EDMD-Causing Emerin Mutant Myogenic Progenitors Exhibit Impaired Differentiation Using Similar Mechanisms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Lentiviral Transduction
2.3. Myogenic Differentiation Immunofluorescence Assay
2.4. Western Blots
2.5. RNAseq
2.6. Data Sharing Statement
3. Results
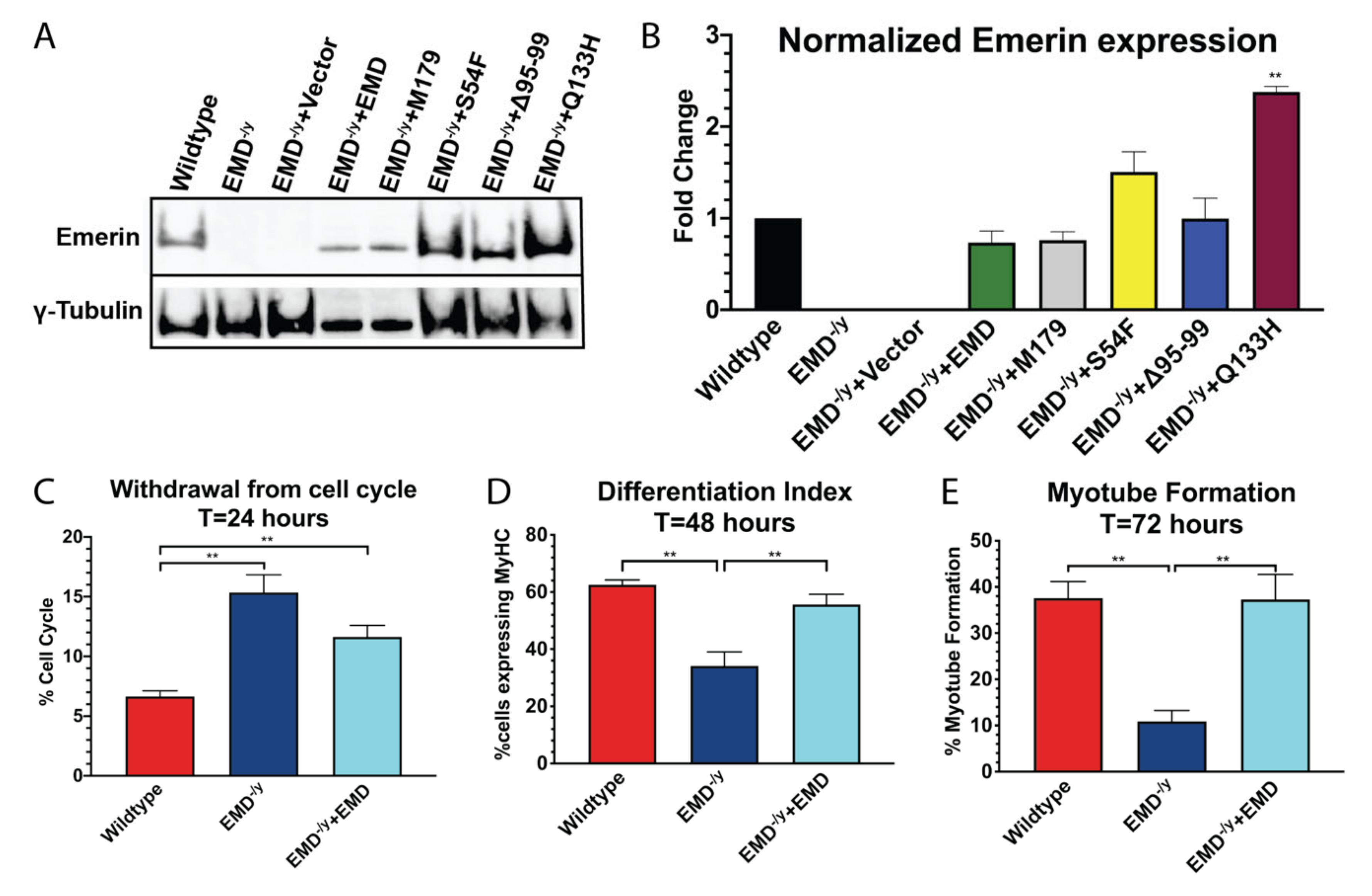
3.1. Generation of Stable Myogenic Progenitor Cell Lines Expressing EDMD-Causing Emerin Mutants
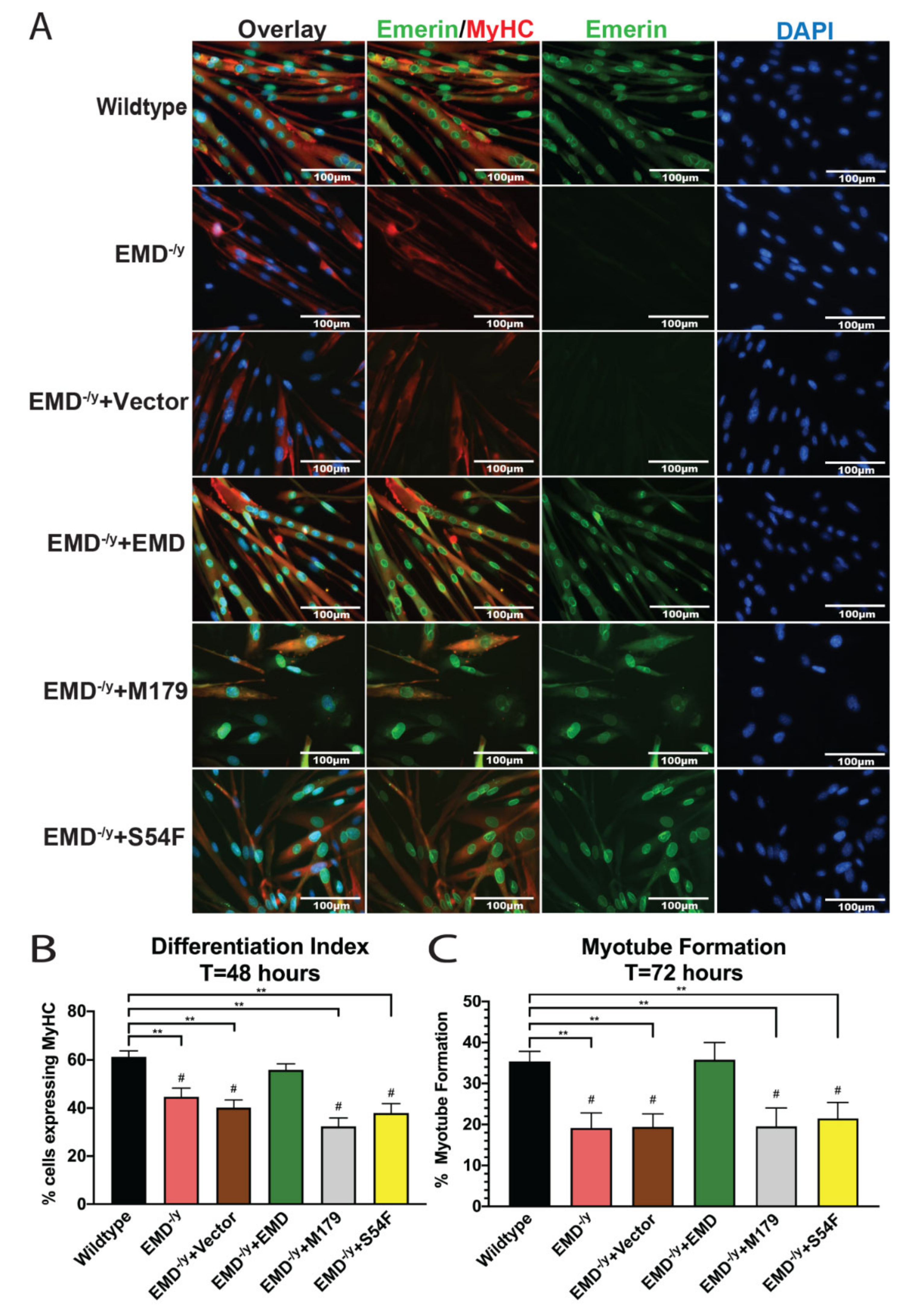
3.2. Myogenic Progenitors Expressing S54F and M179 Exhibit Impaired Differentiation
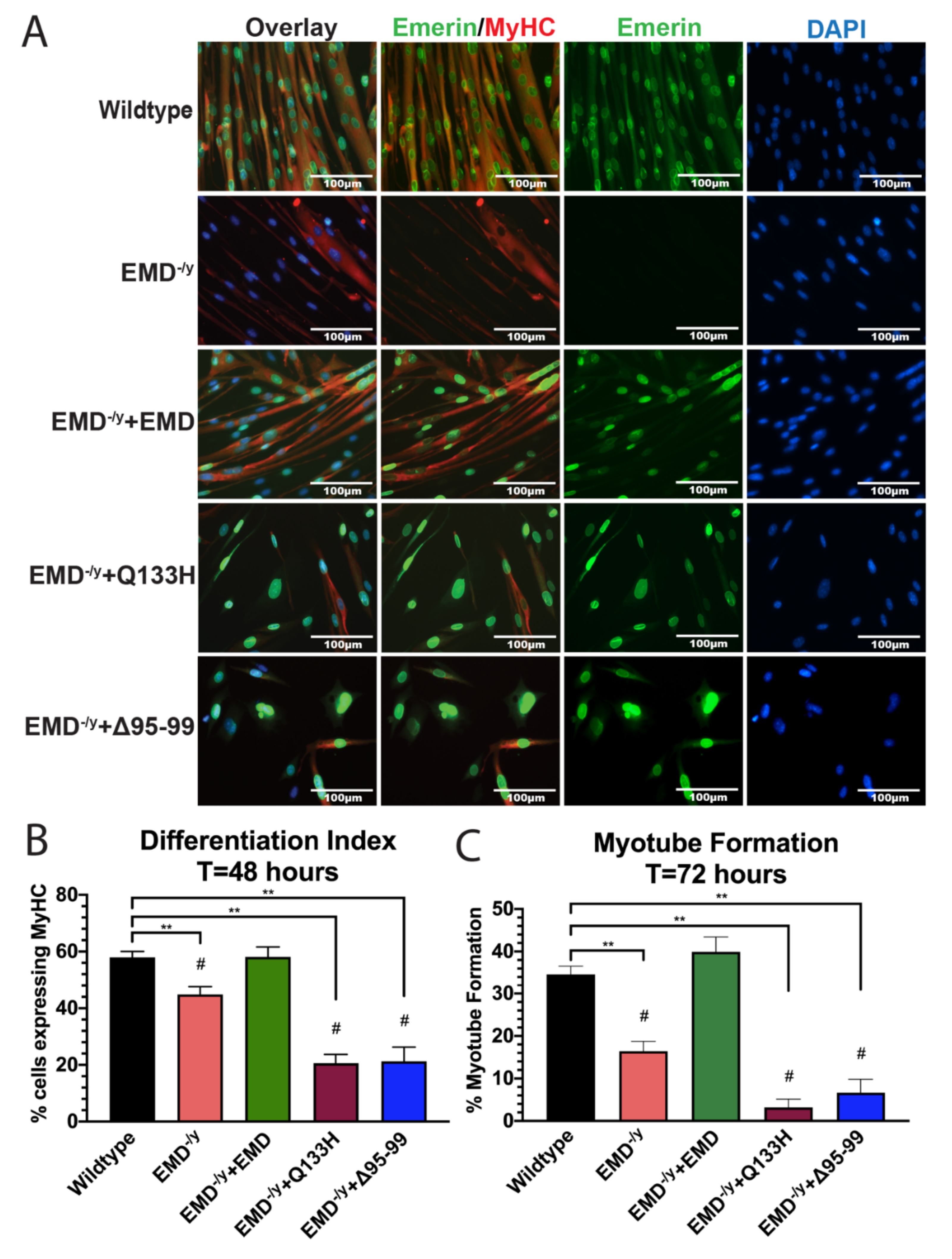
3.3. Q133H and Δ95–99 Mutant Myogenic Progenitors Exhibit Impaired Differentiation
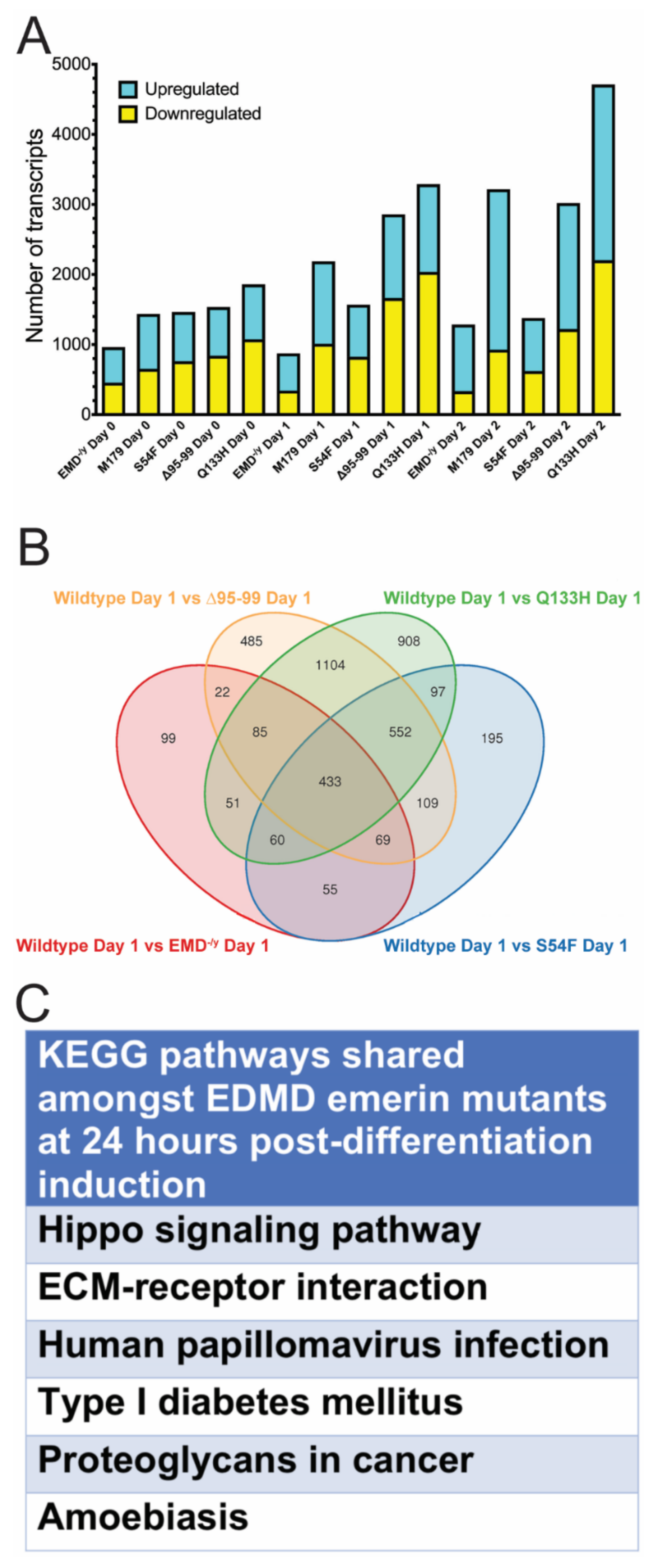
3.4. Identification of Pathways Shared Between all EDMD-Causing Emerin Mutants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 1994, 8, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Méndez-López, I.; Worman, H.J. Inner nuclear membrane proteins: Impact on human disease. Chromosoma 2012, 121, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Vlcek, S.; Foisner, R. Lamins and lamin-associated proteins in aging and disease. Curr. Opin. Cell Boil. 2007, 19, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J. Nuclear lamins and laminopathies. J. Pathol. 2011, 226, 316–325. [Google Scholar] [CrossRef]
- Yates, J.R.; Wehnert, M. The Emery-Dreifuss Muscular Dystrophy Mutation Database. Neuromuscul. Disord. 1999, 9, 199. [Google Scholar]
- Manilal, S.; Man, N.T.; Sewry, C.A.; Morris, G.E. The Emery-Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum. Mol. Genet. 1996, 5, 801–808. [Google Scholar] [CrossRef]
- Manilal, S.; Manila, S.; Recan, D.; Sewry, C.A.; Hoeltzenbein, M.; Llense, S.; Leturcq, F.; Deburgrave, N.; Barbot, J.-C.; Man, N.T.; et al. Mutations in Emery-Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum. Mol. Genet. 1998, 7, 855–864. [Google Scholar] [CrossRef]
- Manilal, S.; Sewry, C.A.; Man, N.T.; Muntoni, F.; Morris, G.E. Diagnosis of X-linked Emery-Dreifuss muscular dystrophy by protein analysis of leucocytes and skin with monoclonal antibodies. Neuromuscul. Disord. 1997, 7, 63–66. [Google Scholar] [CrossRef]
- Nagano, A.; Koga, R.; Ogawa, M.; Kurano, Y.; Kawada, J.; Okada, R.; Hayashi, Y.K.; Tsukahara, T.; Arahata, K. Emerin deficiency at the nuclear membrane in patients with Emery-Dreif uss muscular dystrophy. Nat. Genet. 1996, 12, 254–259. [Google Scholar] [CrossRef]
- Ellis, A.J.; Craxton, M.; Yates, J.R.; Kendrick-Jones, J. Aberrant intracellular targeting and cell cycle-dependent phosphorylation of emerin contribute to the Emery-Dreifuss muscular dystrophy phenotype. J. Cell Sci. 1998, 111, 781–792. [Google Scholar]
- Mora, M.; Cartegni, L.; Di Blasi, C.; Barresi, R.; Bione, S.; Di Barletta, M.R.; Morandi, L.; Merlini, L.; Nigro, V.; Politano, L.; et al. X-linked emery-dreifuss muscular dystrophy can be diagnosed from skin biopsy or blood sample. Ann. Neurol. 1997, 42, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Bagshaw, J.; Aksmanovic, V.M.; Coomber, E.; McMahon, R.; Whittaker, J.L.; Morrison, P.J.; Kendrick-Jones, J.; Ellis, A.J. Genotype-phenotype analysis in X-linked Emery-Dreifuss muscular dystrophy and identification of a missense mutation associated with a milder phenotype. Neuromuscul. Disord. 1999, 9, 159–165. [Google Scholar] [CrossRef]
- Berk, J.M.; Simon, D.N.; Jenkins-Houk, C.R.; Westerbeck, J.W.; Grønning-Wang, L.M.; Carlson, C.R.; Wilson, K.L. The molecular basis of emerin-emerin and emerin-BAF interactions. J. Cell Sci. 2014, 127, 3956–3969. [Google Scholar] [CrossRef] [PubMed]
- Herrada, I.; Bourgeois, B.; Samson, C.; Buendia, B.; Worman, H.J.; Zinn-Justin, S. Purification and Structural Analysis of LEM-Domain Proteins. Methods Enzymol. 2016, 569, 43–61. [Google Scholar] [CrossRef]
- Demmerle, J.; Koch, A.J.; Holaska, J.M. The Nuclear Envelope Protein Emerin Binds Directly to Histone Deacetylase 3 (HDAC3) and Activates HDAC3 Activity. J. Boil. Chem. 2012, 287, 22080–22088. [Google Scholar] [CrossRef]
- Mislow, J.M.; Holaska, J.M.; Kim, M.S.; Lee, K.K.; Segura-Totten, M.; Wilson, K.L.; McNally, E.M. Nesprin-1α self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett. 2002, 525, 135–140. [Google Scholar] [CrossRef]
- Haraguchi, T.; Holaska, J.M.; Yamane, M.; Koujin, T.; Hashiguchi, N.; Mori, C.; Wilson, K.L.; Hiraoka, Y. Emerin binding to Btf, a death-promoting transcriptional repressor, is disrupted by a missense mutation that causes Emery-Dreifuss muscular dystrophy. JBIC J. Boil. Inorg. Chem. 2004, 271, 1035–1045. [Google Scholar] [CrossRef]
- Holaska, J.M.; Lee, K.K.; Kowalski, A.K.; Wilson, K.L. Transcriptional Repressor Germ Cell-less (GCL) and Barrier to Autointegration Factor (BAF) Compete for Binding to Emerin in Vitro. J. Boil. Chem. 2002, 278, 6969–6975. [Google Scholar] [CrossRef]
- Holaska, J.M.; Rais-Bahrami, S.; Wilson, K.L. Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum. Mol. Genet. 2006, 15, 3459–3472. [Google Scholar] [CrossRef]
- Holaska, J.M.; Kowalski, A.K.; Wilson, K.L. Emerin Caps the Pointed End of Actin Filaments: Evidence for an Actin Cortical Network at the Nuclear Inner Membrane. PLoS Boil. 2004, 2, e231. [Google Scholar] [CrossRef]
- Markiewicz, E.; Tilgner, K.; Barker, N.; van de Wetering, M.; Clevers, H.; Dorobek, M.; Hausmanowa-Petrusewicz, I.; Ramaekers, F.; Broers, J.; Blankesteijn, W.M.; et al. The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006, 25, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Salpingidou, G.; Smertenko, A.; Hausmanowa-Petrucewicz, I.; Hussey, P.J.; Hutchison, C. A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. J. Cell Boil. 2007, 178, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Fairley, E.A.; Kendrick-Jones, J.; Ellis, J.A. The Emery-Dreifuss muscular dystrophy phenotype arises from aberrant targeting and binding of emerin at the inner nuclear membrane. J. Cell Sci. 1999, 112, 2571–2582. [Google Scholar] [PubMed]
- Ellis, J.A.; Yates, J.R.; Kendrick-Jones, J.; Brown, C.A. Changes at P183 of emerin weaken its protein-protein interactions resulting in X-linked Emery-Dreifuss muscular dystrophy. Qual. Life Res. 1999, 104, 262–268. [Google Scholar] [CrossRef]
- Holt, I.; Clements, L.; Manilal, S.; Morris, G.E. How does a g993t mutation in the emerin gene cause Emery-Dreifuss muscular dystrophy? Biochem. Biophys. Res. Commun. 2001, 287, 1129–1133. [Google Scholar] [CrossRef]
- Herrada, I.; Samson, C.; Velours, C.; Renault, L.; Östlund, C.; Chervy, P.; Puchkov, D.; Worman, H.J.; Buendia, B.; Zinn-Justin, S. Muscular Dystrophy Mutations Impair the Nuclear Envelope Emerin Self-assembly Properties. ACS Chem. Boil. 2015, 10, 2733–2742. [Google Scholar] [CrossRef]
- Melcon, G.; Kozlov, S.; Cutler, D.A.; Sullivan, T.; Hernandez, L.; Zhao, P.; Mitchell, S.; Nader, G.; Bakay, M.; Rottman, J.N.; et al. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum. Mol. Genet. 2006, 15, 637–651. [Google Scholar] [CrossRef]
- Ozawa, R.; Hayashi, Y.K.; Ogawa, M.; Kurokawa, R.; Matsumoto, H.; Noguchi, S.; Nonaka, I.; Nishino, I. Emerin-Lacking Mice Show Minimal Motor and Cardiac Dysfunctions with Nuclear-Associated Vacuoles. Am. J. Pathol. 2006, 168, 907–917. [Google Scholar] [CrossRef]
- Bakay, M.; Wang, Z.; Melcon, G.; Schiltz, L.; Xuan, J.; Zhao, P.; Sartorelli, V.; Seo, J.; Pegoraro, E.; Angelini, C.; et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb–MyoD pathways in muscle regeneration. Brain 2006, 129, 996–1013. [Google Scholar] [CrossRef]
- Collins, C.M.; Ellis, J.A.; Holaska, J.M. MAPK signaling pathways and HDAC3 activity are disrupted during differentiation of emerin-null myogenic progenitor cells. Dis. Model. Mech. 2017, 10, 385–397. [Google Scholar] [CrossRef]
- Frock, R.L.; Kudlow, B.A.; Evans, A.M.; Jameson, S.A.; Hauschka, S.D.; Kennedy, B.K. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genome Res. 2006, 20, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.D.; Guan, T.; Gerace, L. Overlapping Functions of Nuclear Envelope Proteins NET25 (Lem2) and Emerin in Regulation of Extracellular Signal-Regulated Kinase Signaling in Myoblast Differentiation. Mol. Cell. Boil. 2009, 29, 5718–5728. [Google Scholar] [CrossRef] [PubMed]
- Dedeic, Z.; Cetera, M.; Cohen, T.; Holaska, J.M. Emerin inhibits Lmo7 binding to the Pax3 and MyoD promoters and expression of myoblast proliferation genes. J. Cell Sci. 2011, 124, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Koch, A.J.; Holaska, J.M. Expression Profiling of Differentiating Emerin-Null Myogenic Progenitor Identifies Molecular Pathways Implicated in Their Impaired Differentiation. Cells 2017, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.J.; Holaska, J.M. Loss of Emerin Alters Myogenic Signaling and miRNA Expression in Mouse Myogenic Progenitors. PLoS ONE 2012, 7, e37262. [Google Scholar] [CrossRef] [PubMed]
- Demmerle, J.; Koch, A.J.; Holaska, J.M. Emerin and histone deacetylase 3 (HDAC3) cooperatively regulate expression and nuclear positions of MyoD, Myf5, and Pax7 genes during myogenesis. Chromosom. Res. 2013, 21, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Holaska, J.M.; Wilson, K.L. An Emerin “Proteome”: Purification of Distinct Emerin-Containing Complexes from HeLa Cells Suggests Molecular Basis for Diverse Roles Including Gene Regulation, mRNA Splicing, Signaling, Mechanosensing, and Nuclear Architecture. Biochemistry 2007, 46, 8897–8908. [Google Scholar] [CrossRef]
- Bossone, K.A.; Ellis, J.A.; Holaska, J.M. Histone acetyltransferase inhibition rescues differentiation of emerin-deficient myogenic progenitors. Muscle Nerve 2020. [Google Scholar] [CrossRef]
- Toyoshima, K.; Vogt, P.K. Enhancement and inhibition of avian sarcoma viruses by polycations and polyanions. Virology 1969, 38, 414–426. [Google Scholar] [CrossRef]
- Coelen, R.J.; Jose, D.G.; May, J.T. The effect of hexadimethrine bromide (polybrene) on the infection of the primate retroviruses SSV 1/SSAV 1 and BaEV. Arch. Virol. 1983, 75, 307–311. [Google Scholar] [CrossRef]
- Davis, H.E.; Morgan, J.R.; Yarmush, M.L. Polybrene increases retrovirus gene transfer efficiency by enhancing receptor-independent virus adsorption on target cell membranes. Biophys. Chem. 2002, 97, 159–172. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; Muñoz-Canoves, P.; Perdiguero, E. Regulation of skeletal muscle stem cells through epigenetic mechanisms. Toxicol. Mech. Methods 2011, 21, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Zullo, J.M.; Bertolino, E.; Singh, H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 2008, 452, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Juan, A.H.; Wang, S.; Ko, K.D.; Zare, H.; Tsai, P.-F.; Feng, X.; Vivanco, K.O.; Ascoli, A.M.; Gutierrez-Cruz, G.; Krebs, J.; et al. Roles of H3K27me2 and H3K27me3 Examined During Fate Specification of Embryonic Stem Cells. Cell Rep. 2016, 17, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Kind, J.; Pagie, L.; Ortabozkoyun, H.; Boyle, S.; De Vries, S.S.; Janssen, H.; Amendola, M.; Nolen, L.D.; Bickmore, W.A.; Van Steensel, B. Single-Cell Dynamics of Genome-Nuclear Lamina Interactions. Cell 2013, 153, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Zullo, J.M.; Demarco, I.A.; Pique-Regi, R.; Gaffney, D.J.; Epstein, C.B.; Spooner, C.J.; Luperchio, T.R.; Bernstein, B.E.; Pritchard, J.K.; Reddy, K.; et al. DNA Sequence-Dependent Compartmentalization and Silencing of Chromatin at the Nuclear Lamina. Cell 2012, 149, 1474–1487. [Google Scholar] [CrossRef]
- Faralli, H.; Wang, C.; Nakka, K.; Benyoucef, A.; Sebastian, S.; Zhuang, L.; Chu, A.; Palii, C.G.; Liu, C.; Camellato, B.; et al. UTX demethylase activity is required for satellite cell-mediated muscle regeneration. J. Clin. Investig. 2016, 126, 1555–1665. [Google Scholar] [CrossRef]
- Puri, P.L.; Iezzi, S.; Stiegler, P.; Chen, T.-T.; Schiltz, R.; Muscat, G.; Giordano, A.; Kedes, L.; Wang, J.Y.; Sartorelli, V. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell 2001, 8, 885–897. [Google Scholar] [CrossRef]
- Caretti, G.; Di Padova, M.; Micales, B.; Lyons, G.E.; Sartorelli, V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genome Res. 2004, 18, 2627–2638. [Google Scholar] [CrossRef]
- Mal, A.; Harter, M.L. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 1735–1739. [Google Scholar] [CrossRef]
- Ohkawa, Y.; Marfella, C.G.; Imbalzano, A.N. Skeletal muscle specification by myogenin and Mef2D via the SWI/SNF ATPase Brg1. EMBO J. 2006, 25, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yao, Z.; Sarkar, D.; Lawrence, M.; Sanchez, G.J.; Parker, M.H.; MacQuarrie, K.; Davison, J.; Morgan, M.; Ruzzo, W.L.; et al. Genome-wide MyoD Binding in Skeletal Muscle Cells: A Potential for Broad Cellular Reprogramming. Dev. Cell 2010, 18, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Peng, J.; Jiang, S. The epigenetic regulation of embryonic myogenesis and adult muscle regeneration by histone methylation modification. Biochem. Biophys. Rep. 2016, 6, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.M.; Tifft, E.K.; Wilson, K.L. The nuclear envelope LEM-domain protein emerin. Nucleus 2013, 4, 298–314. [Google Scholar] [CrossRef] [PubMed]
- De La Luna, S.; Allen, K.; Mason, S.L.; La Thangue, N.B. Integration of a growth-suppressing BTB/POZ domain protein with the DP component of the E2F transcription factor. EMBO J. 1999, 18, 212–228. [Google Scholar] [CrossRef]
- Pardee, A. G1 events and regulation of cell proliferation. Science 1989, 246, 603–608. [Google Scholar] [CrossRef]
- Pardee, A.B. A Restriction Point for Control of Normal Animal Cell Proliferation. Proc. Natl. Acad. Sci. USA 1974, 71, 1286–1290. [Google Scholar] [CrossRef]
- DeGregori, J.; Kowalik, T.; Nevins, J.R. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 1995, 15, 4215–4224. [Google Scholar] [CrossRef]
- Asano, M.; Nevins, J.R.; Wharton, R.P. Ectopic E2F expression induces S phase and apoptosis in Drosophila imaginal discs. Genes Dev. 1996, 10, 1422–1432. [Google Scholar] [CrossRef]
- Dyson, N.J. The regulation of E2F by pRB-family proteins. Genome Res. 1998, 12, 2245–2262. [Google Scholar] [CrossRef]
- Lukas, J.; Petersen, O.B.; Holm, K.; Bartek, J.; Helin, K. Deregulated expression of E2F family members induces S-phase entry and overcomes p16INK4A-mediated growth suppression. Mol. Cell. Boil. 1996, 16, 1047–1057. [Google Scholar] [CrossRef]
- Chen, H.-Z.; Tsai, S.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef]
- Johnson, D.G. Role of E2F in cell cycle control and cancer. Front. Biosci. 1998, 3, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Pollard, T.D.; Earnshaw, W.C.; Lippincott-Schwartz, J.; Johnson, G.T. Cell Biology, 3rd ed.; Earnshaw, W.C., Ed.; Elsevier: Philadelphia, PA, USA, 2017. [Google Scholar]
- A Henley, S.; Dick, F.A. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Uchida, C. Roles of pRB in the Regulation of Nucleosome and Chromatin Structures. BioMed Res. Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bandara, L.R.; Adamczewski, J.P.; Hunt, T.; La Thangue, N.B. Cyclin A and the retinoblastoma gene product complex with a common transcription factor. Nature 1991, 352, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Chellappan, S.P.; Hiebert, S.; Mudryj, M.; Horowitz, J.M.; Nevins, J.R. The E2F transcription factor is a cellular target for the RB protein. Cell 1991, 65, 1053–1061. [Google Scholar] [CrossRef]
- Chittenden, T.; Livingston, D.M.; Kaelin, W.G. The T/E1A-binding domain of the retinoblastoma product can interact selectively with a sequence-specific DNA-binding protein. Cell 1991, 65, 1073–1082. [Google Scholar] [CrossRef]
- Hiebert, S.W.; Horowitz, J.M.; Chellappan, S.P.; Nevins, J.R. The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes Dev. 1992, 6, 177–185. [Google Scholar] [CrossRef]
- Lee, K.K.; Haraguchi, T.; Lee, R.S.; Koujin, T.; Hiraoka, Y.; Wilson, K.L. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001, 114, 4567–4573. [Google Scholar]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of a-Type Lamin Expression Compromises Nuclear Envelope Integrity Leading to Muscular Dystrophy. J. Cell Boil. 1999, 147, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genome Res. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, T.; Misteli, T. The lamin protein family. Genome Boil. 2011, 12, 222. [Google Scholar] [CrossRef]
- Shin, J.-Y.; Méndez-López, I.; Wang, Y.; Hays, A.P.; Tanji, K.; Lefkowitch, J.H.; Schulze, P.C.; Worman, H.J.; Dauer, W.T. Lamina-associated polypeptide-1 interacts with the muscular dystrophy protein emerin and is essential for skeletal muscle maintenance. Dev. Cell 2013, 26, 591–603. [Google Scholar] [CrossRef]
- Koch, A.J.; Holaska, J.M. Emerin in health and disease. Semin. Cell Dev. Boil. 2013, 29, 95–106. [Google Scholar] [CrossRef]
- Leach, J.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260–264. [Google Scholar] [CrossRef]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef]
- Wackerhage, H.; Del Re, M.P.; Judson, R.N.; Sudol, M.; Sadoshima, J. The Hippo signal transduction network in skeletal and cardiac muscle. Sci. Signal. 2014, 7, re4. [Google Scholar] [CrossRef]
- Hansen, C.; Moroishi, T.; Guan, K.-L. YAP and TAZ: A nexus for Hippo signaling and beyond. Trends Cell Boil. 2015, 25, 499–513. [Google Scholar] [CrossRef]
- Harvey, K.F.; Tapon, N. The Salvador–Warts–Hippo pathway—An emerging tumour-suppressor network. Nat. Rev. Cancer 2007, 7, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Judson, R.N.; Tremblay, A.M.; Knopp, P.; White, R.; Urcia, R.; De Bari, C.; Zammit, P.S.; Camargo, F.D.; Wackerhage, H. The Hippo pathway member Yap plays a key role in influencing fate decisions in muscle satellite cells. J. Cell Sci. 2012, 125, 6009–6019. [Google Scholar] [CrossRef] [PubMed]
- Park, G.H.; Jeong, H.; Jeong, M.-G.; Jang, E.J.; Bae, M.A.; Lee, Y.-L.; Kim, N.J.; Hong, J.-H.; Hwang, E.S. Novel TAZ modulators enhance myogenic differentiation and muscle regeneration. Br. J. Pharmacol. 2014, 171, 4051–4061. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.M.; Missiaglia, E.; Galli, G.G.; Hettmer, S.; Urcia, R.; Carrara, M.; Judson, R.N.; Thway, K.; Nadal, G.; Selfe, J.L.; et al. The Hippo Transducer YAP1 Transforms Activated Satellite Cells and Is a Potent Effector of Embryonal Rhabdomyosarcoma Formation. Cancer Cell 2014, 26, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Watt, K.I.; Judson, R.; Medlow, P.; Reid, K.; Kurth, T.B.; Burniston, J.G.; Ratkevicius, A.; De Bari, C.; Wackerhage, H. Yap is a novel regulator of C2C12 myogenesis. Biochem. Biophys. Res. Commun. 2010, 393, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Bae, S.; An, S.Y.; Byun, M.R.; Hwang, J.-H.; Yaffe, M.B.; Hong, J.-H.; Hwang, E.S. TAZ as a novel enhancer of MyoD-mediated myogenic differentiation. FASEB J. 2010, 24, 3310–3320. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Sun, C.; De Mello, V.; Selfe, J.L.; Missiaglia, E.; Shipley, J.; Murray, I.G.; Zammit, P.S.; Wackerhage, H. The Hippo effector TAZ (WWTR1) transforms myoblasts and TAZ abundance is associated with reduced survival in embryonal rhabdomyosarcoma. J. Pathol. 2016, 240, 3–14. [Google Scholar] [CrossRef]
- Sun, C.; De Mello, V.; Mohamed, A.; Quiroga, H.P.O.; Garcia-Munoz, A.; Al Bloshi, A.; Tremblay, A.M.; Von Kriegsheim, A.; Collie-Duguid, E.; Vargesson, N.; et al. Common and Distinctive Functions of the Hippo Effectors Taz and Yap in Skeletal Muscle Stem Cell Function. STEM CELLS 2017, 35, 1958–1972. [Google Scholar] [CrossRef]
- Owens, D.J.; Fischer, M.; Jabre, S.; Moog, S.; Mamchaoui, K.; Butler-Browne, G.; Coirault, C. Lamin Mutations Cause Increased YAP Nuclear Entry in Muscle Stem Cells. Cells 2020, 9, 816. [Google Scholar] [CrossRef]
- Hay, E.D. Cell Biology of Extracellular Matrix; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Thomas, K.; Engler, A.J.; Meyer, G.A. Extracellular matrix regulation in the muscle satellite cell niche. Connect. Tissue Res. 2014, 56, 1–8. [Google Scholar] [CrossRef]
- Cornelison, D.; Filla, M.S.; Stanley, H.M.; Rapraeger, A.C.; Olwin, B.B. Syndecan-3 and Syndecan-4 Specifically Mark Skeletal Muscle Satellite Cells and Are Implicated in Satellite Cell Maintenance and Muscle Regeneration. Dev. Boil. 2001, 239, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Cornelison, D.; Wilcox-Adelman, S.A.; Goetinck, P.F.; Rauvala, H.; Rapraeger, A.C.; Olwin, B.B. Essential and separable roles for Syndecan-3 and Syndecan-4 in skeletal muscle development and regeneration. Genome Res. 2004, 18, 2231–2236. [Google Scholar] [CrossRef] [PubMed]
- Osses, N.; Brandan, E. ECM is required for skeletal muscle differentiation independently of muscle regulatory factor expression. Am. J. Physiol. Physiol. 2002, 282, C383–C394. [Google Scholar] [CrossRef] [PubMed]
- Osses, N.; Casar, J.C.; Brandan, E. Inhibition of extracellular matrix assembly induces the expression of osteogenic markers in skeletal muscle cells by a BMP-2 independent mechanism. BMC Cell Boil. 2009, 10, 73. [Google Scholar] [CrossRef]
- Allen, R.E.; Boxhorn, L.K. Regulation of skeletal muscle satellite cell proliferation and differentiation by transforming growth factor-beta, insulin-like growth factor I, and fibroblast growth factor. J. Cell. Physiol. 1989, 138, 311–315. [Google Scholar] [CrossRef]
- Gutieérrez, J.; Brandan, E. A Novel Mechanism of Sequestering Fibroblast Growth Factor 2 by Glypican in Lipid Rafts, Allowing Skeletal Muscle Differentiation. Mol. Cell. Boil. 2010, 30, 1634–1649. [Google Scholar] [CrossRef]
- Jenniskens, G.J.; Hafmans, T.; Veerkamp, J.H.; Van Kuppevelt, T.H. Spatiotemporal distribution of heparan sulfate epitopes during myogenesis and synaptogenesis: A study in developing mouse intercostal muscle. Dev. Dyn. 2002, 225, 70–79. [Google Scholar] [CrossRef]
- Liu, X.; Nestor, K.; McFarland, D.C.; Velleman, S.G. Developmental expression of skeletal muscle heparan sulfate proteoglycans in turkeys with different growth rates. Poult. Sci. 2002, 81, 1621–1628. [Google Scholar] [CrossRef]
- Sanes, J.R. The Basement Membrane/Basal Lamina of Skeletal Muscle. J. Boil. Chem. 2003, 278, 12601–12604. [Google Scholar] [CrossRef]
- Miura, T.; Kishioka, Y.; Wakamatsu, J.; Hattori, A.; Nishimura, T. Interaction between myostatin and extracellular matrix components. Anim. Sci. J. 2010, 81, 102–107. [Google Scholar] [CrossRef]
- Yasaka, N.; Wakamatsu, J.; Suzuki, K.; Kishioka, Y.; Nishimura, T. Laminin binds to myostatin and attenuates its signaling. Anim. Sci. J. 2013, 84, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.V. Skeletal Muscle Stem Cells from Animals I. Basic Cell Biology. Int. J. Boil. Sci. 2010, 6, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Besse-Patin, A.; Montastier, E.; Vinel, C.; Castan-Laurell, I.; Louche, K.; Dray, C.; Daviaud, D.; Mir, L.; Marques, M.-A.; Thalamas, C.; et al. Effect of endurance training on skeletal muscle myokine expression in obese men: Identification of apelin as a novel myokine. Int. J. Obes. 2013, 38, 707–713. [Google Scholar] [CrossRef]
- Yamamoto, T.; Habata, Y.; Matsumoto, Y.; Yasuhara, Y.; Hashimoto, T.; Hamajyo, H.; Anayama, H.; Fujii, R.; Fuse, H.; Shintani, Y.; et al. Apelin-transgenic mice exhibit a resistance against diet-induced obesity by increasing vascular mass and mitochondrial biogenesis in skeletal muscle. Biochim. Biophys. Acta (BBA) Gen. Subj. 2011, 1810, 853–862. [Google Scholar] [CrossRef]
- Hamai, N.; Nakamura, M.; Asano, A. Inhibition of Mitochondrial Protein Synthesis Impaired C2C12 Myoblast Differentiation. Cell Struct. Funct. 1997, 22, 421–431. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Herzberg, N.H.; Middelkoop, E.; Adorf, M.; Dekker, H.L.; Van Galen, M.J.; Berg, M.V.D.; Bolhuis, A.P.; Bogert, C.V.D. Mitochondria in cultured human muscle cells depleted of mitochondrial DNA. Eur. J. Cell Boil. 1993, 61, 400–408. [Google Scholar]
- Korohoda, W.; Pietrzkowski, Z.; Reiss, K. Chloramphenicol, an inhibitor of mitochondrial protein synthesis, inhibits myoblast fusion and myotube differentiation. Folia Histochem. Cytobiol. 1993, 31, 9–13. [Google Scholar]
- Rochard, P.; Rodier, A.; Casas, F.; Cassar-Malek, I.; Marchal-Victorion, S.; Daury, L.; Wrutniak, C.; Cabello, G. Mitochondrial activity is involved in the regulation of myoblast differentiation through myogenin expression and activity of myogenic factors. J. Boil. Chem. 2000, 275, 2733–2744. [Google Scholar] [CrossRef]
- Seyer, P.; Grandemange, S.; Busson, M.; Carazo, Á.; Gamaléri, F.; Pessemesse, L.; Casas, F.; Cabello, G.; Wrutniak-Cabello, C. Mitochondrial activity regulates myoblast differentiation by control of c-Myc expression. J. Cell. Physiol. 2006, 207, 75–86. [Google Scholar] [CrossRef]
- Barbieri, E.; Battistelli, M.; Casadei, L.; Vallorani, L.; Piccoli, G.; Guescini, M.; Gioacchini, A.M.; Polidori, E.; Zeppa, S.; Ceccaroli, P.; et al. Morphofunctional and Biochemical Approaches for Studying Mitochondrial Changes during Myoblasts Differentiation. J. Aging Res. 2011, 2011, 1–16. [Google Scholar] [CrossRef]
- Moyes, C.; Mathieu-Costello, O.A.; Tsuchiya, N.; Filburn, C.; Hansford, R.G. Mitochondrial biogenesis during cellular differentiation. Am. J. Physiol. Content 1997, 272, C1345–C1351. [Google Scholar] [CrossRef] [PubMed]
- Remels, A.H.V.; Langen, R.; Schrauwen, P.; Schaart, G.; Schols, A.; Gosker, H.R. Regulation of mitochondrial biogenesis during myogenesis. Mol. Cell. Endocrinol. 2010, 315, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Dzeja, P.P.; Faustino, R.S.; Perez-Terzic, C.; Behfar, A.; Terzic, A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat. Clin. Pr. Neurol. 2007, 4, S60–S67. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Dzeja, P.P.; Faustino, R.S.; Terzic, A. Developmental restructuring of the creatine kinase system integrates mitochondrial energetics with stem cell cardiogenesis. Ann. N. Y. Acad. Sci. 2008, 1147, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Fink, E.; Fortin, D.; Serrurier, B.; Ventura-Clapier, R.; Bigard, A. Recovery of contractile and metabolic phenotypes in regenerating slow muscle after notexin-induced or crush injury. J. Muscle Res. Cell Motil. 2003, 24, 421–429. [Google Scholar] [CrossRef]
- Wagatsuma, A.; Kotake, N.; Yamada, S. Muscle regeneration occurs to coincide with mitochondrial biogenesis. Mol. Cell. Biochem. 2010, 349, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Duguez, S.; Féasson, L.; Denis, C.; Freyssenet, D. Mitochondrial biogenesis during skeletal muscle regeneration. Am. J. Physiol. Metab. 2002, 282, E802–E809. [Google Scholar] [CrossRef]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia Requires Notch Signaling to Maintain the Undifferentiated Cell State. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Lee, D.S.M.; Skuli, N.; Mesquita, R.C.; Kim, M.N.; Yodh, A.G.; Nguyen-McCarty, M.; Li, B.; Simon, M.C. HIF modulation of Wnt signaling regulates skeletal myogenesis in vivo. J. Cell Sci. 2015, 128, 2405–2412. [Google Scholar] [CrossRef]
- Ono, Y.; Sensui, H.; Sakamoto, Y.; Nagatomi, R. Knockdown of hypoxia-inducible factor-1α by siRNA inhibits C2C12 myoblast differentiation. J. Cell. Biochem. 2006, 98, 642–649. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iyer, A.; Holaska, J.M. EDMD-Causing Emerin Mutant Myogenic Progenitors Exhibit Impaired Differentiation Using Similar Mechanisms. Cells 2020, 9, 1463. https://doi.org/10.3390/cells9061463
Iyer A, Holaska JM. EDMD-Causing Emerin Mutant Myogenic Progenitors Exhibit Impaired Differentiation Using Similar Mechanisms. Cells. 2020; 9(6):1463. https://doi.org/10.3390/cells9061463
Chicago/Turabian StyleIyer, Ashvin, and James M. Holaska. 2020. "EDMD-Causing Emerin Mutant Myogenic Progenitors Exhibit Impaired Differentiation Using Similar Mechanisms" Cells 9, no. 6: 1463. https://doi.org/10.3390/cells9061463
APA StyleIyer, A., & Holaska, J. M. (2020). EDMD-Causing Emerin Mutant Myogenic Progenitors Exhibit Impaired Differentiation Using Similar Mechanisms. Cells, 9(6), 1463. https://doi.org/10.3390/cells9061463