The Molecular Landscape of Hürthle Cell Thyroid Cancer Is Associated with Altered Mitochondrial Function—A Comprehensive Review


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Search Strategy and Selection Criteria
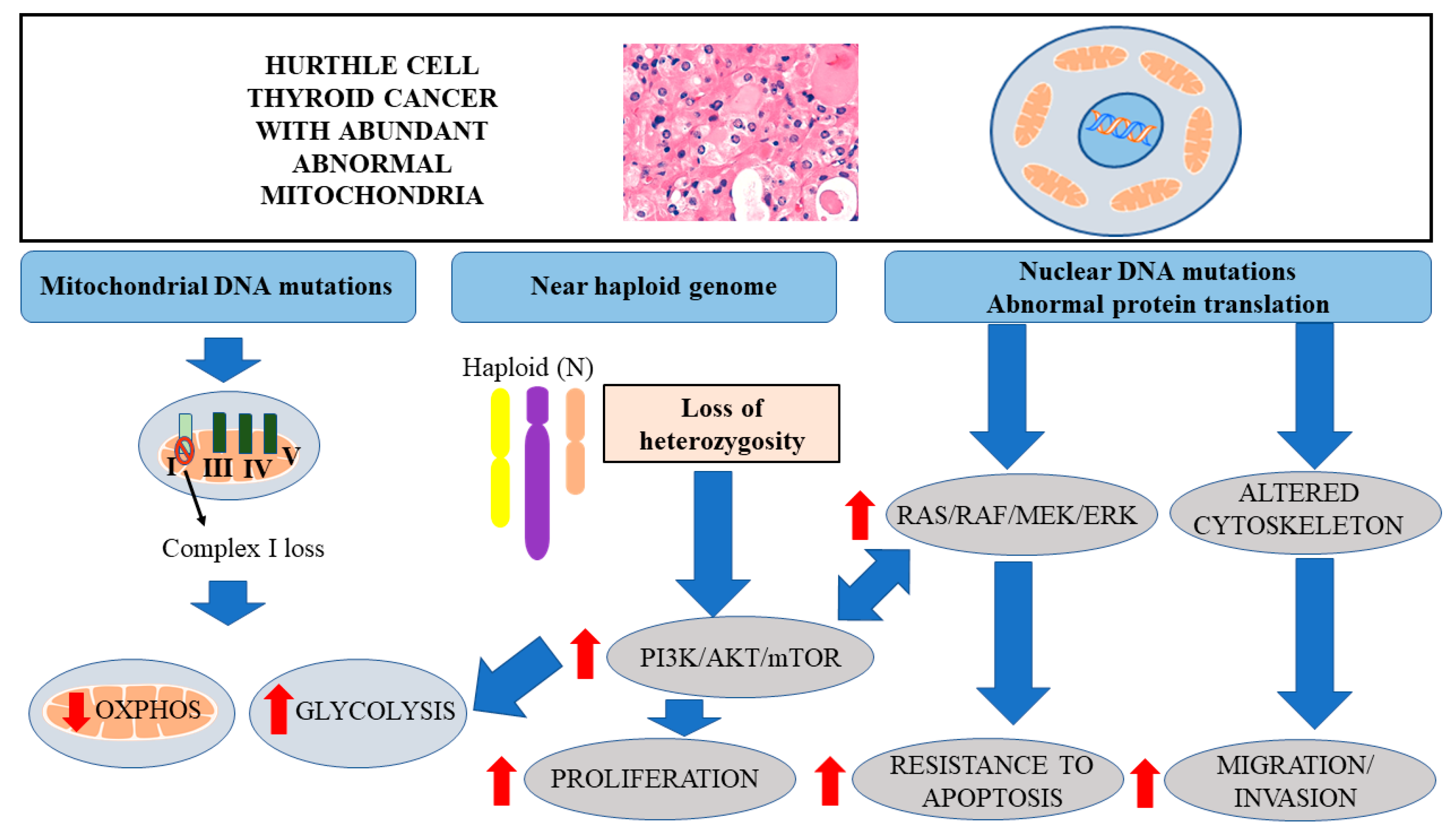
3. Genetic Alterations in HTC
4. LOH and Activation of AKT/mTOR Signaling
5. Distinct Metabolic Profile of Cancer Cells
6. Mitochondrial DNA Mutations in HTC Leading to Decreased OXPHOS
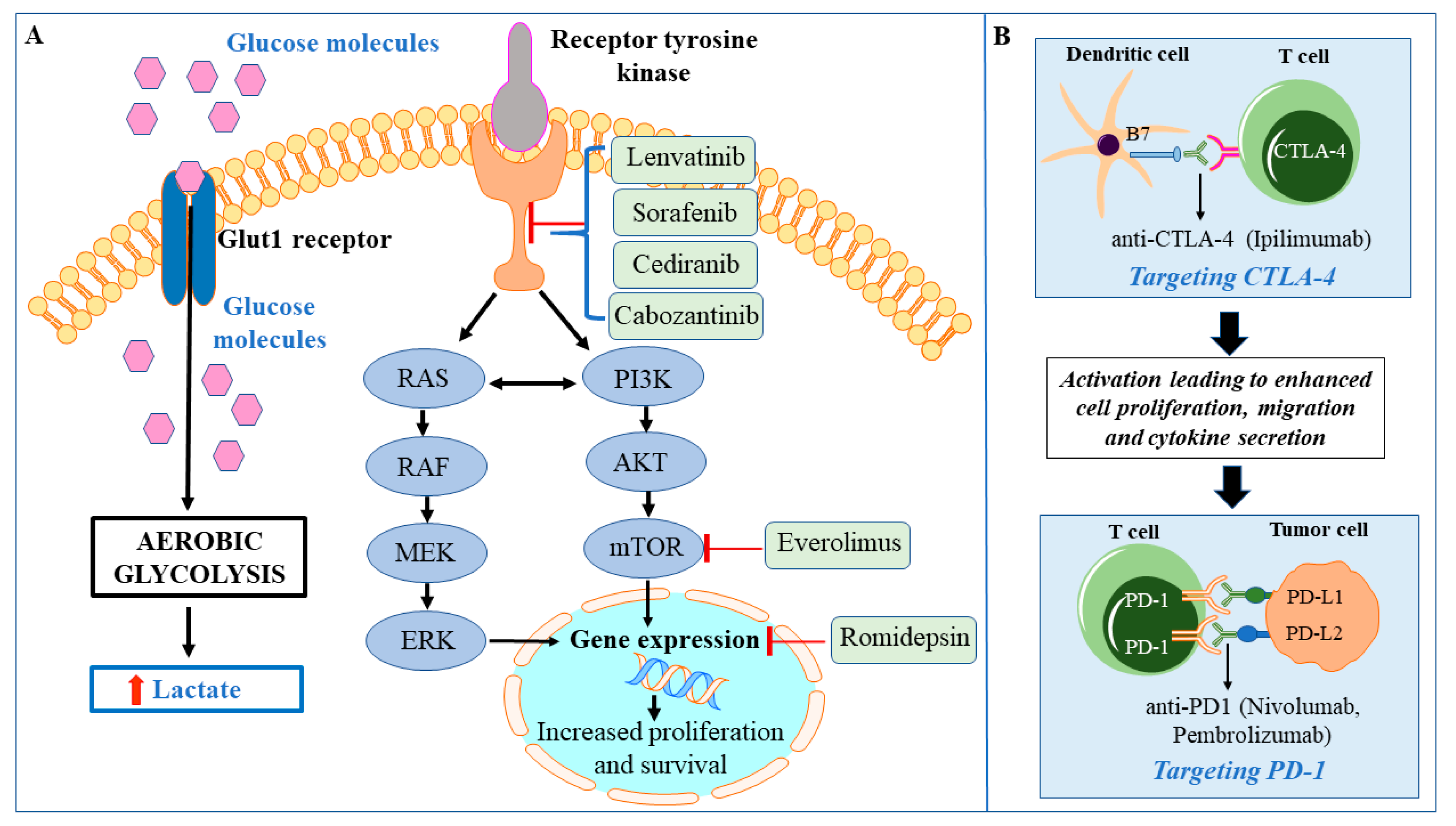
7. Enhanced Glycolysis in HTC
8. Targeted Therapies for HTC
9. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DTC | Differentiated thyroid carcinoma |
| FTC | Follicular thyroid carcinoma |
| FDG-PET | F-18-fluorodeoxy-glucose positron emission tomography |
| PTC | Papillary thyroid carcinoma |
| FDA | Food and drug administration |
| MEK | Mitogen-activated protein kinase |
| ERK | Extracellular signal-regulated kinase |
| HK | Hexokinase |
| RAI | Radioactive iodine |
| ROS | Reactive oxygen species |
| ETC | Electron transport chain |
| TKI | Tyrosine kinase inhibitors |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | PD ligand 1 |
| CTLA-4 | Cytotoxic T lymphocyte associated antigen 4 |
| PDGFR | Platelet-derived growth factor receptor |
| VEGF | Vascular endothelial growth factor |
References
- Tan, G.H.; Gharib, H. Thyroid Incidentalomas: Management Approaches to Nonpalpable Nodules Discovered Incidentally on Thyroid Imaging. Ann. Intern. Med. 1997, 126, 226. [Google Scholar] [CrossRef]
- Morganti, S.; Ceda, G.P.; Saccani, M.; Milli, B.; Ugolotti, D.; Prampolini, R.; Maggio, M.; Valenti, G.; Ceresini, G. Thyroid disease in the elderly: Sex-related differences in clinical expression. J. Endocrinol. Investig. 2005, 28, 101–104. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Haugen, B.R. 2015 American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American Thyroid Association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Gopal, R.K.; Kübler, K.; Calvo, S.E.; Polak, P.; Livitz, D.G.; Rosebrock, D.; Sadow, P.M.; Campbell, B.; Donovan, S.E.; Amin, S.; et al. Widespread Chromosomal Losses and Mitochondrial DNA Alterations as Genetic Drivers in Hürthle Cell Carcinoma. Cancer Cell 2018, 34, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Ganly, I.; Makarov, V.; Deraje, S.; Dong, Y.; Reznik, E.; Seshan, V.; Nanjangud, G.J.; Eng, S.; Bose, P.; Kuo, F.; et al. Integrated Genomic Analysis of Hürthle Cell Cancer Reveals Oncogenic Drivers, Recurrent Mitochondrial Mutations, and Unique Chromosomal Landscapes. Cancer Cell 2018, 34, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Chindris, A.-M.; Casler, J.D.; Bernet, V.; Rivera, M.; Thomas, C.; Kachergus, J.M.; Necela, B.M.; Hay, I.D.; Westphal, S.A.; Grant, C.S.; et al. Clinical and Molecular Features of Hürthle Cell Carcinoma of the Thyroid. J. Clin. Endocrinol. Metab. 2015, 100, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Nabhan, F.; Angell, T.E.; Harrell, M.; Nasr, C.; Wei, S.; Sipos, J.A. Use of Molecular Diagnostic Tests in Thyroid Nodules with Hürthle Cell-Dominant Cytology. Thyroid 2020. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.I.; Nasr, C.; Bischoff, L.; Busaidy, N.L.; Byrd, D.; Callender, G.; Dickson, P.; Duh, Q.-Y.; Ehya, H.; Goldner, W.; et al. NCCN Guidelines Insights: Thyroid Carcinoma, Version 2.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 1429–1440. [Google Scholar] [CrossRef]
- Grani, G.; Lamartina, L.; Durante, C.; Filetti, S.; Cooper, D.S. Follicular thyroid cancer and Hürthle cell carcinoma: Challenges in diagnosis, treatment, and clinical management. Lancet Diabetes Endocrinol. 2018, 6, 500–514. [Google Scholar] [CrossRef]
- Ganly, I.; McFadden, D.G. Short Review: Genomic Alterations in Hürthle Cell Carcinoma. Thyroid 2019, 29, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Pearlstein, S.; Lahouti, A.H.; Opher, E.; Nikiforov, Y.E.; Kuriloff, D.B. Thyroseq V3 Molecular Profiling for Tailoring the Surgical Management of Hürthle Cell Neoplasms. Case Rep. Endocrinol. 2018, 2018, 9329035. [Google Scholar] [CrossRef] [PubMed]
- Klubo-Gwiezdzinska, J.; Wartofsky, L. The Role of Molecular Diagnostics in the Management of Indeterminate Thyroid Nodules. J. Clin. Endocrinol. Metab. 2018, 103, 3507–3510. [Google Scholar] [CrossRef] [PubMed]
- Schatz-Siemers, N.; Brandler, T.C.; Oweity, T.; Sun, W.; Hernandez, A.; Levine, P. Hürthle cell lesions on thyroid fine needle aspiration cytology: Molecular and histologic correlation. Diagn. Cytopathol. 2019, 47, 977–985. [Google Scholar] [CrossRef]
- Hao, Y.; Duh, Q.-Y.; Kloos, R.T.; Babiarz, J.; Harrell, M.; Traweek, S.T.; Kim, S.Y.; Fedorowicz, G.; Walsh, P.S.; Sadow, P.M.; et al. Identification of Hürthle cell cancers: Solving a clinical challenge with genomic sequencing and a trio of machine learning algorithms. BMC Syst. Boil. 2019, 13, 27. [Google Scholar] [CrossRef]
- Corver, W.E.; Ruano, D.; Weijers, K.; den Hartog, W.C.E.; van Nieuwenhuizen, M.P. Genome haploidisation with chromosome 7 retention in oncocytic follicular thyroid carcinoma. PLoS ONE 2012, 7, e38287. [Google Scholar] [CrossRef]
- San Martin, V.T.; Lawrence, L.; Bena, J.; Madhun, N.Z.; Berber, E.; Elsheikh, T.M.; Nasr, C.E. Real-world Comparison of Afirma GEC and GSC for the Assessment of Cytologically Indeterminate Thyroid Nodules. J. Clin. Endocrinol. Metab. 2020, 105, e428–e435. [Google Scholar] [CrossRef]
- Alexander, E.K.; Kennedy, G.C.; Baloch, Z.W.; Cibas, E.S.; Chudova, D.; Diggans, J.; Friedman, L.; Kloos, R.T.; LiVolsi, V.A.; Mandel, S.J.; et al. Preoperative diagnosis of benign thyroid nodules with indeterminate cytology. N Engl. J. Med. 2012, 367, 705–715. [Google Scholar] [CrossRef]
- Steward, D.L.; Carty, S.E.; Sippel, R.S.; Yang, S.P.; Sosa, J.A.; Sipos, J.A.; Figge, J.J.; Mandel, S.; Haugen, B.R.; Burman, K.D. Performance of a Multigene Genomic Classifier in Thyroid Nodules With Indeterminate Cytology: A Prospective Blinded Multicenter Study. JAMA Oncol. 2019, 5, 204–212. [Google Scholar] [CrossRef]
- Abubaker, J.; Jehan, Z.; Bavi, P.; Sultana, M.; Al-Harbi, S.; Ibrahim, M.; Al-Nuaim, A.; Ahmed, M.; Amin, T.; Al-Fehaily, M.; et al. Clinicopathological Analysis of Papillary Thyroid Cancer withPIK3CAAlterations in a Middle Eastern Population. J. Clin. Endocrinol. Metab. 2008, 93, 611–618. [Google Scholar] [CrossRef]
- Ghossein, R.A.; Hiltzik, D.H.; Carlson, D.L.; Patel, S.G.; Shaha, A.; Shah, J.P.; Tuttle, R.M.; Singh, B. Prognostic factors of recurrence in encapsulated Hurthle cell carcinoma of the thyroid gland. Cancer 2006, 106, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Shaha, A.R.; Shah, J.P.; Loree, T.R. Patterns of nodal and distant metastasis based on histologic varieties in differentiated carcinoma of the thyroid. Am. J. Surg. 1996, 172, 692–694. [Google Scholar] [CrossRef]
- Grossman, Z.; Clark, O.H. Hurthle Cell Carcinoma: Total thyroidectomy with central neck lymph node dissection is the therapy of choice for patients with Hürthle cell carcinoma. Cancer Control 1997, 4, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Penabad, L.; Chiu, A.C.; Hoff, A.O.; Schultz, P.; Gaztambide, S.; Ordòñez, N.G.; Sherman, S.I. Prognostic factors in patients with Hürthle cell neoplasms of the thyroid. Cancer 2003, 97, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Besic, N.; Vidergar-Kralj, B.; Frkovic-Grazio, S.; Movrin-Stanovnik, T.; Auersperg, M. The Role of Radioactive Iodine in the Treatment of Hürthle Cell Carcinoma of the Thyroid. Thyroid 2003, 13, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Carcangiu, M.L.; Bianchi, S.; Savino, D.; Voynick, I.M.; Rosai, J. Follicular Hurthle cell tumors of the thyroid gland. Cancer 1991, 68, 1944–1953. [Google Scholar] [CrossRef]
- Kushchayeva, Y.; Duh, Q.-Y.; Kebebew, E.; Clark, O.H. Prognostic Indications for Hurthle Cell Cancer. World J. Surg. 2004, 28, 1266–1270. [Google Scholar] [CrossRef]
- Máximo, V.; Lima, J.; Prazeres, H.; Soares, P.; Sobrinho-Simões, M. The biology and the genetics of Hürthle cell tumors of the thyroid. Endocrine-Relat. Cancer 2012, 19, R131–R147. [Google Scholar] [CrossRef]
- Máximo, V.; Sobrinho-Simões, M. Hürthle cell tumours of the thyroid. A review with emphasis on mitochondrial abnormalities with clinical relevance. Virchows Archiv 2000, 437, 107–115. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.; Shah, R.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Martin, M.; Maßhöfer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; van de Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, L.; Tang, X.; Zhang, X.; Qiao, Y.; Shi, Y.; Xu, Y.; Wang, Z.; Yu, Y.; Sun, F. Doxorubicin induces apoptosis by targeting Madcam1 and AKT and inhibiting protein translation initiation in hepatocellular carcinoma cells. Oncotarget 2015, 6, 24075–24091. [Google Scholar] [CrossRef] [PubMed]
- Kunstman, J.W.; Juhlin, C.C.; Goh, G.; Brown, T.C.; Stenman, A.; Healy, J.; Rubinstein, J.C.; Choi, M.; Kiss, N.; Nelson-Williams, C.; et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum. Mol. Genet. 2015, 24, 2318–2329. [Google Scholar] [CrossRef]
- Cercek, A.; Braghiroli, M.I.; Chou, J.F.; Hechtman, J.; Kemeny, N.; Saltz, L.; Capanu, M.; Yaeger, R. Clinical Features and Outcomes of Patients with Colorectal Cancers Harboring NRAS Mutations. Clin. Cancer Res. 2017, 23, 4753–4760. [Google Scholar] [CrossRef]
- Haas, M.; Ormanns, S.; Baechmann, S.; Remold, A.; Kruger, S.; Westphalen, C.B.; Siveke, J.T.; Wenzel, P.; Schlitter, A.M.; Esposito, I.; et al. Extended RAS analysis and correlation with overall survival in advanced pancreatic cancer. Br. J. Cancer 2017, 116, 1462–1469. [Google Scholar] [CrossRef]
- Mishra, V.; Kowtal, P.; Rane, P.; Sarin, R. Genetic risk association of CDKN1A and RET gene SNPs with medullary thyroid carcinoma: Results from the largest MTC cohort and meta-analysis. Cancer Med. 2019, 8, 6151–6161. [Google Scholar] [CrossRef]
- Cetinkaya, A.C.; Eraslan, S.; Kirdar, B. Transcriptomic response of yeast cells to ATX1 deletion under different copper levels. BMC Genom. 2016, 17, 489. [Google Scholar] [CrossRef]
- Bae, J.; Kim, Y.; Jeon, S.; Kim, S.H.; Kim, T.-J.; Lee, S.; Kim, M.-H.; Lim, D.-J.; Lee, Y.S.; Jung, C.K. Clinical utility of TERT promoter mutations and ALK rearrangement in thyroid cancer patients with a high prevalence of the BRAF V600E mutation. Diagn. Pathol. 2016, 11, 21. [Google Scholar] [CrossRef]
- Donatini, G.; Beaulieu, A.; Castagnet, M.; Kraimps, J.-L.; Levillain, P.; Fromont, G. Thyroid Hürthle cell tumors: Research of potential markers of malignancy. J. Endocrinol. Investig. 2015, 39, 153–158. [Google Scholar] [CrossRef]
- Máximo, V.; Botelho, T.; Capela, J.; Soares, P.; Lima, J.; Taveira, A.; Amaro, T.; Barbosa, A.P.; Preto, A.; Harach, H.R.; et al. Somatic and germline mutation in GRIM-19, a dual function gene involved in mitochondrial metabolism and cell death, is linked to mitochondrion-rich (Hürthle cell) tumours of the thyroid. Br. J. Cancer 2005, 92, 1892–1898. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Porcelli, A.M.; Bonora, E.; Pennisi, L.F.; Toller, M.; Iommarini, L.; Ghelli, A.; Moretti, M.; Betts, C.M.; Martinelli, G.N.; et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 9001–9006. [Google Scholar] [CrossRef] [PubMed]
- Boot, A.; Oosting, J.; de Miranda, N.; Zhang, Y.; Corver, W.E.; van de Water, B.; Morreau, H.; van Wezel, T. Imprinted survival genes preclude loss of heterozygosity of chromosome 7 in cancer cells. J. Pathol. 2016, 240, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Chen, X.-S.; Li, L.-Y.; Guan, Y.-D.; Yang, J.-M.; Cheng, Y. Anticancer strategies based on the metabolic profile of tumor cells: Therapeutic targeting of the Warburg effect. Acta Pharmacol. Sin. 2016, 37, 1013–1019. [Google Scholar] [CrossRef]
- Xie, C.-H.; Naito, A.; Mizumachi, T.; Evans, T.T.; Douglas, M.G.; Cooney, C.; Fan, C.-Y.; Higuchi, M. Mitochondrial regulation of cancer associated nuclear DNA methylation. Biochem. Biophys. Res. Commun. 2007, 364, 656–661. [Google Scholar] [CrossRef]
- Ma, Y.; Bai, R.-K.; Trieu, R.; Wong, L.-J.C. Mitochondrial dysfunction in human breast cancer cells and their transmitochondrial cybrids. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 29–37. [Google Scholar] [CrossRef]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA mutations in human cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef]
- Jerónimo, C.; Nomoto, S.; Caballero, O.L.; Usadel, H.; Henrique, R.; Varzim, G.; Oliveira, J.; Lopes, C.; Fliss, M.S.; Sidransky, D. Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene 2001, 20, 5195–5198. [Google Scholar] [CrossRef]
- Jones, J.B.; Song, J.J.; Hempen, P.M.; Parmigiani, G.; Hruban, R.H.; Kern, S.E. Detection of mitochondrial DNA mutations in pancreatic cancer offers a "mass"-ive advantage over detection of nuclear DNA mutations. Cancer Res. 2001, 61, 1299–1304. [Google Scholar] [PubMed]
- Polyak, K.; Li, Y.; Zhu, H.; Lengauer, C.; Willson, J.K.; Markowitz, S.D.; Trush, M.A.; Kinzler, K.W.; Vogelstein, B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998, 20, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Habano, W.; Nakamura, S.-i.; Sugai, T. Microsatellite instability in the mitochondrial DNA of colorectal carcinomas: Evidence for mismatch repair systems in mitochondrial genome. Oncogene 1998, 17, 1931–1937. [Google Scholar] [CrossRef] [PubMed]
- Fliss, M.S.; Usadel, H.; Caballero, O.L.; Wu, L.; Buta, M.R.; Eleff, S.M.; Jen, J.; Sidransky, D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 2000, 287, 2017–2019. [Google Scholar] [CrossRef]
- Sanchez-Cespedes, M.; Parrella, P.; Nomoto, S.; Cohen, D.; Xiao, Y.; Esteller, M.; Jeronimo, C.; Jordan, R.C.K.; Nicol, T.; Koch, W.M.; et al. Identification of a mononucleotide repeat as a major target for mitochondrial DNA alterations in human tumors. Cancer research, 61(19), 7015-7019.., et al., Identification of a mononucleotide repeat as a major target for mitochondrial DNA alterations in human tumors. Cancer Res. 2001, 61, 7015–7019. [Google Scholar]
- Wong, L.J.C.; Lueth, M.; Li, X.N.; Lau, C.C.; Vogel, H. Detection of mitochondrial DNA mutations in the tumor and cerebrospinal fluid of medulloblastoma patients. Cancer Res. 2003, 63, 3866–3871. [Google Scholar]
- Savagner, F.; Franc, B.; Guyetant, S.; Rodien, P.; Reynier, P.A.; Malthiery, Y. Defective mitochondrial ATP synthesis in oxyphilic thyroid tumors. J. Clin. Endocrinol. Metab. 2001, 86, 4920–4925. [Google Scholar] [CrossRef]
- Bonora, E.; Porcelli, A.M.; Gasparre, G.; Biondi, A.; Ghelli, A.; Carelli, V.; Baracca, A.; Tallini, G.; Martinuzzi, A.; Lenaz, G.; et al. Defective Oxidative Phosphorylation in Thyroid Oncocytic Carcinoma Is Associated with Pathogenic Mitochondrial DNA Mutations Affecting Complexes I and III. Cancer Res. 2006, 66, 6087–6096. [Google Scholar] [CrossRef]
- Stankov, K.; Biondi, A.; D’Aurelio, M.; Gasparre, G.; Falasca, A.; Romeo, G.; Lenaz, G. Mitochondrial Activities of a Cell Line Derived from Thyroid Hürthle Cell Tumors. Thyroid 2006, 16, 325–331. [Google Scholar] [CrossRef]
- Gui, Y.; Zhang, L.; Lv, W.; Zhang, W.; Zhao, J.; Hu, X. NFE2L2 variations reduce antioxidant response in patients with Parkinson disease. Oncotarget 2016, 7, 10756–10764. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D. Mechanistic Studies of the Nrf2-Keap1 Signaling Pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D. The Nrf2-Keap1-ARE Signaling Pathway: The Regulation and Dual Function of Nrf2 in Cancer. Antioxid. Redox Signal. 2010, 13, 1623–1626. [Google Scholar] [CrossRef] [PubMed]
- Krhin, B.; Goricar, K.; Gazic, B.; Dolzan, V.; Besic, N. Functional polymorphisms in antioxidant genes in Hurthle cell thyroid neoplasm—An association of GPX1 polymorphism and recurrent Hurthle cell thyroid carcinoma. Radiol. Oncol. 2016, 50, 289–296. [Google Scholar] [CrossRef]
- Liemburg-Apers, D.C.; Willems, P.H.; Koopman, W.J.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Sleeman, M.W.; Wortley, K.E.; Lai, K.M.V.; Gowen, L.C.; Kintner, J.; Kline, W.O.; Garcia, K.; Stitt, T.N.; Yancopoulos, G.D.; Wiegand, S.J.; et al. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat. Med. 2005, 11, 199–205. [Google Scholar] [CrossRef]
- Samih, N.; Hovsepian, S.; Aouani, A.; Lombardo, D.; Fayet, G. Glut-1 Translocation in FRTL-5 Thyroid Cells: Role of Phosphatidylinositol 3-Kinase and N-Glycosylation. Endocrinology 2000, 141, 4146–4155. [Google Scholar] [CrossRef]
- Melstrom, L.G.; Salabat, M.R.; Ding, X.-Z.; Milam, B.M.; Strouch, M.; Pelling, J.C.; Bentrem, D.J. Apigenin Inhibits the GLUT-1 Glucose Transporter and the Phosphoinositide 3-Kinase/Akt Pathway in Human Pancreatic Cancer Cells. Pancreas 2008, 37, 426–431. [Google Scholar] [CrossRef]
- Kim, H.M.; Koo, J.S. Differential Expression of Glycolysis-Related Proteins in Follicular Neoplasms versus Hürthle Cell Neoplasms: A Retrospective Analysis. Dis. Markers 2017, 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Marin-Hernandez, A.; Gallardo-Perez, J.C.; Ralph, S.J.; Rodriguez-Enriquez, S.; Moreno-Sanchez, R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef]
- Klaus, A.; Fathi, O.; Tatjana, T.W.; Bruno, N.; Oskar, K. Expression of Hypoxia-Associated Protein HIF-1α in Follicular Thyroid Cancer is Associated with Distant Metastasis. Pathology Oncology Res. 2018, 24, 289–296. [Google Scholar] [CrossRef]
- Cao, D.; Hou, M.; Guan, Y.S.; Jiang, M.; Yang, Y.; Gou, H.F. Expression of HIF-1alpha and VEGF in colorectal cancer: association with clinical outcomes and prognostic implications. BMC Cancer 2009, 9, 432. [Google Scholar] [CrossRef]
- Huang, S.M.; Lee, J.C.; Wu, T.J.; Chow, N.H. Clinical relevance of vascular endothelial growth factor for thyroid neoplasms. World J. Surg. 2001, 25, 302–306. [Google Scholar] [CrossRef]
- Pathak, A.; Klonisch, T.; Nason, R.W.; Leslie, W.D. FDG-PET characteristics of Hürthle cell and follicular adenomas. Ann. Nucl. Med. 2016, 30, 506–509. [Google Scholar] [CrossRef]
- Pryma, D.A.; Schöder, H.; Gönen, M.; Robbins, R.J.; Larson, S.M.; Yeung, H.W.D. Diagnostic accuracy, and prognostic value of 18F-FDG PET in Hürthle cell thyroid cancer patients. J. Nucl. Med. 2006, 47, 1260–1266. [Google Scholar]
- Schmidbauer, B.; Menhart, K.; Hellwig, D.; Grosse, J. Differentiated Thyroid Cancer—Treatment: State of the Art. Int. J. Mol. Sci. 2017, 18, 1292. [Google Scholar] [CrossRef]
- Chalan, P.; di Dalmazi, G.; Pani, F.; de Remigis, A.; Corsello, A.; Caturegli, P.P. Thyroid dysfunctions secondary to cancer immunotherapy. J. Endocrinol. Investig. 2017, 41, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; He, Z.; Wang, X.; Li, H.; Liu, X.S. Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. Elife 2019, 8, e49020. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumari, S.; Adewale, R.; Klubo-Gwiezdzinska, J. The Molecular Landscape of Hürthle Cell Thyroid Cancer Is Associated with Altered Mitochondrial Function—A Comprehensive Review. Cells 2020, 9, 1570. https://doi.org/10.3390/cells9071570
Kumari S, Adewale R, Klubo-Gwiezdzinska J. The Molecular Landscape of Hürthle Cell Thyroid Cancer Is Associated with Altered Mitochondrial Function—A Comprehensive Review. Cells. 2020; 9(7):1570. https://doi.org/10.3390/cells9071570
Chicago/Turabian StyleKumari, Sonam, Ruth Adewale, and Joanna Klubo-Gwiezdzinska. 2020. "The Molecular Landscape of Hürthle Cell Thyroid Cancer Is Associated with Altered Mitochondrial Function—A Comprehensive Review" Cells 9, no. 7: 1570. https://doi.org/10.3390/cells9071570
APA StyleKumari, S., Adewale, R., & Klubo-Gwiezdzinska, J. (2020). The Molecular Landscape of Hürthle Cell Thyroid Cancer Is Associated with Altered Mitochondrial Function—A Comprehensive Review. Cells, 9(7), 1570. https://doi.org/10.3390/cells9071570