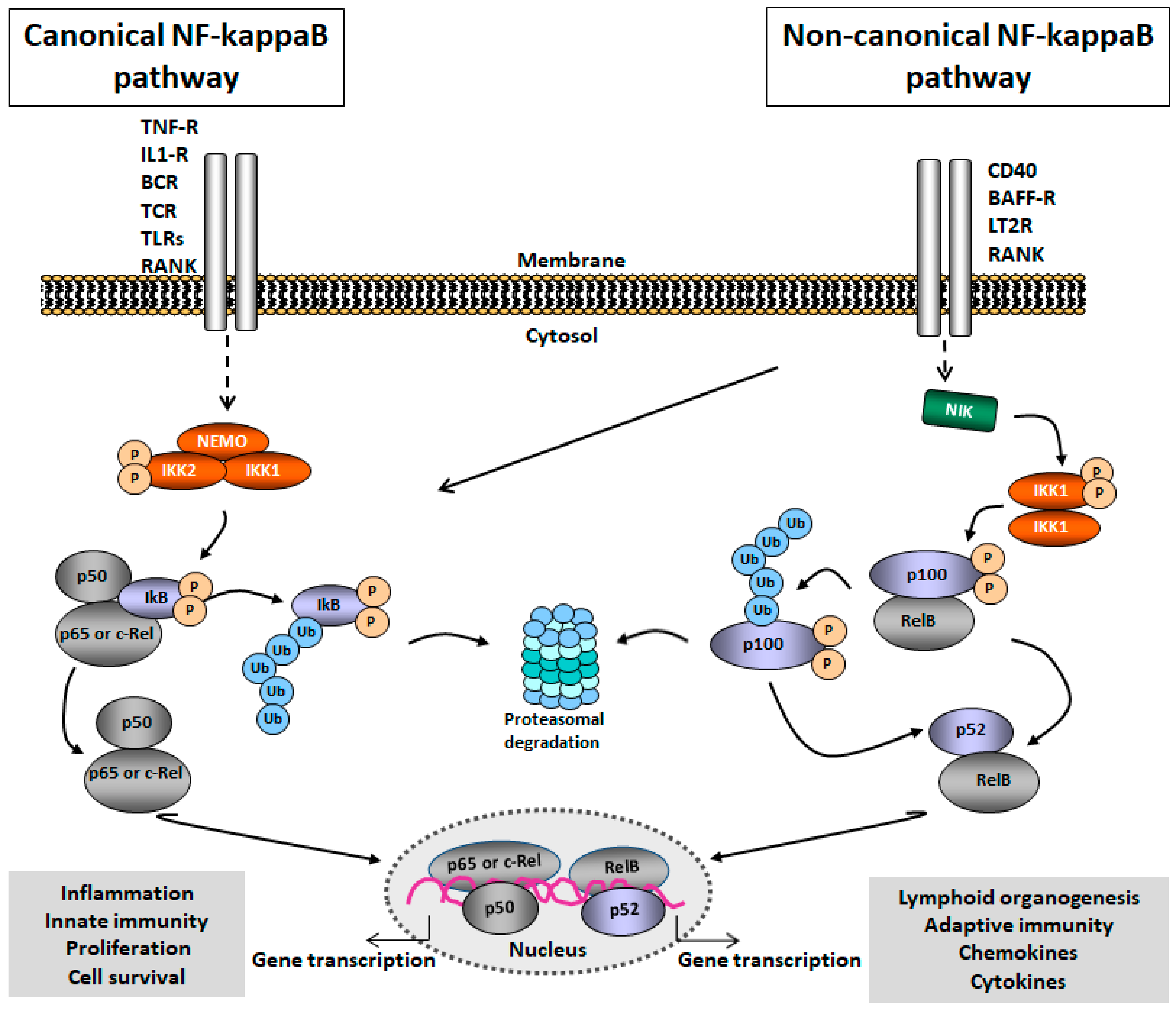
2.1. NF-kappaB Signaling
2.2. Role of NF-kappaB in RA
2.3. Role of NF-KappaB in MS
3. Delivery of Substances into the Cell’s Interior
3.1. Passive Targeting
3.2. Active Targeting
4. Approaches for NF-kappaB Inhibition
4.1. NF-kappaB Inhibition by NSAIDs and GC’s
4.2. Other Inhibitors of NF-kappaB
5. The “Sneaking Ligand” (SL) Approach for NF-kappaB Inhibition
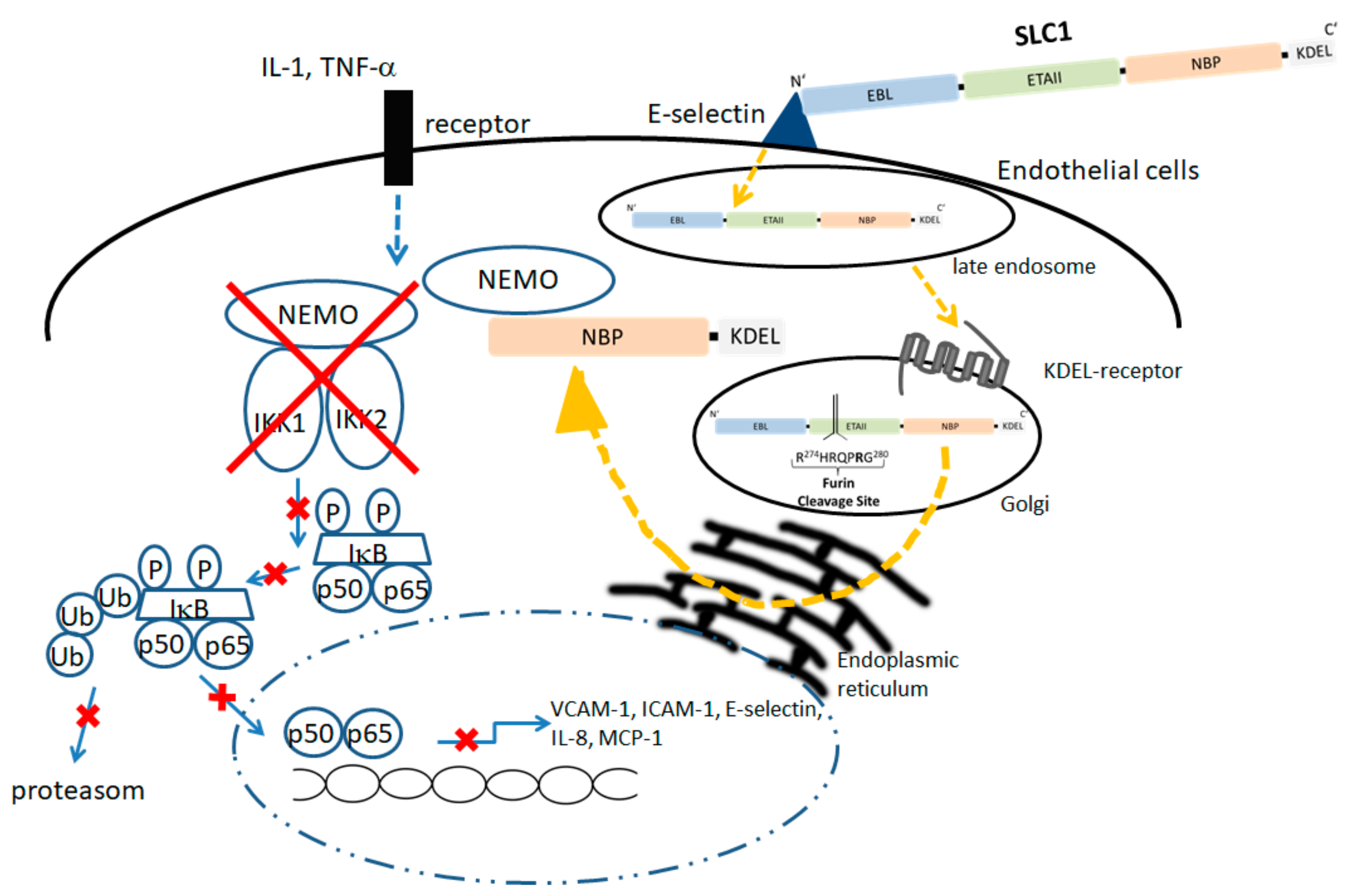
5.1. Modular Structure and Mode of Action of an Endothelium-Specific NF-KappaB Inhibitor
5.2. Biological Effects of SLC1 In Vitro and Vivo
5.3. Outlook for the Application of SCL1
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Pires, B.R.B.; Silva, R.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two Sides of the Same Coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Yue, Y.; Stone, S.; Lin, W. Role of nuclear factor kappaB in multiple sclerosis and experimental autoimmune encephalomyelitis. Neural. Regen. Res. 2018, 13, 1507–1515. [Google Scholar] [CrossRef]
- Simmonds, R.E.; Foxwell, B.M. Signalling, inflammation and arthritis: NF-kappaB and its relevance to arthritis and inflammation. Rheumatology 2008, 47, 584–590. [Google Scholar] [CrossRef]
- Makarov, S.S. NF-kappa B in rheumatoid arthritis: A pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthr. Res. 2001, 3, 200–206. [Google Scholar] [CrossRef]
- Miagkov, A.V.; Kovalenko, D.V.; Brown, C.E.; Didsbury, J.R.; Cogswell, J.P.; Stimpson, S.A.; Baldwin, A.S.; Makarov, S.S. NF-kappaB activation provides the potential link between inflammation and hyperplasia in the arthritic joint. Proc. Natl. Acad. Sci. USA 1998, 95, 13859–13864. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Amos, C.I.; Lee, A.T.; Lu, Y.; Remmers, E.F.; Kastner, D.L.; Seldin, M.F.; Criswell, L.A.; Plenge, R.M.; Holers, V.M.; et al. REL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat. Genet. 2009, 41, 820–823. [Google Scholar] [CrossRef]
- Mc Guire, C.; Prinz, M.; Beyaert, R.; van Loo, G. Nuclear factor kappa B (NF-kappaB) in multiple sclerosis pathology. Trends Mol. Med. 2013, 19, 604–613. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, H.; Liang, L.; Zhan, Z.; Yang, X.; Yu, X.; Ye, Y.; Sun, L. Antiinflammatory effect of Rho kinase blockade via inhibition of NF-kappaB activation in rheumatoid arthritis. Arthr. Rheum. 2008, 58, 3366–3376. [Google Scholar] [CrossRef]
- Jimi, E.; Aoki, K.; Saito, H.; D’Acquisto, F.; May, M.J.; Nakamura, I.; Sudo, T.; Kojima, T.; Okamoto, F.; Fukushima, H.; et al. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat. Med. 2004, 10, 617–624. [Google Scholar] [CrossRef] [PubMed]
- May, M.J.; D’Acquisto, F.; Madge, L.A.; Glockner, J.; Pober, J.S.; Ghosh, S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science 2000, 289, 1550–1554. [Google Scholar] [CrossRef] [PubMed]
- Voll, R.E.; Mikulowska, A.; Kalden, J.R.; Holmdahl, R. Amelioration of type II collagen induced arthritis in rats by treatment with sodium diethyldithiocarbamate. J. Rheumatol. 1999, 26, 1352–1358. [Google Scholar]
- Yamamoto, Y.; Gaynor, R.B. Role of the NF-kappaB pathway in the pathogenesis of human disease states. Curr. Mol. Med. 2001, 1, 287–296. [Google Scholar] [CrossRef]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef]
- Senftleben, U. Anti-inflammatory interventions of NF-kappaB signaling: Potential applications and risks. Biochem. Pharmacol. 2008, 75, 1567–1579. [Google Scholar]
- Yamamoto, Y.; Gaynor, R.B. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J. Clin. Investig. 2001, 107, 135–142. [Google Scholar] [CrossRef]
- Pikarsky, E.; Ben-Neriah, Y. NF-kappaB inhibition: A double-edged sword in cancer? Eur. J. Cancer 2006, 42, 779–784. [Google Scholar] [CrossRef]
- Kanters, E.; Pasparakis, M.; Gijbels, M.J.; Vergouwe, M.N.; Partouns-Hendriks, I.; Fijneman, R.J.; Clausen, B.E.; Forster, I.; Kockx, M.M.; Rajewsky, K.; et al. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J. Clin. Investig. 2003, 112, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Gilroy, D.W.; Colville-Nash, P.R.; Willoughby, D.A. Possible new role for NF-kappaB in the resolution of inflammation. Nat. Med. 2001, 7, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Sehnert, B.; Burkhardt, H.; Wessels, J.T.; Schroder, A.; May, M.J.; Vestweber, D.; Zwerina, J.; Warnatz, K.; Nimmerjahn, F.; Schett, G.; et al. NF-kappaB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-kappaB in immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 16556–16561. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Sung, B. NF-kappaB in cancer: A matter of life and death. Cancer Discov. 2011, 1, 469–471. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-kappaB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef]
- Noort, A.R.; Tak, P.P.; Tas, S.W. Non-canonical NF-kappaB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde? Arthr. Res. Ther. 2015, 17, 15. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-kappaB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. How NF-kappaB is activated: The role of the IkappaB kinase (IKK) complex. Oncogene 1999, 18, 6867–6874. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.R.; Bradley, R.; Ganju, R.K. LPS-induced MCP-1 expression in human microvascular endothelial cells is mediated by the tyrosine kinase, Pyk2 via the p38 MAPK/NF-kappaB-dependent pathway. Mol. Immunol. 2009, 46, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995, 9, 899–909. [Google Scholar] [CrossRef]
- Dong, J.; Jimi, E.; Zeiss, C.; Hayden, M.S.; Ghosh, S. Constitutively active NF-kappaB triggers systemic TNFalpha-dependent inflammation and localized TNFalpha-independent inflammatory disease. Genes Dev. 2010, 24, 1709–1717. [Google Scholar] [CrossRef]
- Monaco, C.; Andreakos, E.; Kiriakidis, S.; Mauri, C.; Bicknell, C.; Foxwell, B.; Cheshire, N.; Paleolog, E.; Feldmann, M. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 5634–5639. [Google Scholar] [CrossRef]
- Mishra, V.; Banga, J.; Silveyra, P. Oxidative stress and cellular pathways of asthma and inflammation: Therapeutic strategies and pharmacological targets. Pharmacol. Ther. 2018, 181, 169–182. [Google Scholar] [CrossRef]
- Fischer, S.; Rath, T.; Neurath, M.F. Inflammatory bowel diseases: Crohn’s disease and ulcerative colitis. Internist 2018, 59, 681–693. [Google Scholar] [CrossRef]
- Deane, K.D.; Demoruelle, M.K.; Kelmenson, L.B.; Kuhn, K.A.; Norris, J.M.; Holers, V.M. Genetic and environmental risk factors for rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 3–18. [Google Scholar] [CrossRef]
- Glant, T.T.; Mikecz, K.; Rauch, T.A. Epigenetics in the pathogenesis of rheumatoid arthritis. BMC Med. 2014, 12, 35. [Google Scholar] [CrossRef]
- Li, S.; Yu, Y.; Yue, Y.; Zhang, Z.; Su, K. Microbial Infection and Rheumatoid Arthritis. J. Clin. Cell Immunol. 2013, 4. [Google Scholar] [CrossRef]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Muller-Ladner, U.; Pap, T.; Gay, R.E.; Neidhart, M.; Gay, S. Mechanisms of disease: The molecular and cellular basis of joint destruction in rheumatoid arthritis. Nat. Clin. Pract. Rheumatol. 2005, 1, 102–110. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Kurowska-Stolarska, M.; Alivernini, S. Synovial tissue macrophages: Friend or foe? RMD Open 2017, 3, e000527. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.N.; Lundy, S.K.; Fox, D.A. Synovial biology and T cells in rheumatoid arthritis. Pathophysiology 2005, 12, 183–189. [Google Scholar] [CrossRef]
- Zabel, U.; Henkel, T.; Silva, M.S.; Baeuerle, P.A. Nuclear uptake control of NF-kappa B by MAD-3, an I kappa B protein present in the nucleus. EMBO J. 1993, 12, 201–211. [Google Scholar] [CrossRef]
- Gilston, V.; Jones, H.W.; Soo, C.C.; Coumbe, A.; Blades, S.; Kaltschmidt, C.; Baeuerle, P.A.; Morris, C.J.; Blake, D.R.; Winyard, P.G. NF-kappa B activation in human knee-joint synovial tissue during the early stage of joint inflammation. Biochem. Soc. Trans. 1997, 25, 518. [Google Scholar] [CrossRef]
- Handel, M.L.; McMorrow, L.B.; Gravallese, E.M. Nuclear factor-kappa B in rheumatoid synovium: Localization of p50 and p65. Arthr. Rheum. 1995, 38, 1762–1770. [Google Scholar] [CrossRef]
- Caplazi, P.; Baca, M.; Barck, K.; Carano, R.A.; DeVoss, J.; Lee, W.P.; Bolon, B.; Diehl, L. Mouse Models of Rheumatoid Arthritis. Vet. Pathol. 2015, 52, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. AP-1 and NF-kappaB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity 1998, 28, 197–208. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Martinez-Munoz, L.; Cascio, G.; Lucas, P.; Pablos, J.L.; Rodriguez-Frade, J.M. T Cell Migration in Rheumatoid Arthritis. Front. Immunol. 2015, 6, 384. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xiu, Y.; Li, J.; Xing, L.; Yao, Z. NF-kappaB-Mediated Regulation of Osteoclastogenesis. Endocrinol. Metab. 2015, 30, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Takakura, N.; Matsuda, M.; Khan, M.; Hiura, F.; Aoki, K.; Hirohashi, Y.; Mori, K.; Yasuda, H.; Hirata, M.; Kitamura, C.; et al. A novel inhibitor of NF-kappaB-inducing kinase prevents bone loss by inhibiting osteoclastic bone resorption in ovariectomized mice. Bone 2020, 135, 115316. [Google Scholar] [CrossRef]
- Nakamura, H.; Aoki, K.; Masuda, W.; Alles, N.; Nagano, K.; Fukushima, H.; Osawa, K.; Yasuda, H.; Nakamura, I.; Mikuni-Takagaki, Y.; et al. Disruption of NF-kappaB1 prevents bone loss caused by mechanical unloading. J. Bone Miner. Res. 2013, 28, 1457–1467. [Google Scholar] [CrossRef]
- Bosello, S.; Youinou, P.; Daridon, C.; Tolusso, B.; Bendaoud, B.; Pietrapertosa, D.; Morelli, A.; Ferraccioli, G. Concentrations of BAFF correlate with autoantibody levels, clinical disease activity, and response to treatment in early rheumatoid arthritis. J. Rheumatol. 2008, 35, 1256–1264. [Google Scholar]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef]
- Smulski, C.R.; Eibel, H. BAFF and BAFF-Receptor in B Cell Selection and Survival. Front. Immunol. 2018, 9, 2285. [Google Scholar] [CrossRef]
- Thompson, N.; Isenberg, D.A.; Jury, E.C.; Ciurtin, C. Exploring BAFF: Its expression, receptors and contribution to the immunopathogenesis of Sjogren’s syndrome. Rheumatology 2016, 55, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Chang, Y.; Wei, W. The role of BAFF in the progression of rheumatoid arthritis. Cytokine 2015, 76, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.J.; Yoon, B.Y.; Jhun, J.Y.; Oh, H.J.; Min, S.W.; Cho, M.L.; Park, S.H.; Kim, H.Y.; Min, J.K. Regulation of B cell activating factor (BAFF) receptor expression by NF-KappaB signaling in rheumatoid arthritis B cells. Exp. Mol. Med. 2011, 43, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Aupperle, K.R.; Bennett, B.L.; Boyle, D.L.; Tak, P.P.; Manning, A.M.; Firestein, G.S. NF-kappa B regulation by I kappa B kinase in primary fibroblast-like synoviocytes. J. Immunol. 1999, 163, 427–433. [Google Scholar]
- Tak, P.P.; Gerlag, D.M.; Aupperle, K.R.; van de Geest, D.A.; Overbeek, M.; Bennett, B.L.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. Inhibitor of nuclear factor kappaB kinase beta is a key regulator of synovial inflammation. Arthr. Rheum. 2001, 44, 1897–1907. [Google Scholar] [CrossRef]
- Kumar, D.R.; Aslinia, F.; Yale, S.H.; Mazza, J.J. Jean-Martin Charcot: The father of neurology. Clin. Med. Res. 2011, 9, 46–49. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2010, 24, 641–651. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef]
- Oh, H.; Ghosh, S. NF-kappaB: Roles and regulation in different CD4(+) T-cell subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef]
- Ransohoff, R.M. Animal models of multiple sclerosis: The good, the bad and the bottom line. Nat. Neurosci. 2012, 15, 1074–1077. [Google Scholar] [CrossRef]
- Greve, B.; Weissert, R.; Hamdi, N.; Bettelli, E.; Sobel, R.A.; Coyle, A.; Kuchroo, V.K.; Rajewsky, K.; Schmidt-Supprian, M. I kappa B kinase 2/beta deficiency controls expansion of autoreactive T cells and suppresses experimental autoimmune encephalomyelitis. J. Immunol. 2007, 179, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, G.; De Lorenzi, R.; Schmidt, H.; Huth, M.; Mildner, A.; Schmidt-Supprian, M.; Lassmann, H.; Prinz, M.R.; Pasparakis, M. Inhibition of transcription factor NF-kappaB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat. Immunol. 2006, 7, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Zhou, X.F.; Yu, J.; Cheng, X.; Sun, S.C. Regulation of Th17 cell differentiation and EAE induction by MAP3K NIK. Blood 2009, 113, 6603–6610. [Google Scholar] [CrossRef] [PubMed]
- Ellrichmann, G.; Thone, J.; Lee, D.H.; Rupec, R.A.; Gold, R.; Linker, R.A. Constitutive activity of NF-kappa B in myeloid cells drives pathogenicity of monocytes and macrophages during autoimmune neuroinflammation. J. Neuroinflamm. 2012, 9, 15. [Google Scholar] [CrossRef]
- Lee, M.J.; Bing, S.J.; Choi, J.; Jang, M.; Lee, G.; Lee, H.; Chang, B.S.; Jee, Y.; Lee, S.J.; Cho, I.H. IKKbeta-mediated inflammatory myeloid cell activation exacerbates experimental autoimmune encephalomyelitis by potentiating Th1/Th17 cell activation and compromising blood brain barrier. Mol. Neurodegen. 2016, 11, 54. [Google Scholar] [CrossRef]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Brambilla, R.; Persaud, T.; Hu, X.; Karmally, S.; Shestopalov, V.I.; Dvoriantchikova, G.; Ivanov, D.; Nathanson, L.; Barnum, S.R.; Bethea, J.R. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J. Immunol. 2009, 182, 2628–2640. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Dvoriantchikova, G.; Barakat, D.; Ivanov, D.; Bethea, J.R.; Shestopalov, V.I. Transgenic inhibition of astroglial NF-kappaB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. J. Neuroinflamm. 2012, 9, 213. [Google Scholar] [CrossRef] [PubMed]
- Shao, X. Protective Role of NF-kappaB in Inflammatory Demyelination. J. Neurosci. 2018, 38, 2416–2417. [Google Scholar] [CrossRef] [PubMed]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: A review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Di Natale, C.; Florio, D.; Marasco, D. Peptides as Therapeutic Agents for Inflammatory-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2714. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.J.; Hinner, M.J. Getting across the cell membrane: An overview for small molecules, peptides, and proteins. Methods Mol. Biol. 2015, 1266, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qin, X.; Kong, F.; Chen, P.; Pan, G. Improving cellular uptake of therapeutic entities through interaction with components of cell membrane. Drug Deliv. 2019, 26, 328–342. [Google Scholar] [CrossRef]
- Crommelin, D.J.; Storm, G.; Jiskoot, W.; Stenekes, R.; Mastrobattista, E.; Hennink, W.E. Nanotechnological approaches for the delivery of macromolecules. J. Control. Release 2003, 87, 81–88. [Google Scholar] [CrossRef]
- Oku, N. Innovations in Liposomal DDS Technology and Its Application for the Treatment of Various Diseases. Biol. Pharm. Bull. 2017, 40, 119–127. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Rahman, M.; Beg, S.; Anwar, F.; Kumar, V.; Ubale, R.; Addo, R.T.; Ali, R.; Akhter, S. Liposome-Based Nanomedicine Therapeutics for Rheumatoid Arthritis. Crit. Rev. Ther. Drug Carr. Syst. 2017, 34, 283–316. [Google Scholar] [CrossRef]
- Fischer, P.M.; Krausz, E.; Lane, D.P. Cellular delivery of impermeable effector molecules in the form of conjugates with peptides capable of mediating membrane translocation. Bioconjug. Chem. 2001, 12, 825–841. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.P.; Arami, H.; Banga, I.; Gupta, J.; Gandhi, S. Cell penetrating peptides in preclinical and clinical cancer diagnosis and therapy. Oncotarget 2018, 9, 37252–37267. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Wadia, J.S.; Dowdy, S.F. Modulation of cellular function by TAT mediated transduction of full length proteins. Curr. Protein Pept. Sci. 2003, 4, 97–104. [Google Scholar] [CrossRef]
- Lindsay, M.A. Peptide-mediated cell delivery: Application in protein target validation. Curr. Opin. Pharmacol. 2002, 2, 587–594. [Google Scholar] [CrossRef]
- Maiolo, J.R.; Ferrer, M.; Ottinger, E.A. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim. Biophys. Acta 2005, 1712, 161–172. [Google Scholar] [CrossRef]
- Habault, J.; Poyet, J.L. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef]
- Jia, L.; Gorman, G.S.; Coward, L.U.; Noker, P.E.; McCormick, D.; Horn, T.L.; Harder, J.B.; Muzzio, M.; Prabhakar, B.; Ganesh, B.; et al. Preclinical pharmacokinetics, metabolism, and toxicity of azurin-p28 (NSC745104) a peptide inhibitor of p53 ubiquitination. Cancer Chem. Pharmacol. 2011, 68, 513–524. [Google Scholar] [CrossRef]
- Warso, M.A.; Richards, J.M.; Mehta, D.; Christov, K.; Schaeffer, C.; Rae Bressler, L.; Yamada, T.; Majumdar, D.; Kennedy, S.A.; Beattie, C.W.; et al. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br. J. Cancer 2013, 108, 1061–1070. [Google Scholar] [CrossRef]
- Hong, K.; Kirpotin, D.B.; Park, J.W.; Shao, Y.; Shalaby, R.; Colbern, G.; Benz, C.C.; Papahadjopoulos, D. Anti-HER2 immunoliposomes for targeted drug delivery. Ann. N. Y. Acad. Sci. 1999, 886, 293–296. [Google Scholar] [CrossRef]
- Paoli, E.E.; Ingham, E.S.; Zhang, H.; Mahakian, L.M.; Fite, B.Z.; Gagnon, M.K.; Tam, S.; Kheirolomoom, A.; Cardiff, R.D.; Ferrara, K.W. Accumulation, internalization and therapeutic efficacy of neuropilin-1-targeted liposomes. J. Control. Release 2014, 178, 108–117. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Allured, V.S.; Collier, R.J.; Carroll, S.F.; McKay, D.B. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc. Natl. Acad. Sci. USA 1986, 83, 1320–1324. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.; Kantardjieff, K.A.; Collier, R.J.; Eisenberg, D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Menestrina, G.; Schiavo, G.; Montecucco, C. Molecular mechanisms of action of bacterial protein toxins. Mol. Asp. Med. 1994, 15, 79–193. [Google Scholar] [CrossRef]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: Optimized by evolution for effective killing. Front. Microbiol. 2015, 6, 963. [Google Scholar] [CrossRef]
- Shapira, A.; Benhar, I. Toxin-based therapeutic approaches. Toxins 2010, 2, 2519–2583. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Harder, S.; Hovelmann, S.; Jager, E.; Al-Batran, S.E.; Loibl, S.; Atmaca, A.; Cimpoiasu, C.; Neumann, A.; Abera, A.; et al. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res. 2005, 7, R617–R626. [Google Scholar] [CrossRef] [PubMed]
- Allahyari, H.; Heidari, S.; Ghamgosha, M.; Saffarian, P.; Amani, J. Immunotoxin: A new tool for cancer therapy. Tumour Biol. 2017, 39, 1010428317692226. [Google Scholar] [CrossRef] [PubMed]
- Hessler, J.L.; Kreitman, R.J. An early step in Pseudomonas exotoxin action is removal of the terminal lysine residue, which allows binding to the KDEL receptor. Biochemistry 1997, 36, 14577–14582. [Google Scholar] [CrossRef]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin-recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef]
- Elkin, S.R.; Lakoduk, A.M.; Schmid, S.L. Endocytic pathways and endosomal trafficking: A primer. Wien. Med. Wochenschr. 2016, 166, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Hristodorov, D.; Mladenov, R.; Huhn, M.; Barth, S.; Thepen, T. Macrophage-targeted therapy: CD64-based immunotoxins for treatment of chronic inflammatory diseases. Toxins 2012, 4, 676–694. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Kung, C.C.; Dai, S.P.; Chiang, H.; Huang, H.S.; Sun, W.H. Temporal expression patterns of distinct cytokines and M1/M2 macrophage polarization regulate rheumatoid arthritis progression. Mol. Biol. Rep. 2020, 47, 3423–3437. [Google Scholar] [CrossRef]
- Tardito, S.; Martinelli, G.; Soldano, S.; Paolino, S.; Pacini, G.; Patane, M.; Alessandri, E.; Smith, V.; Cutolo, M. Macrophage M1/M2 polarization and rheumatoid arthritis: A systematic review. Autoimmun. Rev. 2019. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Schwager, K.; Kaspar, M.; Bootz, F.; Marcolongo, R.; Paresce, E.; Neri, D.; Trachsel, E. Preclinical characterization of DEKAVIL (F8-IL10), a novel clinical-stage immunocytokine which inhibits the progression of collagen-induced arthritis. Arthr. Res. Ther. 2009, 11, 142. [Google Scholar] [CrossRef]
- Galeazzi, M.; Bazzichi, L.; Sebastiani, G.D.; Neri, D.; Garcia, E.; Ravenni, N.; Giovannoni, L.; Wilton, J.; Bardelli, M.; Baldi, C.; et al. A phase IB clinical trial with Dekavil (F8-IL10), an immunoregulatory ‘armed antibody’ for the treatment of rheumatoid arthritis, used in combination wiIh methotrexate. Isr. Med. Assoc. J. 2014, 16, 666. [Google Scholar]
- Murer, P.; Neri, D. Antibody-cytokine fusion proteins: A novel class of biopharmaceuticals for the therapy of cancer and of chronic inflammation. New Biotechnol. 2019, 52, 42–53. [Google Scholar] [CrossRef]
- Neri, D. Antibody-Cytokine Fusions: Versatile Products for the Modulation of Anticancer Immunity. Cancer Immunol. Res. 2019, 7, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Sehnert, B.; Burkhardt, H.; Dubel, S.; Voll, R.E. The “sneaking-ligand” approach: Cell-type specific inhibition of the classical NF-kappaB pathway. Methods Mol. Biol. 2015, 1280, 559–578. [Google Scholar] [CrossRef] [PubMed]
- Sehnert, B.; Burkhardt, H.; May, M.J.; Zwerina, J.; Voll, R.E. Sneaking-ligand fusion proteins attenuate serum transfer arthritis by endothelium-targeted NF-kappaB inhibition. Methods Mol. Biol. 2015, 1280, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Luedde, T.; Schmidt-Supprian, M. Dissection of the NF-kappaB signalling cascade in transgenic and knockout mice. Cell Death Differ. 2006, 13, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Grumont, R.; Gugasyan, R.; Wong, L.; Isomura, I.; Ho, W.; Banerjee, A. Unravelling the complexities of the NF-kappaB signalling pathway using mouse knockout and transgenic models. Oncogene 2006, 25, 6781–6799. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef]
- Herrington, F.D.; Carmody, R.J.; Goodyear, C.S. Modulation of NF-kappaB Signaling as a Therapeutic Target in Autoimmunity. J. Biomol. Screen 2016, 21, 223–242. [Google Scholar] [CrossRef]
- D’Acquisto, F.; May, M.J.; Ghosh, S. Inhibition of nuclear factor kappa B (NF-B): An emerging theme in anti-inflammatory therapies. Mol. Interv. 2002, 2, 22–35. [Google Scholar] [CrossRef]
- Park, M.H.; Hong, J.T. Roles of NF-kappaB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Prescott, J.A.; Cook, S.J. Targeting IKKbeta in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKbeta Inhibitors. Cells 2018, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.; Marschenz, S.; Grittner, U.; Alexander, T.; Hiepe, F.; Meisel, A. Bortezomib in antibody-mediated autoimmune diseases (TAVAB): Study protocol for a unicentric, non-randomised, non-placebo controlled trial. BMJ Open 2019, 9, e024523. [Google Scholar] [CrossRef] [PubMed]
- Neubert, K.; Meister, S.; Moser, K.; Weisel, F.; Maseda, D.; Amann, K.; Wiethe, C.; Winkler, T.H.; Kalden, J.R.; Manz, R.A.; et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat. Med. 2008, 14, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.; Peskar, B.A. Anti-inflammatory effects of aspirin and sodium salicylate. Eur. J. Pharmacol. 2002, 447, 1–9. [Google Scholar] [CrossRef]
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef]
- Vane, J.R.; Mitchell, J.A.; Appleton, I.; Tomlinson, A.; Bishop-Bailey, D.; Croxtall, J.; Willoughby, D.A. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc. Natl. Acad. Sci. USA 1994, 91, 2046–2050. [Google Scholar] [CrossRef]
- Fattahi, M.J.; Mirshafiey, A. Prostaglandins and rheumatoid arthritis. Arthritis 2012, 2012, 239310. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef]
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef]
- Takada, Y.; Bhardwaj, A.; Potdar, P.; Aggarwal, B.B. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-kappaB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene 2004, 23, 9247–9258. [Google Scholar] [CrossRef]
- D’Acquisto, F.; Iuvone, T.; Rombola, L.; Sautebin, L.; Di Rosa, M.; Carnuccio, R. Involvement of NF-kappaB in the regulation of cyclooxygenase-2 protein expression in LPS-stimulated J774 macrophages. FEBS Lett. 1997, 418, 175–178. [Google Scholar] [CrossRef]
- Pierce, J.W.; Read, M.A.; Ding, H.; Luscinskas, F.W.; Collins, T. Salicylates inhibit I kappa B-alpha phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. J. Immunol. 1996, 156, 3961–3969. [Google Scholar] [PubMed]
- Mazzeo, D.; Panina-Bordignon, P.; Recalde, H.; Sinigaglia, F.; D’Ambrosio, D. Decreased IL-12 production and Th1 cell development by acetyl salicylic acid-mediated inhibition of NF-kappaB. Eur. J. Immunol. 1998, 28, 3205–3213. [Google Scholar] [CrossRef]
- Scheuren, N.; Bang, H.; Munster, T.; Brune, K.; Pahl, A. Modulation of transcription factor NF-kappaB by enantiomers of the nonsteroidal drug ibuprofen. Br. J. Pharmacol. 1998, 123, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.J.; Cryer, H.G.; Fu, M.; Lo, F.R. Regulation of macrophage eicosanoid generation is dependent on nuclear factor kappaB. J. Trauma Acute Care Surg. 1998, 45, 19–23. [Google Scholar] [CrossRef]
- De Bosscher, K.; Vanden Berghe, W.; Haegeman, G. Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene 2006, 25, 6868–6886. [Google Scholar] [CrossRef]
- Kavanaugh, A.; Wells, A.F. Benefits and risks of low-dose glucocorticoid treatment in the patient with rheumatoid arthritis. Rheumatology 2014, 53, 1742–1751. [Google Scholar] [CrossRef]
- Crofford, L.J. Use of NSAIDs in treating patients with arthritis. Arthr. Res. Ther. 2013, 15. [Google Scholar] [CrossRef]
- Bijlsma, W.J.; Buttgereit, F. Adverse events of glucocorticoids during treatment of rheumatoid arthritis: Lessons from cohort and registry studies. Rheumatology 2016, 55, ii3–ii5. [Google Scholar] [CrossRef]
- Alexander, T.; Sarfert, R.; Klotsche, J.; Kuhl, A.A.; Rubbert-Roth, A.; Lorenz, H.M.; Rech, J.; Hoyer, B.F.; Cheng, Q.; Waka, A.; et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann. Rheum. Dis. 2015, 74, 1474–1478. [Google Scholar] [CrossRef]
- Meister, S.; Frey, B.; Lang, V.R.; Gaipl, U.S.; Schett, G.; Schlotzer-Schrehardt, U.; Voll, R.E. Calcium channel blocker verapamil enhances endoplasmic reticulum stress and cell death induced by proteasome inhibition in myeloma cells. Neoplasia 2010, 12, 550–561. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, J.H.; Park, Y.B.; Lee, S.K. Bortezomib attenuates murine collagen-induced arthritis. Ann. Rheum. Dis. 2009, 68, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- D’Acquisto, F.; Ialenti, A.; Ianaro, A.; Di Vaio, R.; Carnuccio, R. Local administration of transcription factor decoy oligonucleotides to nuclear factor-kappaB prevents carrageenin-induced inflammation in rat hind paw. Gene Ther. 2000, 7, 1731–1737. [Google Scholar] [CrossRef]
- Lin, Y.Z.; Yao, S.Y.; Veach, R.A.; Torgerson, T.R.; Hawiger, J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J. Biol. Chem. 1995, 270, 14255–14258. [Google Scholar] [CrossRef] [PubMed]
- Torgerson, T.R.; Colosia, A.D.; Donahue, J.P.; Lin, Y.Z.; Hawiger, J. Regulation of NF-kappa B, AP-1, NFAT, and STAT1 nuclear import in T lymphocytes by noninvasive delivery of peptide carrying the nuclear localization sequence of NF-kappa B p50. J. Immunol. 1998, 161, 6084–6092. [Google Scholar]
- Kishore, N.; Sommers, C.; Mathialagan, S.; Guzova, J.; Yao, M.; Hauser, S.; Huynh, K.; Bonar, S.; Mielke, C.; Albee, L.; et al. A selective IKK-2 inhibitor blocks NF-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J. Biol. Chem. 2003, 278, 32861–32871. [Google Scholar] [CrossRef]
- McIntyre, K.W.; Shuster, D.J.; Gillooly, K.M.; Dambach, D.M.; Pattoli, M.A.; Lu, P.; Zhou, X.D.; Qiu, Y.; Zusi, F.C.; Burke, J.R. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthr. Rheum. 2003, 48, 2652–2659. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- May, M.J.; Marienfeld, R.B.; Ghosh, S. Characterization of the Ikappa B-kinase NEMO binding domain. J. Biol. Chem. 2002, 277, 45992–46000. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Cazalla, D.; Sanford, J.R.; Caceres, J.F. A rapid and efficient protocol to purify biologically active recombinant proteins from mammalian cells. Protein Expr. Purif. 2005, 42, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.H. Gene expression in Mammalian cells and its applications. Adv. Pharm. Bull. 2013, 3, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Stahl, S.; Lundeberg, J.; Uhlen, M.; Nygren, P.A. Affinity fusion strategies for detection, purification, and immobilization of recombinant proteins. Protein Expr. Purif. 1997, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Krebber, A.; Bornhauser, S.; Burmester, J.; Honegger, A.; Willuda, J.; Bosshard, H.R.; Pluckthun, A. Reliable cloning of functional antibody variable domains from hybridomas and spleen cell repertoires employing a reengineered phage display system. J. Immunol. Methods 1997, 201, 35–55. [Google Scholar] [CrossRef]
- Muller, W.A. The regulation of transendothelial migration: New knowledge and new questions. Cardiovasc. Res. 2015, 107, 310–320. [Google Scholar] [CrossRef]
- Martin, T.; Cardarelli, P.M.; Parry, G.C.; Felts, K.A.; Cobb, R.R. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur. J. Immunol. 1997, 27, 1091–1097. [Google Scholar] [CrossRef]
- Abbassi, O.; Kishimoto, T.K.; McIntire, L.V.; Anderson, D.C.; Smith, C.W. E-selectin supports neutrophil rolling in vitro under conditions of flow. J. Clin. Investig. 1993, 92, 2719–2730. [Google Scholar] [CrossRef]
- Vestweber, D.; Blanks, J.E. Mechanisms that regulate the function of the selectins and their ligands. Physiol. Rev. 1999, 79, 181–213. [Google Scholar] [CrossRef]
- Martens, C.L.; Cwirla, S.E.; Lee, R.Y.; Whitehorn, E.; Chen, E.Y.; Bakker, A.; Martin, E.L.; Wagstrom, C.; Gopalan, P.; Smith, C.W.; et al. Peptides which bind to E-selectin and block neutrophil adhesion. J. Biol. Chem. 1995, 270, 21129–21136. [Google Scholar] [CrossRef]
- Wick, M.J.; Hamood, A.N.; Iglewski, B.H. Analysis of the structure-function relationship of Pseudomonas aeruginosa exotoxin A. Mol. Microbiol. 1990, 4, 527–535. [Google Scholar] [CrossRef]
- Blanks, J.E.; Moll, T.; Eytner, R.; Vestweber, D. Stimulation of P-selectin glycoprotein ligand-1 on mouse neutrophils activates beta 2-integrin mediated cell attachment to ICAM-1. Eur. J. Immunol. 1998, 28, 433–443. [Google Scholar] [CrossRef]
- Zollner, O.; Lenter, M.C.; Blanks, J.E.; Borges, E.; Steegmaier, M.; Zerwes, H.G.; Vestweber, D. L-selectin from human, but not from mouse neutrophils binds directly to E-selectin. J. Cell Biol. 1997, 136, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Seitz, G.; Warmann, S.W.; Fuchs, J.; Heitmann, H.; Mahrt, J.; Busse, A.C.; Ruck, P.; Hoffman, R.M.; Wessels, J.T. Imaging of cell trafficking and metastases of paediatric rhabdomyosarcoma. Cell Prolif. 2008, 41, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Shcherbo, D.; Merzlyak, E.M.; Chepurnykh, T.V.; Fradkov, A.F.; Ermakova, G.V.; Solovieva, E.A.; Lukyanov, K.A.; Bogdanova, E.A.; Zaraisky, A.G.; Lukyanov, S.; et al. Bright far-red fluorescent protein for whole-body imaging. Nat. Methods 2007, 4, 741–746. [Google Scholar] [CrossRef]
- Christensen, A.D.; Haase, C.; Cook, A.D.; Hamilton, J.A. K/BxN Serum-Transfer Arthritis as a Model for Human Inflammatory Arthritis. Front. Immunol. 2016, 7, 213. [Google Scholar] [CrossRef]
- Ji, H.; Ohmura, K.; Mahmood, U.; Lee, D.M.; Hofhuis, F.M.; Boackle, S.A.; Takahashi, K.; Holers, V.M.; Walport, M.; Gerard, C.; et al. Arthritis critically dependent on innate immune system players. Immunity 2002, 16, 157–168. [Google Scholar] [CrossRef]
- Chatzidionysiou, K.; van Vollenhoven, R.F. When to initiate and discontinue biologic treatments for rheumatoid arthritis? J. Intern. Med. 2011, 269, 614–625. [Google Scholar] [CrossRef]
- Xu, H.; Ye, X.; Steinberg, H.; Liu, S.F. Selective blockade of endothelial NF-kappaB pathway differentially affects systemic inflammation and multiple organ dysfunction and injury in septic mice. J. Pathol. 2010, 220, 490–498. [Google Scholar]
- Collins, T.; Cybulsky, M.I. NF-kappaB: Pivotal mediator or innocent bystander in atherogenesis? J. Clin. Investig. 2001, 107, 255–264. [Google Scholar] [CrossRef]
- Naugler, W.E.; Karin, M. NF-kappaB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef]
- Sethi, G.; Sung, B.; Aggarwal, B.B. Nuclear factor-kappaB activation: From bench to bedside. Exp. Biol. Med. 2008, 233, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Heisler, I.; Keller, J.; Tauber, R.; Sutherland, M.; Fuchs, H. A cleavable adapter to reduce nonspecific cytotoxicity of recombinant immunotoxins. Int. J. Cancer 2003, 103, 277–282. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sehnert, B.; Burkhardt, H.; Dübel, S.; Voll, R.E. Cell-Type Targeted NF-kappaB Inhibition for the Treatment of Inflammatory Diseases. Cells 2020, 9, 1627. https://doi.org/10.3390/cells9071627
Sehnert B, Burkhardt H, Dübel S, Voll RE. Cell-Type Targeted NF-kappaB Inhibition for the Treatment of Inflammatory Diseases. Cells. 2020; 9(7):1627. https://doi.org/10.3390/cells9071627
Chicago/Turabian StyleSehnert, Bettina, Harald Burkhardt, Stefan Dübel, and Reinhard E. Voll. 2020. "Cell-Type Targeted NF-kappaB Inhibition for the Treatment of Inflammatory Diseases" Cells 9, no. 7: 1627. https://doi.org/10.3390/cells9071627
APA StyleSehnert, B., Burkhardt, H., Dübel, S., & Voll, R. E. (2020). Cell-Type Targeted NF-kappaB Inhibition for the Treatment of Inflammatory Diseases. Cells, 9(7), 1627. https://doi.org/10.3390/cells9071627