Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases



Abstract
:1. Introduction
2. The Properties and Role of Iron and Iron Proteins in Human Health
2.1. Basic Properties and Distribution of Iron in the Body
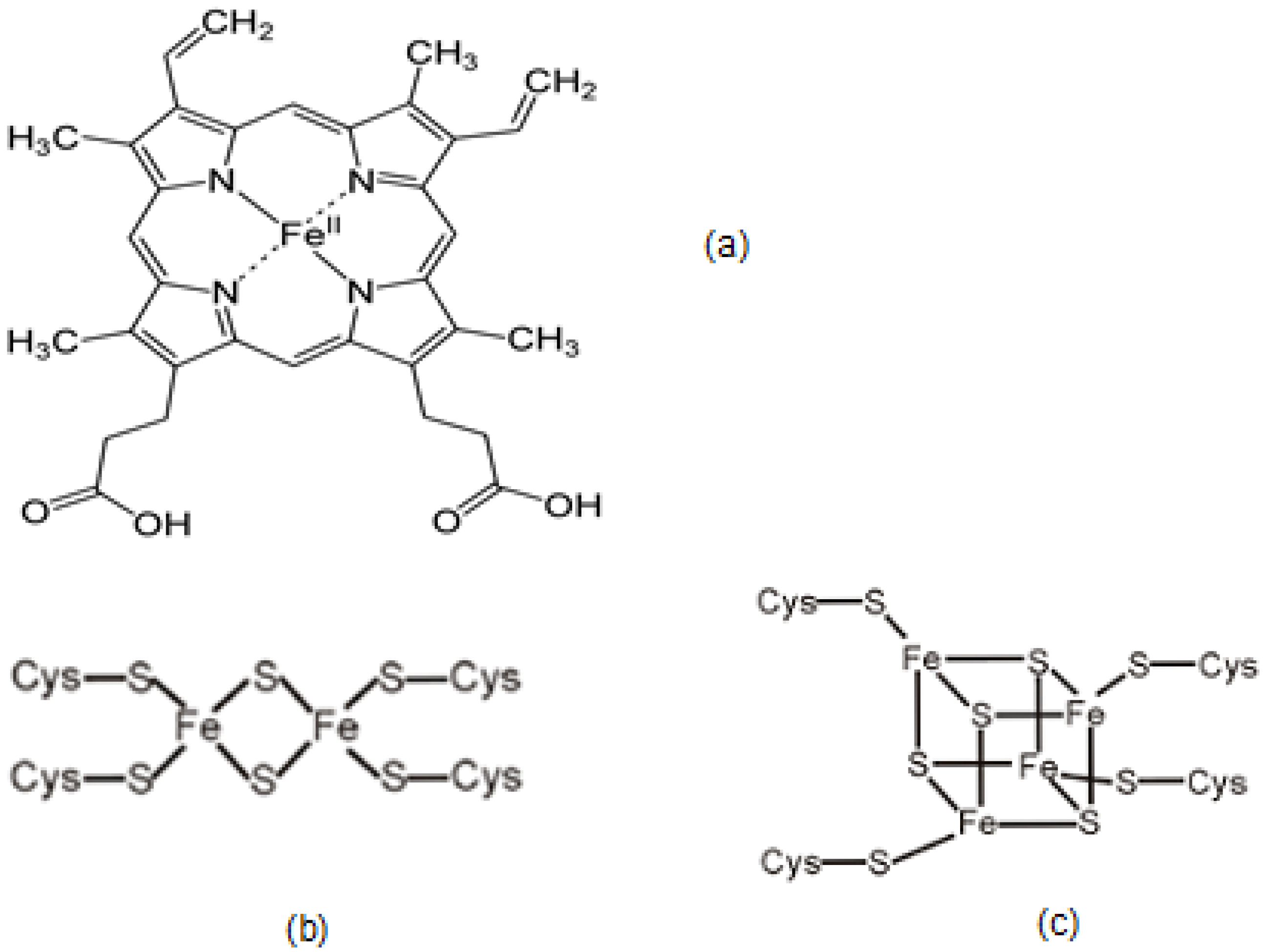
2.2. Iron in Heme, Hemoglobin and Red Blood Cells
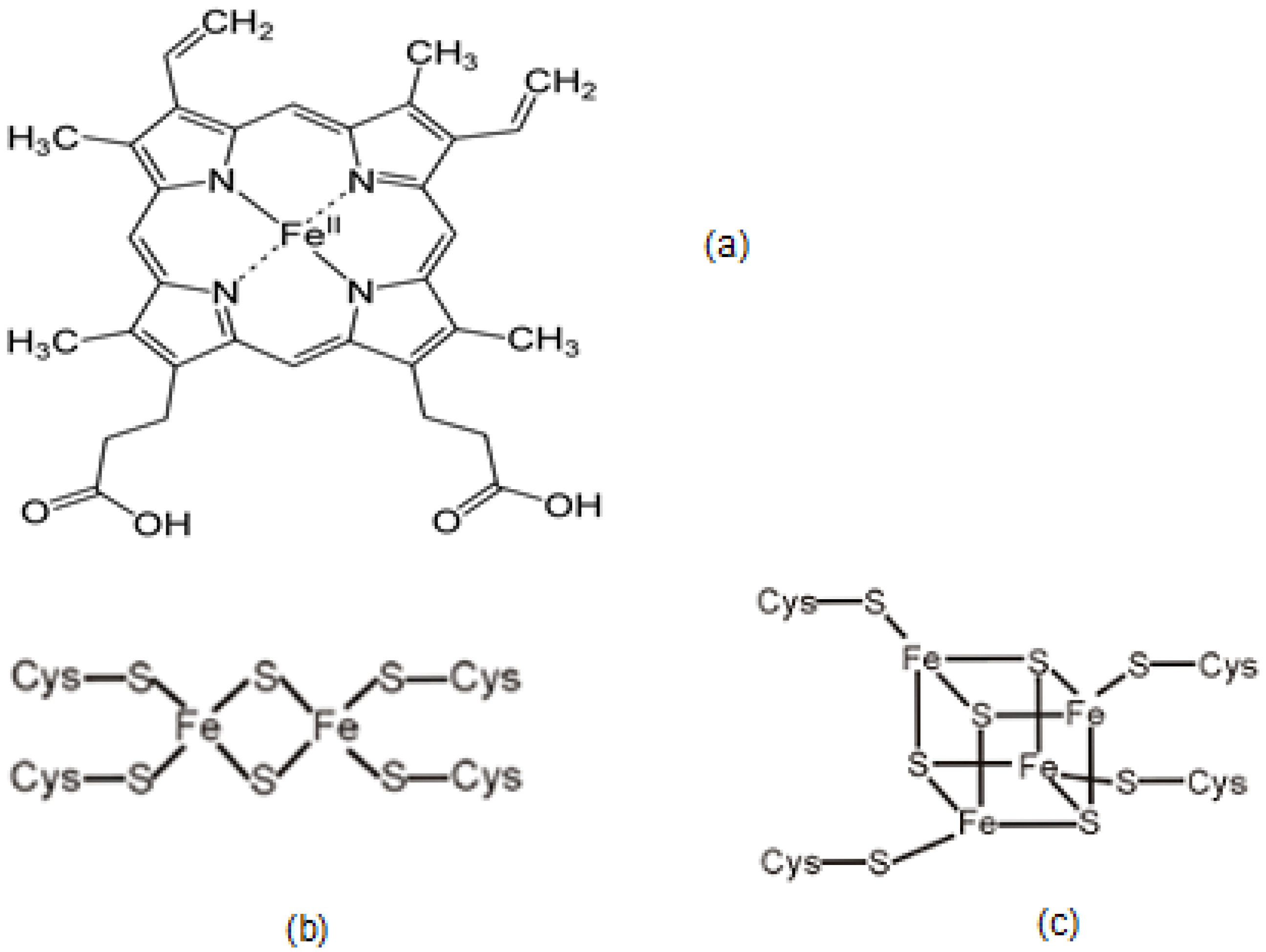
2.3. The Role and Function of Iron-Containing Proteins
2.4. Factors Affecting Iron-Containing Proteins and Implications on Health
3. Ligands and Chelators Binding with Iron
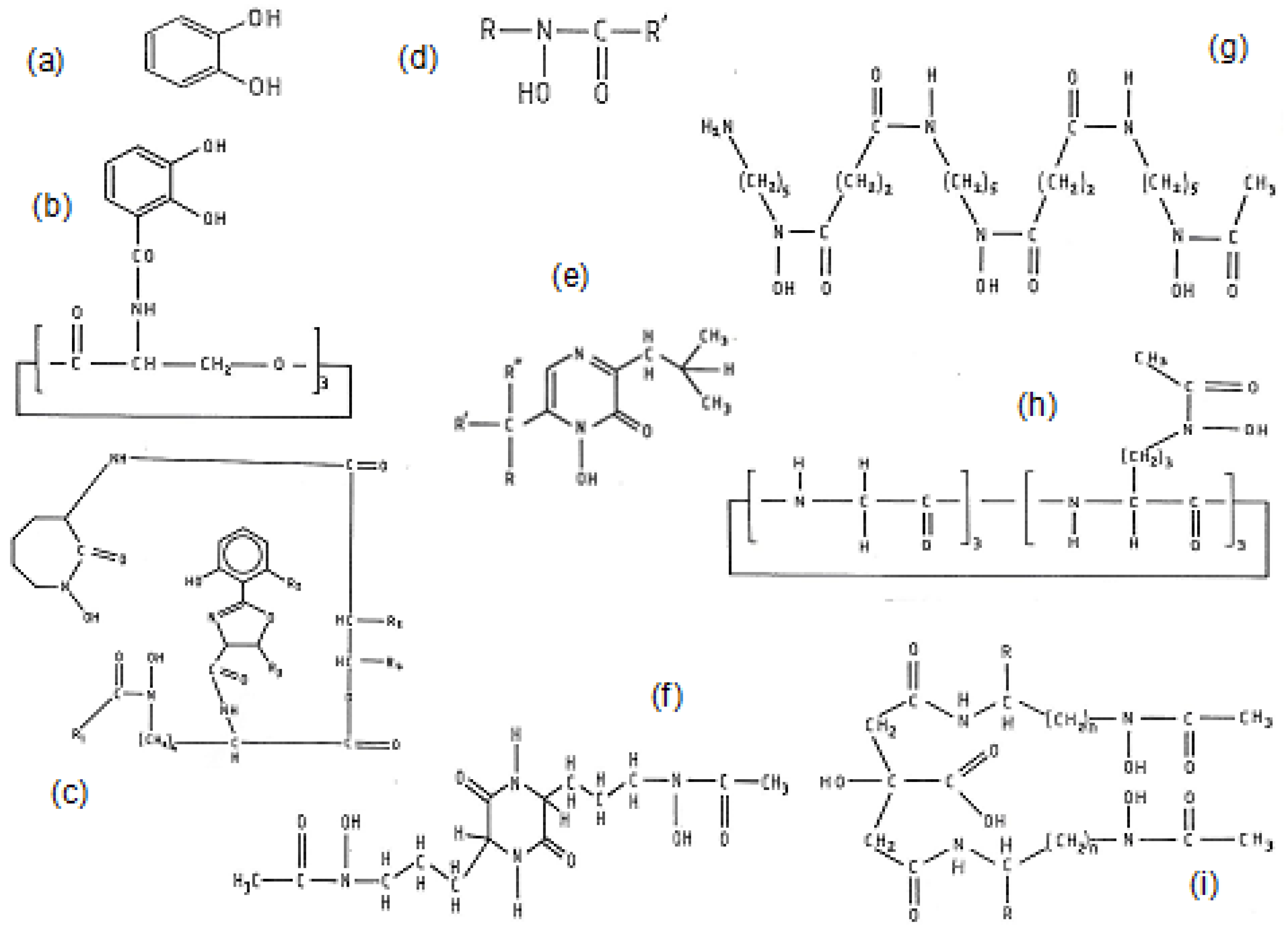
3.1. Naturally Occurring Microbial Chelators (Siderophores)
3.2. Naturally Occurring Plant Chelators (Phytochelators)
3.3. Iron Chelating Drugs in Clinical Use
4. Biologic and Physiological Implications of Interactions with Iron Chelators
4.1. Effects of Chelator and Chelator Iron Complexes on Iron Absorption
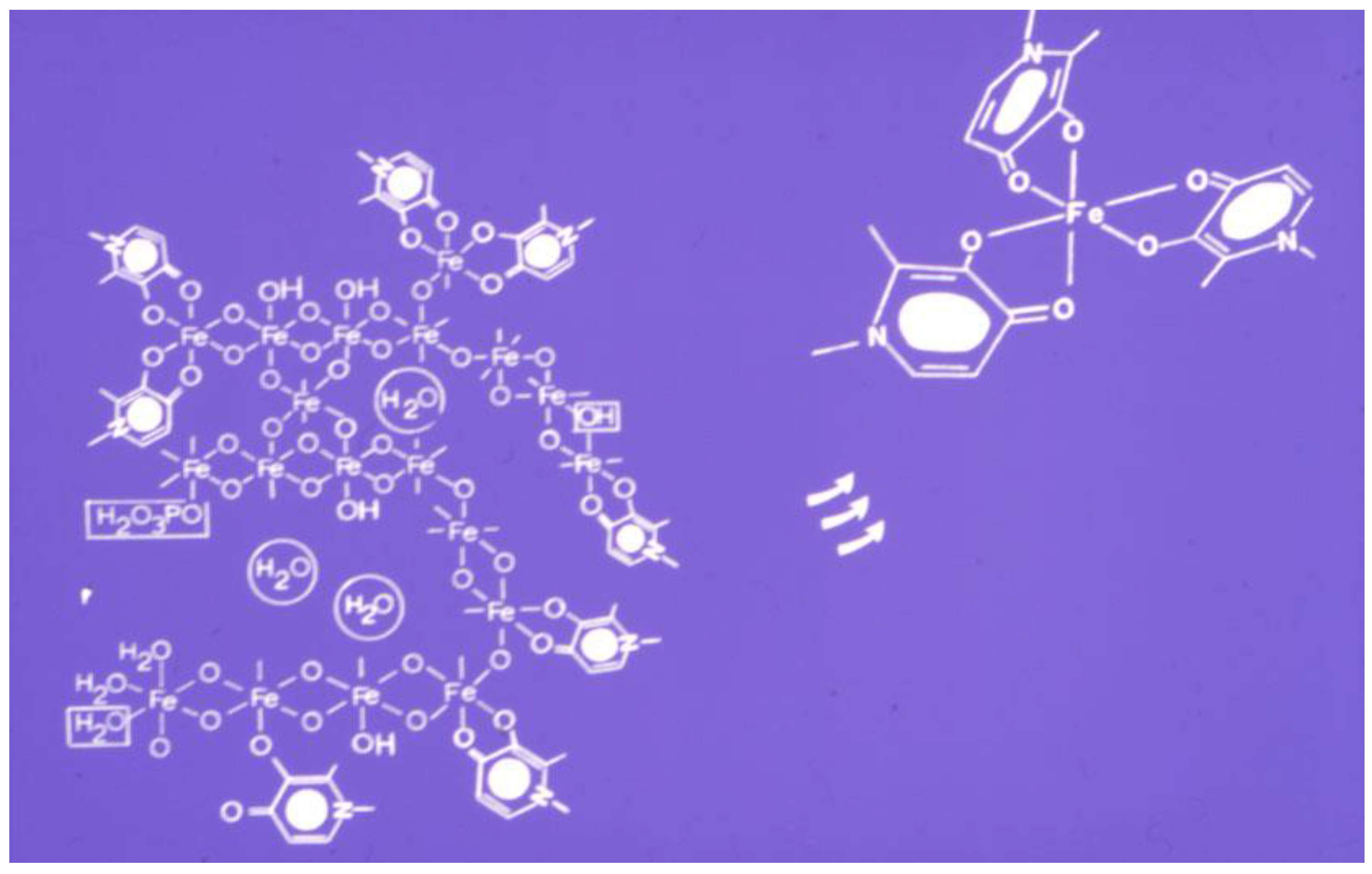
4.2. Iron Removal by Chelators from Ferritin and Hemosiderin and Other Proteins
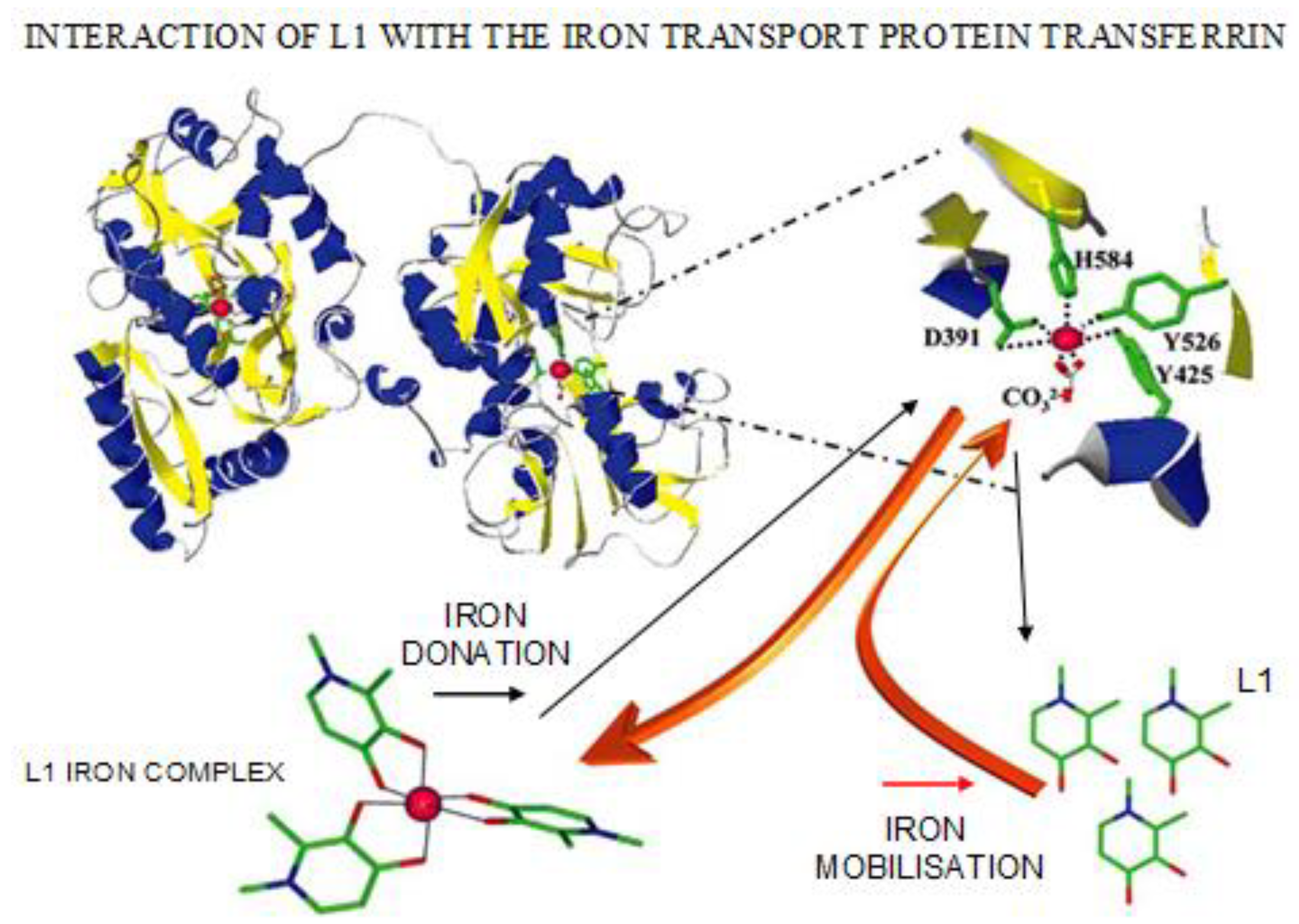
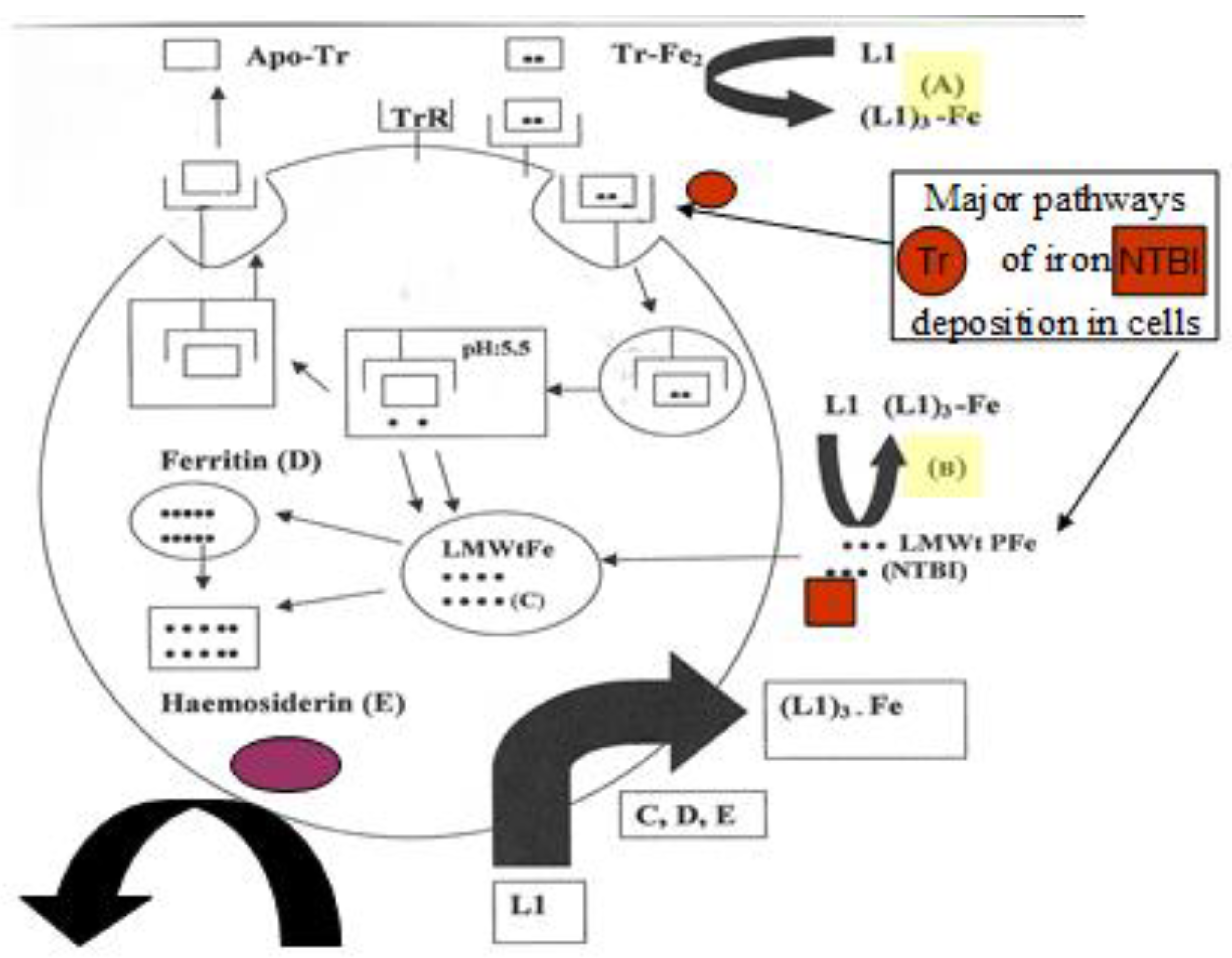
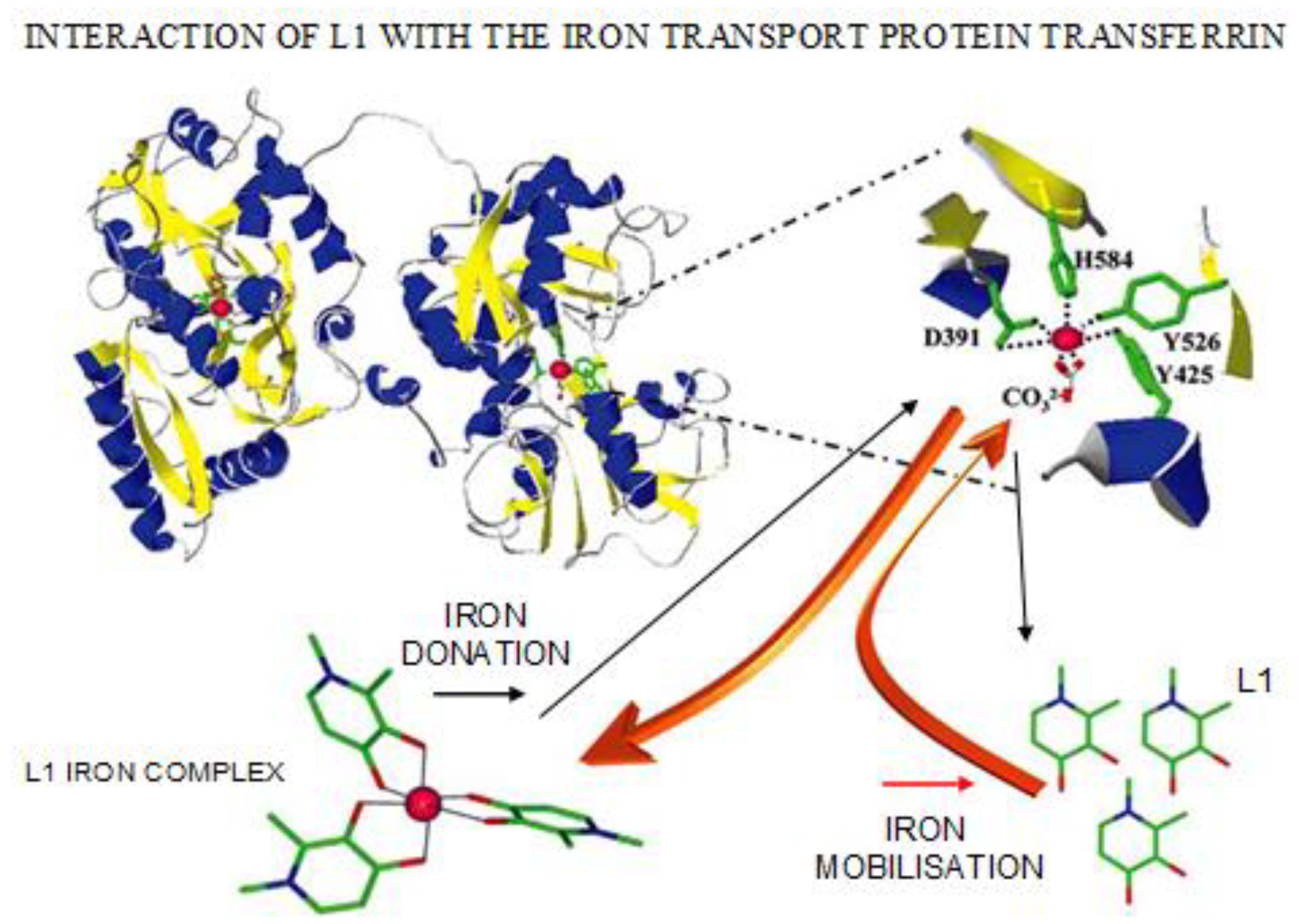
4.3. Transferrin Iron Removal and Other Interactions by Chelators
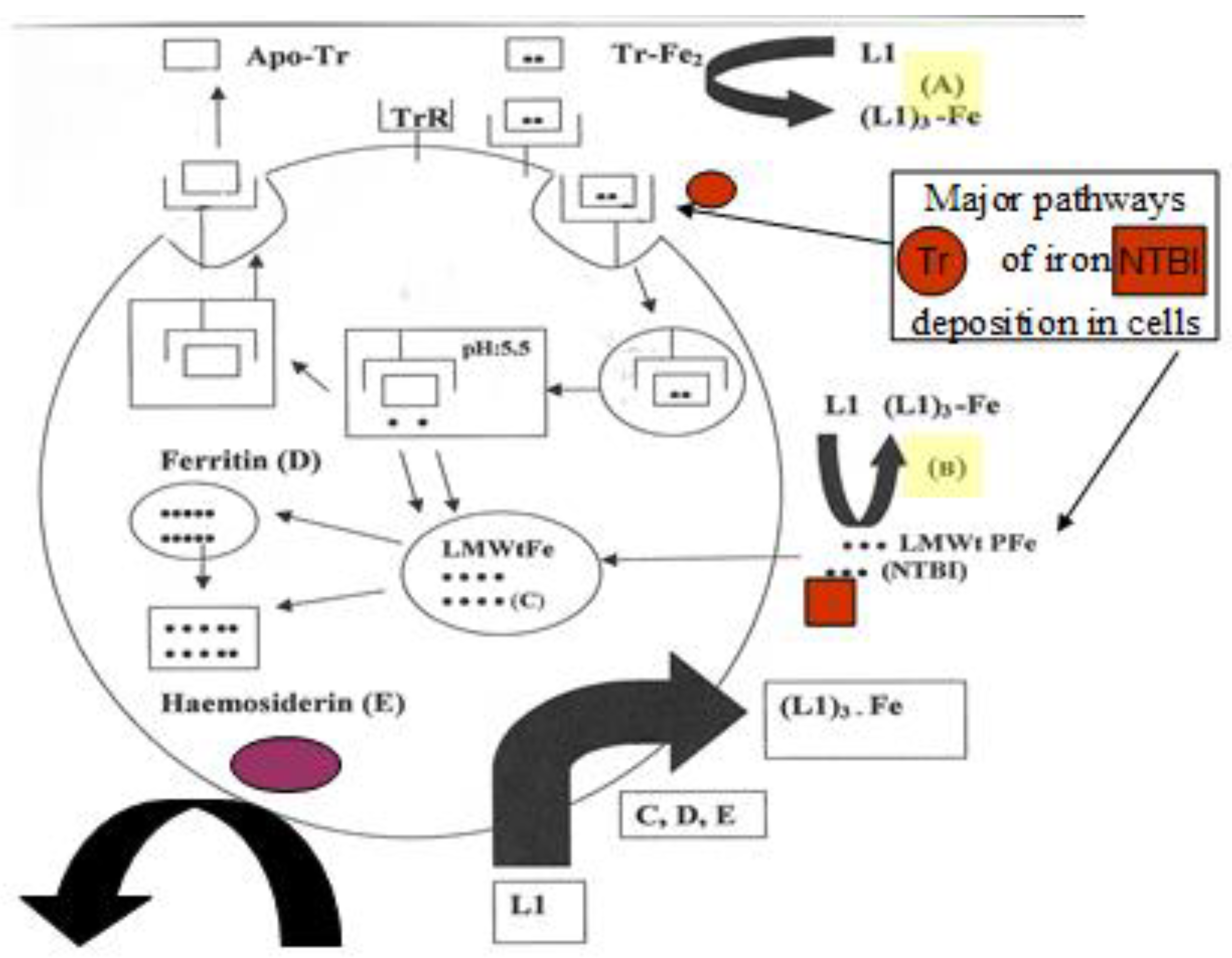
4.4. The Intracellular Low Molecular Weight Iron Pool Changes during Chelation
4.5. Allosteric and Other Interactions of Chelating Drugs with Proteins
5. Interaction of Iron Proteins with Other Metal Ions and the Role of Chelators
6. Chelator Protein Interactions and Free Radical Pathology
7. Prospects for the Clinical Use of Chelators in Infections and Cancer
7.1. Iron, Chelation and Therapeutic Strategies in Infections
7.2. Iron, Chelation and Cancer Therapeutic Strategies
8. Future Prospects
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Prasad, A.S. Zinc: An overview. Nutrition 1995, 11, 93–99. [Google Scholar] [PubMed]
- E Coleman, J. Zinc Proteins: Enzymes, Storage Proteins, Transcription Factors, and Replication Proteins. Annu. Rev. Biochem. 1992, 61, 897–946. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Zinc deficiency. BMJ 2003, 326, 409–410. [Google Scholar] [CrossRef] [PubMed]
- Daniel, K.G.; Harbach, R.H.; Guida, W.C.; Dou, Q.P. Copper storage diseases: Menkes, Wilsons, and cancer. Front. Biosci 2004, 9, 2652–2662. [Google Scholar] [CrossRef]
- Galaris, D.; Pantopoulos, K. Oxidative Stress and Iron Homeostasis: Mechanistic and Health Aspects. Crit. Rev. Clin. Lab. Sci. 2008, 45, 1–23. [Google Scholar] [CrossRef]
- Gozzelino, R.; Arosio, P. Iron Homeostasis in Health and Disease. Int. J. Mol. Sci. 2016, 17, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; De Benoist, B. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993–2005. Public Heal. Nutr. 2008, 12, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Community control of hereditary anaemias: Memorandum from a WHO meeting*. Bull. World Heal. Organ. 1983, 61, 63–80.
- Zurlo, M.; De Stefano, P.; Borgna-Pignatti, C.; Di Palma, A.; Melevendi, C.; Piga, A.; Di Gregorio, F.; Burattini, M.; Terzoli, S. Survival and causes of death in thalassaemia major. Lancet 1989, 334, 27–30. [Google Scholar] [CrossRef]
- Inamoto, Y.; Lee, S.J. Late effects of blood and marrow transplantation. Haematologica 2017, 102, 614–625. [Google Scholar] [CrossRef]
- Gratwohl, A.; Pasquini, M.C.; Aljurf, M.; Atsuta, Y.; Baldomero, H.; Foeken, L.; Gratwohl, M.; Bouzas, L.F.; Confer, D.; Frauendorfer, K.; et al. Worldwide Network for Blood and Marrow Transplantation. (WBMT). One million haemopoietic stem-cell transplants: A retrospective observational study. Lancet Haematol. 2015, 2, e91–e100. [Google Scholar]
- Shenoy, S.; Angelucci, E.; Arnold, S.D.; Baker, K.S.; Bhatia, M.; Bresters, D.; Dietz, A.C.; De La Fuente, J.; Duncan, C.; Gaziev, J.; et al. Current Results and Future Research Priorities in Late Effects after Hematopoietic Stem Cell Transplantation for Children with Sickle Cell Disease and Thalassemia: A Consensus Statement from the Second Pediatric Blood and Marrow Transplant Consortium International Conference on Late Effects after Pediatric Hematopoietic Stem Cell Transplantation. Boil. Blood Marrow Transplant. 2017, 23, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Germing, U.; Schroeder, T.; Kaivers, J.; Kündgen, A.; Kobbe, G.; Gattermann, N. Novel therapies in low- and high-risk myelodysplastic syndrome. Expert Rev. Hematol. 2019, 12, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J. How to manage iron toxicity in post-allogeneic hematopoietic stem cell transplantation? Expert Rev. Hematol. 2020, 13, 299–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontoghiorghes, G.J.; Kontoghiorghe, C.N. Prospects for the introduction of targeted antioxidant drugs for the prevention and treatment of diseases related to free radical pathology. Expert Opin. Investig. Drugs 2019, 28, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta (BBA)-Bioenerg 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Nakamura, T.; Naguro, I.; Ichijo, H.; Nakmura, T. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef]
- Balsano, C.; Porcu, C.; Sideri, S. Is copper a new target to counteract the progression of chronic diseases? Metallomics. 2018, 10, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Denisov, E.T.; Afanas’Ev, I.B. Oxidation and Antioxidants in Organic Chemistry and Biology; CRC Press: Boca Raton, FA, USA, 2005. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.; E Cross, C. Free radicals, antioxidants, and human disease: Where are we now? J. Lab. Clin. Med. 1992, 119, 598–620. [Google Scholar]
- Kontoghiorghes, G.J. Free radicals, oxidant stress and drug action. In Iron Chelation in Biochemistry and Medicine; Rice-Evans, C., Ed.; Rechelieu Press: London, UK, 1978; pp. 277–303. [Google Scholar]
- Kontoghiorghes, G.J. Advances on Chelation and Chelator Metal Complexes in Medicine. Int. J. Mol. Sci. 2020, 21, 2499. [Google Scholar] [CrossRef] [Green Version]
- Development of Iron Chelators for Clinical Us; Anderson, F.W.; Hiller, M.C. (Eds.) DHEW Publication: Bethesda, MD, USA, 1975; pp. 1–275. [Google Scholar]
- Kontoghiorghes, G.J. The Design of Orally Active Iron Chelators for the Treatment of Thalassaemia. Ph.D. Thesis, University of Essex, Colchester, UK, 1982; pp. 1–243. Available online: https://www.pri.ac.cy/files/KGJ_thesis_1982.pdf (accessed on 7 May 2020).
- Kontoghiorghes, G.J.; Pattichis, K.; Neocleous, K.; Kolnagou, A. The design and development of deferiprone (L1) and other iron chelators for clinical use: Targeting methods and application prospects. Curr. Med. Chem. 2004, 11, 2161–2183. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.; Eracleous, E.; Economides, C.; Kolnagou, A. Advances in iron overload therapies. Prospects for effective use of deferiprone (L1), deferoxamine, the new experimental chelators ICL670, GT56-252, L1NAll and their combinations. Curr. Med. Chem. 2005, 12, 2663–2681. [Google Scholar] [CrossRef] [PubMed]
- Weatherall, D.; Clegg, J.B. Inherited haemoglobin disorders: An increasing global health problem. Bull. World Heal. Organ. 2001, 79, 704–712. [Google Scholar]
- Teawtrakul, N.; Chansung, K.; Sirijerachai, C.; Wanitpongpun, C.; Thepsuthammarat, K. The impact and disease burden of thalassemia in Thailand: A population-based study in 2010. J. Med. Assoc. Thail. 2012, 95, 95. [Google Scholar]
- Cairo, G.; Bernuzzi, F.; Recalcati, S. A precious metal: Iron, an essential nutrient for all cells. Genes Nutr. 2006, 1, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Prudent, M.; D’Alessandro, A. Red blood cell storage lesion: Causes and potential clinical consequences. Blood Transfus. 2019, 17, 27–52. [Google Scholar]
- Asaro, R.J.; Zhu, Q.; Cabrales, P. Erythrocyte Aging, Protection via Vesiculation: An Analysis Methodology via Oscillatory Flow. Front. Physiol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Alaarg, A.; Schiffelers, R.M.; Van Solinge, W.W.; Van Wijk, R. Red blood cell vesiculation in hereditary hemolytic anemia. Front. Physiol. 2013, 4, 4. [Google Scholar] [CrossRef]
- Jóźwik, M.; Jóźwik, M.; Józwik, M.; Szczypka, M.; Gajewska, J.; Laskowska-Klita, T. Antioxidant defence of red blood cells and plasma in stored human blood. Clin. Chim. Acta 1997, 267, 129–142. [Google Scholar] [CrossRef]
- Tolosano, E.; Altruda, F. Hemopexin: Structure, Function, and Regulation. DNA Cell Boil. 2002, 21, 297–306. [Google Scholar] [CrossRef]
- Shih, A.W.; McFarlane, A.; Verhovsek, M. Haptoglobin testing in hemolysis: Measurement and interpretation. Am. J. Hematol. 2014, 89, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S. Blood Flow Regulation by S-Nitrosohemoglobin in the Physiological Oxygen Gradient. Science 1997, 276, 2034–2037. [Google Scholar] [CrossRef] [Green Version]
- Postnikova, G.B.; Shekhovtsova, E.A. Myoglobin: Oxygen Depot or Oxygen Transporter to Mitochondria? A Novel Mechanism of Myoglobin Deoxygenation in Cells (review). Biochemistry (Moscow) 2018, 83, 168–183. [Google Scholar] [CrossRef]
- Reeder, B.; Wilson, M.T. Hemoglobin and myoglobin associated oxidative stress: From molecular mechanisms to disease States. Curr. Med. Chem. 2005, 12, 2741–2751. [Google Scholar] [CrossRef] [PubMed]
- Reeder, B.J. Redox and Peroxidase Activities of the Hemoglobin Superfamily: Relevance to Health and Disease. Antioxid. Redox Signal 2017, 26, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Arzola, K.; Velázquez-Cruz, A.; Guerra-Castellano, A.; Casado-Combreras, M.A.; Pérez-Mejías, G.; Díaz-Quintana, A.J.; Díaz-Moreno, I.; De La Rosa, M.A. New moonlighting functions of mitochondrial cytochrome c in the cytoplasm and nucleus. FEBS Lett. 2019, 593, 3101–3119. [Google Scholar] [CrossRef] [Green Version]
- Grevel, A.; Pfanner, N.; Becker, T. Coupling of import and assembly pathways in mitochondrial protein biogenesis. Boil. Chem. 2019, 401, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Vringer, E.; Tait, S.W. Mitochondria and Inflammation: Cell Death Heats Up. Front. Cell Dev. Boil. 2019, 7, 100. [Google Scholar] [CrossRef]
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An extreme multifunctional protein with a key role in cell fate. Int. J. Boil. Macromol. 2019, 136, 1237–1246. [Google Scholar] [CrossRef]
- Ramzan, R.; Vogt, S.; Kadenbach, B. Stress-mediated generation of deleterious ROS in healthy individuals-role of cytochrome c oxidase. J. Mol. Med. 2020, 98, 651–657. [Google Scholar] [CrossRef] [Green Version]
- Timon-Gomez, A.; Nývltová, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Boil. 2018, 76, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Llases, M.; Morgada, M.N.; Vila, A.J. Biochemistry of Copper Site Assembly in Heme-Copper Oxidases: A Theme with Variations. Int. J. Mol. Sci. 2019, 20, 3830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, D.; Kim, S.-M.; Correia, M.A. Cytochrome P450 endoplasmic reticulum-associated degradation (ERAD): Therapeutic and pathophysiological implications. Acta Pharm. Sin. B 2019, 10, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, P.; Nagini, S.; Palrasu, M. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Tornio, A.; Backman, J.T. Cytochrome P450 in Pharmacogenetics: An Update. Adv. Pharmacol. 2018, 3–32. [Google Scholar] [CrossRef] [Green Version]
- Danielson, P.B. The Cytochrome P450 Superfamily: Biochemistry, Evolution and Drug Metabolism in Humans. Curr. Drug Metab. 2002, 3, 561–597. [Google Scholar] [CrossRef]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Nemeth, E. New insights into iron regulation and erythropoiesis. Curr. Opin. Hematol. 2015, 22, 199–205. [Google Scholar] [CrossRef]
- Miller, C.G.; Holmgren, A.; Arnér, E.S.J.; Schmidt, E.E. NADPH-dependent and -independent disulfide reductase systems. Free. Radic. Boil. Med. 2018, 127, 248–261. [Google Scholar] [CrossRef]
- Puig, S.; Ramos-Alonso, L.; Romero, A.M.; Pastor, M.T.M. The elemental role of iron in DNA synthesis and repair. Metallomics 2017, 9, 1483–1500. [Google Scholar] [CrossRef] [Green Version]
- Aye, Y.; Li, M.; Long, M.J.C.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2014, 34, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J.; Neocleous, K.; Kolnagou, A. Benefits and risks of deferiprone in iron overload in Thalassaemia and other conditions: Comparison of epidemiological and therapeutic aspects with deferoxamine. Drug Saf. 2003, 26, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Patrinos, G.P.; Grosveld, F. Pharmacogenomics and Therapeutics of Hemoglobinopathies. Hemoglobin 2008, 32, 229–236. [Google Scholar] [CrossRef]
- Beinert, H.; Holm, R.H.; Münck, E. Iron-Sulfur Clusters: Nature’s Modular, Multipurpose Structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Ciofi-Baffoni, S.; Nasta, V.; Banci, L. Protein networks in the maturation of human iron–sulfur proteins. Metallomics 2018, 10, 49–72. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Rodriguez, M.; Chatzi, A.; Tokatlidis, K. Iron–sulfur clusters: From metals through mitochondria biogenesis to disease. J. Boil. Inorg. Chem. 2018, 23, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabata, H. Transferrin and transferrin receptors update. Free. Radic. Boil. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef]
- Levina, A.; Lay, P.A. Transferrin Cycle and Clinical Roles of Citrate and Ascorbate in Improved Iron Metabolism. ACS Chem. Boil. 2019, 14, 893–900. [Google Scholar] [CrossRef]
- Ott, D.B.; Hartwig, A.; Stillman, M.J. Competition between Al3+ and Fe3+ binding to human transferrin and toxicological implications: Structural investigations using ultra-high resolution ESI MS and CD spectroscopy. Metallomics 2019, 11, 968–981. [Google Scholar] [CrossRef]
- Pratt, R.; Handelman, G.J.; Edwards, T.E.; Gupta, A. Ferric pyrophosphate citrate: Interactions with transferrin. BioMetals 2018, 31, 1081–1089. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Timilsena, Y.; Blanch, E.; Adhikari, B. Lactoferrin: Structure, function, denaturation and digestion. Crit. Rev. Food Sci. Nutr. 2017, 59, 580–596. [Google Scholar] [CrossRef] [PubMed]
- Rosa, L.; Cutone, A.; Lepanto, M.; Paesano, R.; Valenti, P. Lactoferrin: A Natural Glycoprotein Involved in Iron and Inflammatory Homeostasis. Int. J. Mol. Sci. 2017, 18, 1985. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Elia, L.; Poli, M. Ferritin, cellular iron storage and regulation. IUBMB Life 2017, 69, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Saito, H. Storage Iron Turnover from a New Perspective. Acta Haematol. 2019, 141, 201–208. [Google Scholar] [CrossRef] [PubMed]
- La, A.; Nguyen, T.; Sauble, E.; Tu, D.; Gonzalez, A.; Kidane, T.Z.; Soriano, C.; Morgan, J.; Doan, M.; Tran, K.; et al. Mobilization of iron from ferritin: New steps and details. Metallomics 2018, 10, 154–168. [Google Scholar] [CrossRef]
- Wang, W.; Knovich, M.A.; Coffman, L.; Torti, F.M.; Torti, S.V. Serum ferritin: Past, present and future. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2010, 1800, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Sargent, P.J.; Farnaud, S.; Evans, R.W. Structure/function overview of proteins involved in iron storage and transport. Curr. Med. Chem. 2005, 12, 2683–2693. [Google Scholar] [CrossRef]
- Kontoghiorghe, C.N.; Kolnagou, A.; Kontoghiorghes, G.J. Potential clinical applications of chelating drugs in diseases targeting transferrin-bound iron and other metals. Expert Opin. Investig. Drugs 2013, 22, 591–618. [Google Scholar] [CrossRef]
- Mehlenbacher, M.; Poli, M.; Arosio, P.; Santambrogio, P.; Levi, S.; Chasteen, N.D.; Bou-Abdallah, F. Iron Oxidation and Core Formation in Recombinant Heteropolymeric Human Ferritins. Biochemistry 2017, 56, 3900–3912. [Google Scholar] [CrossRef]
- Theil, E.C. Ferritin: The Protein Nanocage and Iron Biomineral in Health and in Disease. Inorg. Chem. 2013, 52, 12223–12233. [Google Scholar] [CrossRef]
- Iancu, T.C. Ferritin and hemosiderin in pathological tissues. Electron Microsc. Rev. 1992, 5, 209–229. [Google Scholar] [CrossRef]
- Jacobs, A. An intracellular transit iron pool. Ciba Found. Symp. 1976, 51, 91–106. [Google Scholar]
- Neilands, J.B. Siderophores: Structure and Function of Microbial Iron Transport Compounds. J. Boil. Chem. 1995, 270, 26723–26726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontoghiorghe, C.N.; Kolnagou, A.; Kontoghiorghes, G.J. Phytochelators Intended for Clinical Use in Iron Overload, Other Diseases of Iron Imbalance and Free Radical Pathology. Molecules 2015, 20, 20841–20872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, B.R.; Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Siderophores in Iron Metabolism: From Mechanism to Therapy Potential. Trends Mol. Med. 2016, 22, 1077–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, M.G.P. The Role of Iron and Siderophores in Infection, and the Development of Siderophore Antibiotics. Clin. Infect. Dis. 2019, 69, S529–S537. [Google Scholar] [CrossRef]
- Huang, H.; Liao, D.; Dong, Y.; Pu, R. Effect of quercetin supplementation on plasma lipid profiles, blood pressure, and glucose levels: A systematic review and meta-analysis. Nutr. Rev. 2020, pii: nuz071. [Google Scholar] [CrossRef] [Green Version]
- Hezaveh, Z.S.; Azarkeivan, A.; Janani, L.; Hosseini, S.; Shidfar, F. The effect of quercetin on iron overload and inflammation in β-thalassemia major patients: A double-blind randomized clinical trial. Complement. Ther. Med. 2019, 46, 24–28. [Google Scholar] [CrossRef]
- Salehi, B.; Stojanović-Radić, Z.; Matejić, J.; Sharifi-Rad, J.; Kumar, N.V.A.; Martins, N.; Sharifi-Rad, J. The therapeutic potential of curcumin: A review of clinical trials. Eur. J. Med. Chem. 2019, 163, 527–545. [Google Scholar] [CrossRef]
- Gillessen, A.; Schmidt, H.H.-J. Silymarin as Supportive Treatment in Liver Diseases: A Narrative Review. Adv. Ther. 2020, 37, 1279–1301. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Zhang, D.; Zhang, J.; Yuan, J. Metabolism, Transport and Drug-Drug Interactions of Silymarin. Molecules 2019, 24, E3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yu, S.; Wang, F.; Yu, H.; Li, X.; Dong, W.; Lin, R.; Liu, Q. Chronic administration of ellagic acid improved the cognition in middle-aged overweight men. Appl. Physiol. Nutr. Metab. 2018, 43, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Kosuru, R.Y.; Roy, A.; Das, S.K.; Bera, S. Gallic Acid and Gallates in Human Health and Disease: Do Mitochondria Hold the Key to Success? Mol. Nutr. Food Res. 2017, 62, 1700699. [Google Scholar] [CrossRef] [PubMed]
- Van Gorkom, G.N.; Lookermans, E.L.; Van Elssen, C.H.; Bos, G.M. The Effect of Vitamin C (Ascorbic Acid) in the Treatment of Patients with Cancer: A Systematic Review. Nutrition 2019, 11, 977. [Google Scholar] [CrossRef] [Green Version]
- Hager, D.N.; Hinson, J.S.; Rothman, R.E. Vitamin C for Sepsis and Acute Respiratory Failure. JAMA 2020, 323, 791–792. [Google Scholar] [CrossRef]
- Nielsen, T.K.; Højgaard, M.; Andersen, J.T.; Poulsen, H.E.; Lykkesfeldt, J.; Mikines, K.J. Elimination of Ascorbic Acid After High-Dose Infusion in Prostate Cancer Patients: A Pharmacokinetic Evaluation. Basic Clin. Pharmacol. Toxicol. 2014, 116, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Martín-Peláez, S.; Covas, M.I.; Fitó, M.; Kušar, A.; Pravst, I. Health effects of olive oil polyphenols: Recent advances and possibilities for the use of health claims. Mol. Nutr. Food Res. 2013, 57, 760–771. [Google Scholar] [CrossRef]
- Visioli, F.; De La Lastra, A.; Andres-Lacueva, C.; Aviram, M.; Calhau, C.; Cassano, A.; D’Archivio, M.; Faria, A.; Favé, G.; Fogliano, V.; et al. Polyphenols and Human Health: A Prospectus. Crit. Rev. Food Sci. Nutr. 2011, 51, 524–546. [Google Scholar] [CrossRef]
- Iriti, M.; Varoni, E.M. Chemopreventive Potential of Flavonoids in Oral Squamous Cell Carcinoma in Human Studies. Nutrients 2013, 5, 2564–2576. [Google Scholar] [CrossRef]
- Leopoldini, M.; Russo, N.; Toscano, M. The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem. 2011, 125, 288–306. [Google Scholar] [CrossRef]
- Perron, N.R.; Brumaghim, J.L. A Review of the Antioxidant Mechanisms of Polyphenol Compounds Related to Iron Binding. Cell Biochem. Biophys. 2009, 53, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J.; Jackson, M.J.; Lunec, J. In Vitro Screening of Iron Chelators Using Models of Free Radical Damage. Free. Radic. Res. Commun. 1986, 2, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Nkhili, E.; Loonis, M.; Mihai, S.; El Hajji, H.; Dangles, O. Reactivity of food phenols with iron and copper ions: Binding, dioxygen activation and oxidation mechanisms. Food Funct. 2014, 5, 1186–1202. [Google Scholar] [CrossRef]
- Korkina, L.G.; Afanas’Ev, I.B. Antioxidant and chelating properties of flavonoids. Adv. Pharmacol. 1997, 38, 151–163. [Google Scholar] [PubMed]
- Kontoghiorghes, G.J. Design, properties and effective use of the oral chelator L1 and other α- ketohydroxypyridines in the treatment of transfusional iron overload in thalassaemia. Ann. N.Y. Acad. Sci. 1990, 612, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Chobot, V.; Drage, S.; Hadacek, F. Redox properties of 8-quinolinol and implications for its mode of action. Nat. Prod. Commun. 2011, 6, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontoghiorghes, G.J.; Piga, A.; Hoffbrand, A. Cytotoxic effects of the lipophilic iron chelator omadine. FEBS Lett. 1986, 204, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Kontoghiorghes, G.J.; Piga, A.; Hoffbrand, A.V. Cytotoxic and DNA-inhibitory effects of iron chelators on human leukaemic cell lines. Hematol. Oncol. 1986, 4, 195–204. [Google Scholar] [CrossRef]
- Kontoghiorghe, C.N.; Kontoghiorghes, G.J. Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with non-transfusion-dependent thalassemia syndromes. Drug Des. Dev. Ther. 2016, 10, 465–481. [Google Scholar] [CrossRef] [Green Version]
- Hershko, C.; Graham, G.; Bates, G.W.; Rachmilewitz, E.A. Non-Specific Serum Iron in Thalassaemia: An Abnormal Serum Iron Fraction of Potential Toxicity. Br. J. Haematol. 1978, 40, 255–263. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; A Aldouri, M.; Hoffbrand, A.V.; Barr, J.; Wonke, B.; Kourouclaris, T.; Sheppard, L. Effective chelation of iron in beta thalassaemia with the oral chelator 1,2-dimethyl-3-hydroxypyrid-4-one. BMJ 1987, 295, 1509–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baksi, A.J.; Pennell, D. Randomized controlled trials of iron chelators for the treatment of cardiac siderosis in thalassaemia major. Front. Pharmacol. 2014, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolnagou, A.; Kleanthous, M.; Kontoghiorghes, G.J. Reduction of body iron stores to normal range levels in thalassaemia by using a deferiprone/deferoxamine combination and their maintenance thereafter by deferiprone monotherapy. Eur. J. Haematol. 2010, 85, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Farmaki, K.; Tzoumari, I.; Pappa, C.; Chouliaras, G.; Berdoukas, V. Normalisation of total body iron load with very intensive combined chelation reverses cardiac and endocrine complications of thalassaemia major. Br. J. Haematol. 2010, 148, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Kolnagou, A.; Kontoghiorghe, C.N.; Kontoghiorghes, G.J. Prevention of Iron Overload and Long Term Maintenance of Normal Iron Stores in Thalassaemia Major Patients using Deferiprone or Deferiprone Deferoxamine Combination. Drug Res. 2017, 67, 404–411. [Google Scholar] [CrossRef]
- Kolnagou, A.; Kontoghiorghes, G.J. Chelation protocols for the elimination and prevention of iron overload in thalassaemia. Front. Biosci. 2018, 23, 1082–1098. [Google Scholar] [CrossRef] [Green Version]
- Kontoghiorghes, G.J.; Kleanthous, M.; Kontoghiorghe, C.N. The History of Deferiprone (L1) and the Paradigm of the Complete Treatment of Iron Overload in Thalassaemia. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020011. [Google Scholar] [CrossRef] [PubMed]
- Pepe, A.; Meloni, A.; Pistoia, L.; Cuccia, L.; Gamberini, M.R.; Lisi, R.; D’Ascola, D.G.; Rosso, R.; Allò, M.; Spasiano, A.; et al. MRI multicentre prospective survey in thalassaemia major patients treated with deferasirox versus deferiprone and desferrioxamine. Br. J. Haematol. 2018, 183, 783–795. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-H.; Chen, X.; Wu, C.-C.; Wu, K.-H.; Song, T.-S.; Weng, T.-F.; Hsieh, Y.-W.; Peng, C.-T. Therapeutic mechanism of combined oral chelation therapy to maximize efficacy of iron removal in transfusion-dependent thalassemia major – a pilot study. Expert Rev. Hematol. 2019, 12, 265–272. [Google Scholar] [CrossRef]
- Eghbali, A.; Shokri, P.; Afzal, R.R.; Bagheri, B. A 1-year randomized trial of deferasirox alone versus deferasirox and deferoxamine combination for the treatment of iron overload in thalassemia major. Transfus. Apher. Sci. 2019, 58, 429–433. [Google Scholar] [CrossRef]
- Karimi, M.; Haghpanah, S.; Bahoush, G.; Ansari, S.; Azarkeivan, A.; Shahsavani, A.; Bazrafshan, A.; Jangjou, A. Evaluation of efficacy, safety, and satisfaction taking deferasirox twice daily versus once daily in patients with transfusion-dependent thalassemia. J. Pediatrics Hematol. 2020, 42, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Binding, A.; Ward, R.; Tomlinson, G.; Kuo, K.H.M. Deferiprone exerts a dose-dependent reduction of liver iron in adults with iron overload. Eur. J. Haematol. 2019, 103, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Muhammad, M.; Patel, J. Deferasirox-a rarer cause of Fanconi syndrome. J. Community Hosp. Intern. Med. Perspect. 2019, 9, 358–359. [Google Scholar] [CrossRef]
- Badeli, H.; Baghersalimi, A.; Eslami, S.; Saadat, F.; Rad, A.H.; Basavand, R.; Papkiadeh, S.R.; Darbandi, B.; Kooti, W.; Peluso, I. Early kidney damage markers after deferasirox treatment in patients with thalassemia major: A case-control study. Oxidative Med. Cell. Longev. 2019, 2019, 5461617-8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parakh, N.; Chandra, J.; Sharma, S.; Dhingra, B.; Jain, R.; Mahto, D. Efficacy and safety of combined oral chelation with deferiprone and deferasirox in children with β-thalassemia major. J. Pediatrics Hematol. 2017, 39, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J. The aim of iron chelation therapy in thalassaemia. Eur. J. Haematol. 2017, 99, 465–466. [Google Scholar] [CrossRef]
- Fredenburg, A.M.; Sethi, R.K.; Allen, D.D.; Yokel, R.A. The pharmacokinetics and blood-brain barrier permeation of the chelators 1,2 dimethly-, 1,2 diethyl-, and 1-[ethan-1’ol]-2-methyl-3-hydroxypyridin-4-one in the rat. Toxicology 1996, 108, 108–191. [Google Scholar] [CrossRef]
- Vreugdenhil, G.; Kontoghiorghes, G.J.; Van Eijk, H.G.; Swaak, A.J. Impaired erythropoietin responsiveness to the anaemia in rheumatoid arthritis. A possible inverse relationship with iron stores and effects of the oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-one. Clin. Exp. Rheumatol. 1991, 9, 35–40. [Google Scholar]
- Vreughtenhil, G.; Kontoghiorghes, G.J.; Van Eijk, H.G.; Swaak, A.J.G. Efficacy and safety of the oral chelator L1 in anaemic rheumadoit arthritis patients. Lancet 1989, II, 1398–1399. [Google Scholar] [CrossRef]
- Boddaert, N.; Sang, K.-H.L.Q.; Rötig, A.; Leroy-Willig, A.; Gallet, S.; Brunelle, F.; Sidi, D.; Thalabard, J.-C.; Munnich, A.; Cabantchik, Z.I. Selective iron chelation in Friedreich ataxia: Biologic and clinical implications. Blood 2007, 110, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, G.; Zibordi, F.; Chiapparini, L.; Bertini, E.; Russo, L.; Piga, A.; Longo, F.; Garavaglia, B.; Aquino, D.; Savoiardo, M.; et al. Iron-related MRI images in patients with pantothenate kinase-associated neurodegeneration (PKAN) treated with deferiprone: Results of a phase II pilot trial. Mov. Disord. 2011, 26, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Forni, G.; Balocco, M.; Cremonesi, L.; Abbruzzese, G.; Parodi, R.C.; Marchese, R. Regression of symptoms after selective iron chelation therapy in a case of neurodegeneration with brain iron accumulation. Mov. Disord. 2008, 23, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.S.J.; Reffitt, D.M.; Doig, L.A.; Meenan, J.; Ellis, R.D.; Thompson, R.P.H.; Powell, J.J. Ferric trimaltol corrects iron deficiency anaemia in patients intolerant of iron. Aliment. Pharmacol. Ther. 1998, 12, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Reffitt, D.M.; Burden, T.J.; Seed, P.T.; Wood, J.; Thompson, R.P.; Powell, J.J. Assessment of iron absorption from ferric trimaltol. Ann. Clin. Biochem. 2000, 37, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Gasche, C.; Ahmad, T.; Tulassay, Z.; Baumgart, D.C.; Bokemeyer, B.; Büning, C.; Howaldt, S.; Stallmach, A. Ferric maltol is effective in correcting iron deficiency anemia in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2015, 21, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Kontoghiorghes, G.J. Chelators affecting iron absorption in mice. Arzneimittelforschung 1990, 40, 1332–1335. [Google Scholar]
- Yamamoto, R.S.; Williams, G.M.; Frankel, H.H.; Weisburger, J.H. 8-hydroxyquinoline: Chronic toxicity and inhibitory effect on the carcinogenicity of N-2- fluorenylacetamide. Toxicol. Appl. Pharmacol. 1971, 19, 687–698. [Google Scholar] [CrossRef]
- Born, T.; Kontoghiorghe, C.N.; Spyrou, A.; Kolnagou, A.; Kontoghiorghes, G.J. EDTA chelation reappraisal following new clinical trials and regular use in millions of patients: Review of preliminary findings and risk/benefit assessment. Toxicol. Mech. Methods 2012, 23, 11–17. [Google Scholar] [CrossRef]
- Taylor, D.M.; Hodgson, S.A.; Stradling, N. Treatment of human contamination with plutonium and americium: Would orally administered Ca- or Zn-DTPA be effective? Radiat. Prot. Dosim. 2007, 127, 469–471. [Google Scholar] [CrossRef]
- Djaldetti, M.; Fishman, P.; Notti, I.; Bessler, H. The effect of tetracycline administration on iron absorption in mice. Biomedicine 1981, 35, 35–150. [Google Scholar]
- Konstantinou, E.; Pashalidis, I.; Kolnagou, A.; Kontoghiorghes, G.J. Interactions of hydroxycarbamide (hydroxyurea) with iron and copper: Implications on toxicity and therapeutic strategies. Hemoglobin 2011, 35, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, L.N.; Kontoghiorghes, G.J. Competition between deferiprone, desferrioxamine and other chelators for iron and the effect of other metals. Arzneimittelforschung 1993, 43, 659–663. [Google Scholar] [PubMed]
- Kontoghiorghes, G.J.; Kolnagou, A. Molecular factors and mechanisms affecting iron and other metal excretion or absorption in health and disease: The role of natural and synthetic chelators. Curr. Med. Chem. 2005, 12, 2695–2709. [Google Scholar] [CrossRef]
- Worwood, M. Inborn errors of metabolism: Iron. Br. Med Bull. 1999, 55, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Senba, M.; Nakamura, T.; Itakura, H. Relationships among iron accumulation, cirrhosis, and hepatitis B virus infection in Bantu siderosis. Ann. Soc. Belg. Med. Trop. 1989, 69, 77–78. [Google Scholar]
- Robertson, A.; Tenenbein, M. Hepatotoxicity in acute iron poisoning. Hum. Exp. Toxicol. 2005, 24, 559–562. [Google Scholar] [CrossRef]
- Baranwal, A.; Singhi, P. Acute iron poisoning: Management guidelines. Indian Pediatrics 2003, 40, 534–540. [Google Scholar]
- Wu, M.-L.; Tsai, W.-J.; Ger, J.; Deng, J.-F. Clinical experience of acute ferric chloride poisoning. Veter-Hum. Toxicol. 2003, 45, 243–246. [Google Scholar]
- Dresow, B.; Fischer, R.; Nielsen, P.; Gabbe, E.E.; Piga, A. Effect of oral iron chelator L1 on iron absorption in man. Ann. N. Y. Acad. Sci. 1998, 850, 466–468. [Google Scholar] [CrossRef]
- Berkovitch, M.; Livne, A.; Lushkov, G.; Segal, M.; Talmor, C.; Bentur, Y.; Klein, J.; Koren, G. The efficacy of oral deferiprone in acute iron poisoning. Am. J. Emerg. Med. 2000, 18, 36–40. [Google Scholar] [CrossRef]
- Iyengar, V.; Pullakhandam, R.; Nair, K.M. Dietary ligands as determinants of iron-zinc interactions at the absorptive enterocyte. J. Food Sci. 2010, 75. [Google Scholar] [CrossRef] [PubMed]
- Petry, N.; Egli, I.; Zeder, C.; Walczyk, T.; Hurrell, R. Polyphenols and phytic acid contribute to the low iron bioavailability from common beans in young women. J. Nutr. 2010, 140, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, A.; González-Osnaya, L.; Sánchez-Chinchillas, A.; Trejo, A. Role of oxate, phytate, tannins and cooking on iron bioavailability from foods commonly consumed in Mexico. Int. J. Food Sci. Nutr. 2009, 61, 29–39. [Google Scholar] [CrossRef]
- Campbell, N.; Hasinoff, B. Iron supplements: A common cause of drug interactions. Br. J. Clin. Pharmacol. 1991, 31, 251–255. [Google Scholar] [CrossRef]
- Jaramillo, A.; Briones, L.; Andrews, M.; Arredondo, M.; Olivares, M.; Brito, A.; Pizarro, F. Effect of phytic acid, tannic acid and pectin on fasting iron bioavailability both in the presence and absence of calcium. J. Trace Elements Med. Boil. 2015, 30, 112–117. [Google Scholar] [CrossRef]
- Bouvard, V.; Loomis, D.; Guyton, K.Z.; Grosse, Y.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Mattock, H.; Straif, K.; International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 2015, 16, 1599–1600. [Google Scholar] [CrossRef] [Green Version]
- Kontoghiorghes, G.J. Orally active alpha-ketohydroxypyridine iron chelators: Effects on iron and other metal mobilisations. Acta Haematol. 1987, 78, 212–216. [Google Scholar] [CrossRef]
- Mavrogeni, S.I.; Gotsis, E.D.; Markussis, V.; Tsekos, N.; Politis, C.; Vretou, E.; Kermastinos, D. T2 relaxation time study of iron overload in b-thalassemia. MAGMA 1998, 6, 7–12. [Google Scholar] [CrossRef]
- Mavrogeni, S.I.; Markussis, V.; Kaklamanis, L.; Tsiapras, D.; Paraskevaidis, I.; Karavolias, G.; Karagiorga, M.; Douskou, M.; Cokkinos, D.V.; Kremastinos, D.T. A comparison of magnetic resonance imaging and cardiac biopsy in the evaluation of heart iron overload in patients with beta-thalassemia major. Eur. J. Haematol. 2005, 75, 241–247. [Google Scholar] [CrossRef]
- Kolnagou, A.; Kontoghiorghe, C.N.; Kontoghiorghes, G.J. New targeted therapies and diagnostic methods for iron overload diseases. Front. Biosci. 2018, 10, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Villeneuve, J.-P.; Bilodeau, M.; Lepage, R.; Côté, J.; Lefebvre, M. Variability in hepatic iron concentration measurement from needle-biopsy specimens. J. Hepatol. 1996, 25, 172–177. [Google Scholar] [CrossRef]
- Kolnagou, A.; Natsiopoulos, K.; Kleanthous, M.; Ioannou, A.; Kontoghiorghes, G.J. Liver iron and serum ferritin levels are misleading for estimating cardiac, pancreatic, splenic and total body iron load in thalassemia patients: Factors influencing the heterogenic distribution of excess storage iron in organs as identified by MRI T2*. Toxicol. Mech. Methods 2012, 23, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.B.; Lynch, D.R.; Fischbeck, K.H. Normal serum iron and ferritin concentrations in patients with Friedreich’s ataxia. Ann Neurol. 1998, 44, 132–134. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J. Iron mobilization from ferritin using α-oxohydroxy heteroaromatic chelators. Biochem. J. 1986, 233, 299–302. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Chambers, S.; Hoffbrand, A.V. Comparative study of iron mobilization from haemosiderin, ferritin and iron(III) precipitates by chelators. Biochem. J. 1987, 241, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Kontoghiorghes, G.J. Decrease solubilisation of ferritin iron and fresh iron(III) precipitate following repeated chelator treatments. Inorg. Chim. Acta 1987, 138, 35–39. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Goddard, J.G.; Bartlett, A.N.; Sheppard, L. Pharmacokinetic studies in humans with the oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-one. Clin. Pharmacol. Ther. 1990, 48, 255–261. [Google Scholar] [CrossRef]
- Olivieri, N.F.; Koren, G.; Matsui, D.; Liu, P.P.; Blendis, L.; Cameron, R.; McClelland, R.A.; Templeton, D.M. Reduction of tissue iron stores and normalization of serum ferritin during treatment with the oral iron chelator L1 in thalassemia intermedia. Blood 1992, 79, 2741–2748. [Google Scholar] [CrossRef] [Green Version]
- Makey, D.G.; Seal, U.S. The detection of four molecular forms of human transferrin during the iron binding process. Biochim. Biophys. Acta (BBA)-Protein Struct. 1976, 453, 250–256. [Google Scholar] [CrossRef]
- Gomme, P.T.; McCann, K.B.; Bertolini, J. Transferrin: Structure, function and potential therapeutic actions. Drug Discov. Today 2005, 10, 267–273. [Google Scholar] [CrossRef]
- Pantopoulos, K. TfR2 links iron metabolism and erythropoiesis. Blood 2015, 125, 1055–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna, B.M.; Ershova, K.; Yan, J.; Ulhaq, A.; Nielsen, T.B.; Hsieh, S.; Pantapalangkoor, P.; Vanscoy, B.; Ambrose, P.; Rudin, S.; et al. Adjunctive transferrin to reduce the emergence of antibiotic resistance in Gram-negative bacteria. J. Antimicrob. Chemother. 2019, 74, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.E.; Weeks, K.; Carter, D.A. Lactoferrin Is Broadly Active against Yeasts and Highly Synergistic with Amphotericin B. Antimicrob. Agents Chemother. 2020, 64, pii: e02284. [Google Scholar] [CrossRef] [Green Version]
- González-Chávez, S.A.; Arévalo-Gallegos, S.; Rascón-Cruz, Q. Lactoferrin: Structure, function and applications. Int. J. Antimicrob. Agents 2009, 33, 301.e1–301.e8. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J. The study of iron mobilisation from transferrin using α-ketohydroxy heteroaromatic chelators. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzym. 1986, 869, 141–146. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J. Iron mobilisation from lactoferrin by chelators at physiological pH. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1986, 882, 267–270. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Sheppard, L. Simple synthesis of the potent iron chelators 1-alkyl-3-hydroxy-2-methylpyrid-4-ones. Inorg. Chim. Acta 1987, 136, L11–L12. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Barr, J.; Nortey, P.; Sheppard, L. Selection of a new generation of orally active α-ketohydroxypyridine iron chelators intended for use in the treatment of iron overload. Am. J. Hematol. 1993, 42, 340–349. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Evans, R.W. Site specificity of iron removal from transferrin by α-ketohydroxypyridine chelators. FEBS Lett. 1985, 189, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Kontoghiorghes, G.J. Iron mobilization from transferrin and non-transferrin bound iron by deferiprone. Implications in the treatment of thalassaemia, anaemia of chronic disease, cancer and other conditions. Hemoglobin 2006, 30, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.M.; Kontoghiorghes, G.J. Mobilisation of plutonium and iron from transferrin and ferritin by α-oxohydroxypyridine chelators. Inorg. Chim. Acta Bioinorg Chem. 1986, 125, L35–L38. [Google Scholar] [CrossRef]
- Evans, R.W.; Sharma, M.; Ogwang, W.; Patel, K.J.; Bartlett, A.N.; Kontoghiorghes, G.J. The effect of α-ketohydroxypyridine chelators on transferrin saturation in vitro and in vivo. Drugs Today 1992, 28 (Suppl. A), 9–23. [Google Scholar]
- Kontoghiorghes, G.J. A New Era in Iron Chelation Therapy: The Design of Optimal, Individually Adjusted Iron Chelation Therapies for the Complete Removal of Iron Overload in Thalassemia and other Chronically Transfused Patients. Hemoglobin 2009, 33, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghe, C.N.; Kontoghiorghes, G.J. New developments and controversies in iron metabolism and iron chelation therapy. World J. Methodol. 2016, 6, 1–19. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; May, A. Uptake and intracellular distribution of iron from transferrin and chelators in erythroid cells. BioMetals 1990, 3, 183–187. [Google Scholar] [CrossRef]
- White, G.P.; Jacobs, A.; Grady, R.W.; Cerami, A. The effect of chelating agents on iron mobilization in Chang cell cultures. Blood 1976, 48, 923–929. [Google Scholar] [CrossRef] [Green Version]
- Forsbeck, K.; Nilsson, K.; Kontoghiorghes, G.J. Variation in iron accumulation, transferrin membrane binding and DNA synthesis in the K-562 and U-937 cell lines induced by chelators and their iron complexes. Eur. J. Haematol. 1987, 39, 318–325. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J. Structure/red blood cell permeability. Activity of iron(III) chelator complexes. Inorg. Chim. Acta 1988, 151, 101–106. [Google Scholar] [CrossRef]
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2010, 1800, 783–792. [Google Scholar] [CrossRef]
- Yang, H.; Yang, M.; Guan, H.; Liu, Z.; Zhao, S.; Takeuchi, S.; Yanagisawa, D.; Tooyama, I. Mitochondrial ferritin in neurodegenerative diseases. Neurosci. Res. 2013, 77, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostert, L.J.; Van Dorst, J.; Koster, J.; Van Eijk, H.G.; Kontoghiorghes, G.J. Free radical and cytotoxic effects of chelators and their iron complexes in the hepatocyte. Free. Radic. Res. Commun. 1987, 3, 379–388. [Google Scholar] [CrossRef]
- Kolnagou, A.; Kleanthous, M.; Kontoghiorghes, G.J. Efficacy, compliance and toxicity factors are affecting the rate of normalization of body iron stores in thalassemia patients using the deferiprone and deferoxamine combination therapy. Hemoglobin 2011, 35, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Bhattacharyya, M. Deferiprone (L1) induced conformation change of hemoglobin: A fluorescence and CD spectroscopic study. Mol. Cell. Biochem. 2000, 204, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Sattarahmady, N.; Heli, H.; Moosavi-Movahedi, A.A.; Karimian, K. Deferiprone: Structural and functional modulating agent of hemoglobin fructation. Mol. Boil. Rep. 2014, 41, 1723–1729. [Google Scholar] [CrossRef]
- Offenbacher, A.R.; Minnihan, E.C.; Stubbe, J.; Barry, B.A. Redox-linked changes to the hydrogen-bonding network of ribonucleotide reductase β2. J. Am. Chem. Soc. 2013, 135, 6380–6383. [Google Scholar] [CrossRef] [Green Version]
- Iman, M.; Khansefid, Z.; Davood, A. Modeling and proposed molecular mechanism of hydroxyurea through docking and molecular dynamic simulation to curtail the action of ribonucleotide reductase. Recent Patents Anti-Cancer Drug Discov. 2016, 11, 461–468. [Google Scholar] [CrossRef]
- Lavelle, D.; Engel, J.D.; Saunthararajah, Y. Fetal hemoglobin induction by epigenetic drugs. Semin. Hematol. 2018, 55, 60–67. [Google Scholar] [CrossRef]
- Ehsani, M.A.; Hedayati-Asl, A.A.; Bagheri, A.; Zeinali, S.; Rashidi, A. Hydroxyurea-induced hematological response in transfusion-independent beta-thalassemia intermedia: Case series and review of literature. Pediatrics Hematol. Oncol. 2009, 26, 560–565. [Google Scholar] [CrossRef]
- Ballas, S.K.; Darbari, D.S. Review/overview of pain in sickle cell disease. Complement. Ther. Med. 2020, 49, 102327. [Google Scholar] [CrossRef]
- Shah, F.; Dwivedi, M. Pathophysiology and recent therapeutic insights of sickle cell disease. Ann. Hematol. 2020, 99, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Nardo-Marino, A.; Brousse, V.; Rees, D.C. Emerging therapies in sickle cell disease. Br. J. Haematol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Carden, M.A.; Little, J.A. Emerging disease-modifying therapies for sickle cell disease. Haematologica 2019, 104, 1710–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, A.; Bansal, D. Thalassemia: Common clinical queries in management. Indian J. Pediatrics 2019, 87, 75–81. [Google Scholar] [CrossRef]
- Shah, S.; Sheth, R.; Shah, K.; Patel, K. Safety and effectiveness of thalidomide and hydroxyurea combination in β-thalassaemia intermedia and major: A retrospective pilot study. Br. J. Haematol. 2019, 188, e18–e21. [Google Scholar] [CrossRef]
- Fraser, D.I.; Liu, K.T.; Reid, B.J.; Hawkins, E.; Sevier, A.; Pyle, M.; Robinson, J.W.; Ouellette, P.H.R.; Ballantyne, J.S. Widespread natural occurrence of hydroxyurea in animals. PLoS ONE 2015, 10, e0142890. [Google Scholar] [CrossRef] [Green Version]
- Manaresi, E.; Gallinella, G. Advances in the development of antiviral strategies against parvovirus B19. Viruses 2019, 11, 659. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, D.; Mascarenhas, J.O. Novel therapies in polycythemia vera. Curr. Hematol. Malign-Rep. 2020, 15, 133–140. [Google Scholar] [CrossRef]
- Rainsford, K.D. Ibuprofen: Pharmacology, efficacy and safety. Inflammopharmacology 2009, 17, 275–342. [Google Scholar] [CrossRef]
- Dos Santos, P.R.; Pich, C.T.; Back, D.; Smiderle, F.; Dumas, F.; Moura, S. Synthesis, chemical characterization and DNA interaction study of new diclofenac and ibuprofen zinc (II)-nicotinamide ternary complexes as cyclooxygenase inhibitor prototypes. J. Inorg. Biochem. 2020, 206, 111046. [Google Scholar] [CrossRef]
- Vane, J.; Botting, R. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258. [Google Scholar] [CrossRef]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Van De Putte, L.B.A.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeremy, J.Y.; Kontoghiorghes, G.J.; Hoffbrand, A.V.; Dandona, P. The iron chelators desferrioxamine and 1-alkyl-2-methyl-3-hydroxypyrid-4-ones inhibit vascular prostacyclin synthesis in vitro. Biochem. J. 1988, 254, 239–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villeneuve, J.-P.; Pichette, V. Cytochrome P450 and liver diseases. Curr. Drug Metab. 2004, 5, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Speer, R.E.; Karuppagounder, S.S.; Basso, M.; Sleiman, S.F.; Kumar, A.; Brand, D.; Smirnova, N.; Gazaryan, I.; Khim, S.J.; Ratan, R.R. Hypoxia-inducible factor prolyl hydroxylases as targets for neuroprotection by "antioxidant" metal chelators: From ferroptosis to stroke. Free. Radic. Boil. Med. 2013, 62, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, S.J.; Vallabhajosula, S. Clinically Proven Radiopharmaceuticals for Infection Imaging: Mechanisms and Applications. Semin. Nucl. Med. 2009, 39, 2–10. [Google Scholar] [CrossRef]
- Zak, O.; Aisen, P. Spectroscopic and thermodynamic studies on the binding of gadolinium(III) to human serum transferrin. Biochemistry 1988, 27, 1075–1080. [Google Scholar] [CrossRef]
- Smith, T.; Perkins, A.C.; Walton, P.H. 99mTc-labelled human serum transferrin for tumour imaging: An in vitro and in vivo study of the complex. Nucl. Med. Commun. 2004, 25, 387–391. [Google Scholar] [CrossRef]
- Tsopelas, C. A study of radiogallium aqueous chemistry: In vitro and in vivo characterisation of 67 Ga-hydrolysed-stannous fluoride particles. J. Label. Compd. Radiopharm. 2016, 59, 197–204. [Google Scholar] [CrossRef]
- E Dahlqvist, G.; Jamar, F.; Zech, F.; Geubel, A.P. In-111 transferrin scintigraphy in cirrhosis with hypoalbuminemia: Evidence for protein-losing enteropathy in a small group of selected cases. Scand. J. Gastroenterol. 2012, 47, 1247–1252. [Google Scholar] [CrossRef]
- Collery, P.; Keppler, B.K.; Madoulet, C.; Desoize, B. Gallium in cancer treatment. Crit. Rev. Oncol. 2002, 42, 283–296. [Google Scholar] [CrossRef]
- Sava, G.; Pacor, S.; Bregant, F.; Ceschia, V. Metal complexes of ruthenium: A potential class of selective anticancer drugs. Anticancer. Res. 1991, 11, 1103–1107. [Google Scholar] [PubMed]
- Peng, H.; Jin, H.; Zhuo, H.; Huang, H. Enhanced antitumor efficacy of cisplatin for treating ovarian cancer in vitro and in vivo via transferrin binding. Oncotarget 2017, 8, 45597–45611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.; Ham, K.; Dawborn, J.; Xipell, J. Treatment of dialysis osteomalacia with desferrioxamine. Lancet 1982, 320, 343–345. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Barr, J.; Baillod, R.A. Studies of aluminium mobilisation in renal dialysis patients using the oral chelator 1, 2-dimethyl-3-hydroxypyrid-4-one. Arzn Forsch Drug Res 1994, 44, 522–526. [Google Scholar]
- Volf, V.; Kontoghiorghes, G.J. Retention of injected plutonium and americium in mice and rats after oral treatment with DTPA, desferrioxamine and alpha-ketohydroxypyridines. Drugs Today 1992, 28 (Suppl. A), 169–172. [Google Scholar]
- Pashalidis, I.; Kontoghiorghes, G.J. Effective complex formation in the interaction of I,2-dimethyl-3-hydroxypyrid-4-one (Deferiprone or L1) with Uranium (VI). J. Radioanalyt. Nucl. Chem. 1999, 242, 181–184. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J. Prospects for introducing deferiprone as potent pharmaceutical antioxidant. Front. Biosci. (Elite Ed) 2009, 1, 161–178. [Google Scholar]
- Kontoghiorghe, C.N.; Kolnagou, A.; Kontoghiorghes, G.J. Antioxidant targeting by deferiprone in diseases related to oxidative damage. Front. Biosci. (Landmark Ed) 2014, 19, 862–885. [Google Scholar] [CrossRef] [Green Version]
- Forman, H.J. Redox signaling: An evolution from free radicals to aging. Free Radic Biol Med. 2016, 97, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Stangherlin, A.; Reddy, A.B. Regulation of circadian clocks by redox homeostasis. J. Boil. Chem. 2013, 288, 26505–26511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arevalo, J.A.; Vazquez-Medina, J.P. The role of peroxiredoxin 6 in cell signaling. Antioxidants. 2018, 7, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ďuračková, Z. Some current insights into oxidative stress. Physiol. Res. 2009, 59, 459–469. [Google Scholar] [PubMed]
- Gutteridge, J.M. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin. Chem. 1995, 41, 1819–1828. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 25, 207–218. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Boil. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Central Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Lewerenz, J.; Ates, G.; Methner, A.; Conrad, M.; Maher, P. Oxytosis/ferroptosis—(re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front. Mol. Neurosci. 2018, 12, 214. [Google Scholar] [CrossRef]
- Hao, S.; Liang, B.; Huang, Q.; Dong, S.; Wu, Z.; He, W.; Shi, M. Metabolic networks in ferroptosis. Oncol. Lett. 2018, 15, 5405–5411. [Google Scholar] [CrossRef]
- Eybl, V.; Caisová, D.; Koutenský, J.; Kontoghiorghes, G.J. Influence of iron chelators, 1,2-dialkyl-3-hydroxypyridin-4-ones, on the lipid peroxidation and glutathione level in the liver of mice. Arch. Toxicol. Suppl. 1991, 14, 185–187. [Google Scholar] [CrossRef]
- Maher, P.; Kontoghiorghes, G.J. Characterization of the neuroprotective potential of derivatives of the iron chelating drug deferiprone. Neurochem. Res. 2015, 40, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Rajapurkar, M.M.; Hegde, U.; Bhattacharya, A.; Alam, M.G.; Shah, S.V. Effect of deferiprone, an oral iron chelator, in diabetic and non-diabetic glomerular disease. Toxicol. Mech. Methods 2012, 23, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Wallis, L.I.; Paley, M.N.; Graham, J.M.; Grünewald, R.A.; Wignall, E.L.; Joy, H.M.; Griffiths, P.D. MRI assessment of basal ganglia iron deposition in Parkinson’s disease. J. Magn. Reson. Imaging 2008, 28, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Brar, S.; Henderson, D.; Schenck, J.; Zimmerman, E.A. Iron accumulation in the substantia nigra of patients with Alzheimer disease and parkinsonism. Arch. Neurol. 2009, 66, 371–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldvogel, D.; van Gelderen, P.; Hallett, M. Increased iron in the dentate nucleus of patients with Friedrich’s ataxia. Ann Neurol. 1999, 46, 123–125. [Google Scholar] [CrossRef]
- Chan, S.; Lian, Q.; Chen, M.-P.; Jiang, D.; Ho, J.T.; Cheung, Y.-F.; Chan, G.C.-F. Deferiprone inhibits iron overload-induced tissue factor bearing endothelial microparticle generation by inhibition oxidative stress induced mitochondrial injury, and apoptosis. Toxicol. Appl. Pharmacol. 2018, 338, 148–158. [Google Scholar] [CrossRef]
- Weinberg, E.D. Iron depletion: A defense against intracellular infection and neoplasm. Life Sci. 1992, 50, 1289–1297. [Google Scholar] [CrossRef]
- Andrews, S.C.; Robinson, A.K.; Rodríguez-Quiñones, F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.; Weinberg, E. Iron: Mammalian defense systems, mechanisms of disease, and chelation therapy approaches. Blood Rev. 1995, 9, 33–45. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Kolnagou, A.; Skiada, A.; Petrikkos, G. The role of iron and chelators on infections in iron overload and non iron loaded conditions: Prospects for the design of new antimicrobial therapies. Hemoglobin 2010, 34, 227–239. [Google Scholar] [CrossRef]
- Kontoghiorghe, C.N.; Andreou, N.; Constantinou, K.; Kontoghiorghes, G.J. World health dilemmas: Orphan and rare diseases, orphan drugs and orphan patients. World J. Methodol. 2014, 4, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Korenromp, E.; Eisele, T.; Newby, H.; Steketee, R.; Kachur, S.P.; Nahlen, B.; Bhattarai, A.; Yoon, S.; MacArthur, J.; et al. New global estimates of malaria deaths. Lancet 2012, 380, 559. [Google Scholar] [CrossRef]
- Uhlemann, A.-C.; Fidock, D.A. Loss of malarial susceptibility to artemisinin in Thailand. Lancet 2012, 379, 1928–1930. [Google Scholar] [CrossRef]
- Gordeuk, V.R.; Thuma, P.E.; Brittenham, G.M.; Zulu, S.; Simwanza, G.; Mhangu, A.; Flesch, G.; Parry, D. Iron chelation with desferrioxamine B in adults with asymptomatic P. Falciparum parasitemia. Blood 1992, 79, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Heppner, D.G.; Hallaway, P.E.; Kontoghiorghes, G.J.; Eaton, J.W. Antimalarial properties of orally active iron chelators. Blood 1988, 72, 358–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrandrea, S.; Carvajal, J.L.; Kaeda, J.S.; Kontoghiorghes, G.J.; Luzzato, L. Growth inhibition of Plasmodium Falciparum by orally active iron chelators. Drugs Today. 1992, 28 (Suppl. A), 25–27. [Google Scholar]
- Goudeau, C.; Loyevsky, M.; Kassim, O.O.; Gordeuk, V.R.; Nick, H. Assessment of antimalarial effect of ICL670A on in vitro cultures of Plasmodium falciparum. Br. J. Haematol. 2001, 115, 918–923. [Google Scholar] [CrossRef]
- Mohanty, D.; Ghosh, K.; Pathare, A.V.; Karnad, D. Deferiprone (L1) as an adjuvant therapy for Plasmodium falciparum malaria. Indian J. Med Res. 2002, 115, 17–21. [Google Scholar] [PubMed]
- Robins-Browne, R.; Prpic, J. Desferrioxamine and systemic yersiniosis. Lancet 1983, 322, 1372. [Google Scholar] [CrossRef]
- Boelaert, J.R.; Fenves, A.Z.; Coburn, J.W. Deferoxamine therapy and mucormycosis in dialysis patients: Report of an international registry. Am. J. Kidney Dis. 1991, 18, 660–667. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Edwards, J.E.; Fu, Y.; Spellberg, B. Deferiprone iron chelation as a novel therapy for experimental mucormycosis. J. Antimicrob. Chemother. 2006, 58, 1070–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, A.S.; Gebermariam, T.; Fu, Y.; Lin, L.; Husseiny, M.I.; French, S.W.; Schwartz, J.; Skory, C.D.; Edwards, J.E.; Spellberg, B.J. The iron chelator deferasirox protects mice from mucormycosis through iron starvation. J. Clin. Investig. 2007, 117, 2649–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock, J.H.; Licéaga, J.; Kontoghiorghes, G.J. The effect of synthetic iron chelators on bacterial growth in human serum. FEMS Microbiol. Immunol. 1988, 1, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Jue, S.; Jue, S.G.; Dawson, G.W.; Brogden, R.N. Ciclopirox olamine 1% cream a preliminary review of its antimicrobial activity and therapeutic use. Drugs 1985, 29, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.R. Zinc pyrithione: A topical antimicrobial with complex pharmaceutics. J. Drugs Dermatol. 2016, 15, 140–144. [Google Scholar] [PubMed]
- Kontoghiorghes, G.J.; Efstathiou, A.; Ioannou-Loucaides, S.; Kolnagou, A. Chelators controlling metal metabolism and toxicity pathways: Applications in cancer prevention, diagnosis and treatment. Hemoglobin 2008, 32, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Voest, E.E.; Vreugdenhil, G.; Marx, J.J.M. Iron-chelating agents in non-iron overload conditions. Ann. Intern. Med. 1994, 120, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef]
- Lui, G.Y.L.; Kovacevic, Z.; Richardson, V.; Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Targeting cancer by binding iron: Dissecting cellular signaling pathways. Oncotarget 2015, 6, 18748–18779. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Park, K.C.; Fouani, L.; Jansson, P.; Wooi, D.; Sahni, S.; Lane, D.; Palanimuthu, D.; Lok, H.C.; Kovacevic, Z.; Huang, M.L.-H.; et al. Copper and conquer: Copper complexes of di-2-pyridylketone thiosemicarbazones as novel anti-cancer therapeutics. Metallomics 2016, 8, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Nutting, C.; Van Herpen, C.M.L.; Miah, A.B.; Bhide, S.A.; Machiels, J.-P.; Buter, J.; Kelly, C.; De Raucourt, D.; Harrington, K.J. Phase II study of 3-AP Triapine in patients with recurrent or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2009, 20, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R. Molecular mechanisms of iron uptake by cells and the use of iron chelators for the treatment of cancer. Curr. Med. Chem. 2005, 12, 2711–2729. [Google Scholar] [CrossRef] [PubMed]
- Ganeshaguru, K.; Lally, J.M.; Piga, A.; Hoffbrand, A.V.; Kontoghiorghes, G.J. Cytotoxic mechanisms of iron chelators. Drugs Today 1992, 28 (Suppl. A), 29–34. [Google Scholar]
- Eybl, V.; Svihovcova, P.; Koutensky., J.; Kontoghiorghes, G.J. Interaction of L1, L1NAll and desferoxamine with Gallium in vivo. Drugs Today. 1992, 28 (Suppl. A), 173–175. [Google Scholar]
- Chitambar, C.R. Gallium-containing anticancer compounds. Futur. Med. Chem. 2012, 4, 1257–1272. [Google Scholar] [CrossRef] [Green Version]
- Jeremy, J.Y.; Gill, J.; Prior, T.; Sifaksi, G.; Barradas, M.A.; Kontoghiorghes, G.J. Inhibition of cycloxygenase and lipoxygenase activity by iron chelators:possible use in the treatment of eicosanoid-related disorders. Drugs Today. 1992, 28 (Suppl. A), 35–43. [Google Scholar]
- Donfrancesco, A.; De Bernardi, B.; Carli, M.; Mancini, A.; Nigro, M.; De Sio, L.; Casale, F.; Bagnulo, S.; Helson, L.; Deb, G. Deferoxamine followed by cyclophosphamide, etoposide, carboplatin, thiotepa, induction regimen in advanced neuroblastoma: Preliminary results. Eur. J. Cancer 1995, 31, 612–615. [Google Scholar] [CrossRef]
- Kolnagou, A.; Kontoghiorghe, C.N.; Kontoghiorghes, G.J. Transition of Thalassaemia and Friedreich ataxia from fatal to chronic diseases. World J. Methodol. 2014, 4, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Timoshnikov, V.A.; Kobzeva, T.; Polyakov, N.E.; Kontoghiorghes, G.J. Redox interactions of vitamin C and iron: Inhibition of the pro-oxidant activity by deferiprone. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Badu-Boateng, C.; Naftalin, R.J. Ascorbate and ferritin interactions: Consequences for iron release in vitro and in vivo and implications for inflammation. Free. Radic. Boil. Med. 2019, 133, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Dziuba, N.; Hardy, J.; Lindahl, P.A. Low-molecular-mass iron complexes in blood plasma of iron-deficient pigs do not originate directly from nutrient iron. Metallomics 2019, 11, 1900–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dichtl, S.; Haschka, D.; Nairz, M.; Seifert, M.; Volani, C.; Lutz, O.; Weiss, G. Dopamine promotes cellular iron accumulation and oxidative stress responses in macrophages. Biochem. Pharmacol. 2018, 148, 193–201. [Google Scholar] [CrossRef]
- Parquet, M.D.C.; Savage, K.A.; Allan, D.S.; Davidson, R.J.; Holbein, B. Novel iron-chelator DIBI inhibits Staphylococcus aureus growth, suppresses experimental MRSA infection in mice and enhances the activities of diverse antibiotics in vitro. Front. Microbiol. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Kolnagou, A.; Economides, C.; Eracleous, E.; Kontoghiorghes, G.J. Long term comparative studies in thalassemia patients treated with deferoxamine or a deferoxamine/deferiprone combination. identification of effective chelation therapy protocols. Hemoglobin 2008, 32, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Klopstock, T.; Tricta, F.; Neumayr, L.; Karin, I.; Zorzi, G.; Fradette, C.; Kmieć, T.; Büchner, B.; E Steele, H.; Horvath, R.; et al. Safety and efficacy of deferiprone for pantothenate kinase-associated neurodegeneration: A randomised, double-blind, controlled trial and an open-label extension study. Lancet Neurol. 2019, 18, 631–642. [Google Scholar] [CrossRef]
- Selim, M.H.; Foster, L.D.; Moy, C.S.; Xi, G.; Hill, M.D.; Morgenstern, L.B.; Greenberg, S.M.; James, M.L.; Singh, V.; Clark, W.M.; et al. Deferoxamine mesylate in patients with intracerebral haemorrhage (i-DEF): A multicentre, randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 2019, 18, 428–438. [Google Scholar] [CrossRef]
- Agrawal, S.; Fox, J.; Thyagarajan, B.; Fox, J.H. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free. Radic. Biol. Med. 2018, 120, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Tosato, M.; Di Marco, V. Metal Chelation Therapy and Parkinson’s Disease: A Critical Review on the Thermodynamics of Complex Formation between Relevant Metal Ions and Promising or Established Drugs. Biomolecules 2019, 9, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in neurodegeneration–cause or consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, D.; Spino, M.; Tricta, F.; Connelly, J.; Cracchiolo, B.M.; Hanauske, A.-R.; Gandolfi, D.D.; Mathews, M.B.; Karn, J.; Holland, B.; et al. Drug-based lead discovery: The novel ablative antiretroviral profile of deferiprone in HIV-1-infected cells and in HIV-infected treatment-naive subjects of a double-blind, placebo-controlled, randomized exploratory trial. PLoS ONE 2016, 11, e0154842. [Google Scholar] [CrossRef] [PubMed]
- Leftin, A.; Zhao, H.; Turkekul, M.; De Stanchina, E.; Manova, K.; Koutcher, J.A. Iron deposition is associated with differential macrophage infiltration and therapeutic response to iron chelation in prostate cancer. Sci. Rep. 2017, 7, 11632. [Google Scholar] [CrossRef] [PubMed]
- Brock, J.H.; Licéaga, J.; Arthur, H.M.; Kontoghiorghes, G.J. Effect of novel 1-alkyl-3-hydroxy-2-methylpyrid-4-one chelators on uptake and release of iron from macrophages. Am. J. Hematol. 1990, 34, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Gaur, K.; Vázquez, A.M.; Duran-Camacho, G.; Dominguez-Martinez, I.; Benjamín-Rivera, J.A.; Fernández-Vega, L.; Sarabia, L.C.; García, A.C.; Pérez-Deliz, F.; Román, J.M.; et al. Iron and Copper Intracellular Chelation as an Anticancer Drug Strategy. Inorganics 2018, 6, 126. [Google Scholar] [CrossRef] [Green Version]
- Kuang, Y.; Wang, Q. Iron and lung cancer. Cancer Lett. 2019, 464, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, L.; Ding, J.; Chen, Y. Iron Metabolism in Cancer. Int. J. Mol. Sci. 2018, 20, 95. [Google Scholar] [CrossRef] [Green Version]
- Heffeter, P.; Pape, V.F.; Enyedy, É.A.; Keppler, B.K.; Szakacs, G.; Kowol, C.R.; Szakas, G. Anticancer thiosemicarbazones: Chemical properties, interaction with iron metabolism, and resistance development. Antioxid. Redox Signal. 2019, 30, 1062–1082. [Google Scholar] [CrossRef]
- Mody, K.; Mansfield, A.S.; Vemireddy, L.; Nygren, P.; Gulbo, J.; Borad, M. A phase I study of the safety and tolerability of VLX600, an iron chelator, in patients with refractory advanced solid tumors. Investig. New Drugs 2018, 37, 684–692. [Google Scholar] [CrossRef]
- De Luca, A.; Barile, A.; Arciello, M.; Rossi, L. Copper homeostasis as target of both consolidated and innovative strategies of anti-tumor therapy. J. Trace Elem. Med. Boil. 2019, 55, 204–213. [Google Scholar] [CrossRef]
- Huang, Y.-F.; Kuo, M.T.; Liu, Y.-S.; Cheng, Y.-M.; Wu, P.-Y.; Chou, C.-Y. A Dose escalation study of trientine plus carboplatin and pegylated liposomal doxorubicin in women with a first relapse of epithelial ovarian, tubal, and peritoneal cancer within 12 months after platinum-based chemotherapy. Front. Oncol. 2019, 9, 437. [Google Scholar] [CrossRef] [PubMed]
- Timoshnikov, V.A.; Kobzeva, T.; Selyutina, O.Y.; Polyakov, N.E.; Kontoghiorghes, G.J. Effective inhibition of copper-catalyzed production of hydroxyl radicals by deferiprone. J. Boil. Inorg. Chem. 2019, 24, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Hagn, G.; Westhofen, R.; Burkovskiy, I.; Holbein, B.; Zhou, J.; Lehmann, C. Iron chelation as novel treatment for interstitial cystitis. Pharmacology 2019, 103, 159–162. [Google Scholar] [CrossRef]
- Kobayashi, M.; Suhara, T.; Baba, Y.; Kawasaki, N.K.; Higa, J.K.; Matsui, T. Pathological roles of iron in cardiovascular disease. Curr. Drug Targets 2018, 19, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Rawal, S. Dietary iron intake, iron status, and gestational diabetes. Am. J. Clin. Nutr. 2017, 106, 1672S–1680S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, P.; Geevarghese, P.; Khaire, P.; Joshi, T.; Suryawanshi, A.; Mundada, S.; Pawar, S.; Farookh, A. Comparison of therapeutic efficacy of ferrous ascorbate and iron polymaltose complex in iron deficiency anemia in children: A randomized controlled trial. Indian J. Pediatrics 2019, 86, 1112–1117. [Google Scholar] [CrossRef]
- Chandra, J. Treating iron deficiency anemia. Indian J. Pediatrics 2019, 86, 1085–1086. [Google Scholar] [CrossRef] [Green Version]
- Węgier, L.P.; Kubiak, M.; Liebert, A.; Clavel, T.; Montagne, A.; Stennevin, A.; Roye, S.; Boudribila, A. Ferrous sulfate oral solution in young children with iron deficiency anemia. Pediatrics Int. 2020. [Google Scholar] [CrossRef]
- Stoffel, N.U.; Zeder, C.; Brittenham, G.M.; Moretti, D.; Zimmermann, M.B. Iron absorption from supplements is greater with alternate day than with consecutive day dosing in iron-deficient anemic women. Haematologica 2019, 105, 1232–1239. [Google Scholar] [CrossRef] [Green Version]
- Enyedy, É.A.; Dömötör, O.; Bali, K.; Hetényi, A.; Tuccinardi, T.; Keppler, B.K. Interaction of the anticancer gallium(III) complexes of 8-hydroxyquinoline and maltol with human serum proteins. J. Boil. Inorg. Chem. 2014, 20, 77–88. [Google Scholar] [CrossRef]
- Tanzey, S.S.; Thompson, S.; Scott, P.J.; Brooks, A.F. Gallium-68: Methodology and novel radiotracers for positron emission tomography (2012–2017). Pharm. Pat. Anal. 2018, 7, 193–227. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lin, H.; Huang, J.; Li, A.; Sun, C.; Richmond, J.; Gao, J. A gadolinium-complex-based theranostic prodrug for in vivo tumour-targeted magnetic resonance imaging and therapy. Chem. Commun. 2019, 55, 4546–4549. [Google Scholar] [CrossRef] [PubMed]
- Blanusa, M.; Varnai, V.M.; Piasek, M.; Kostial, K. Chelators as antidotes of metal toxicity: Therapeutic and experimental aspects. Curr. Med. Chem. 2005, 12, 2771–2794. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.S.; Mittal, M.; Mehta, A. Heavy metal induced oxidative stress & its possible reversal by chelation therapy. Indian J. Med Res. 2008, 128, 501–523. [Google Scholar] [PubMed]
- Kontoghiorghes, G.J. Regulatory molecules and chelators used for the control of essential and toxic metals in health and disease: From molecular interactions to clinical effects and applications. Curr. Med. Chem. 2005, 12, 2661–2662. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Iron Complex Prosthetic Group | Function |
|---|---|---|
| Hemoglobin | Heme | Oxygen transport |
| Myoglobin | Heme | Oxygen transport |
| Cytochromes | Heme | Electron transport. Respiration |
| Cytochrome P450 | Heme | Drug detoxification |
| Ribonucleotide reductase | Amino acids | DNA synthesis |
| Proline hydroxylase | Amino acids | Collagen synthesis |
| Phenylalanine hydroxylase | Amino acids | Degradation of phenylalanine |
| Tryptophan 2,3-dioxygenage | Heme | Degradation of tryptophan |
| Homogentisic acid 2,3-dioxygenase | Amino acids | Detection of alkaptonuria |
| Peroxidases | Heme | Decomposition of hydroperoxides |
| Catalase | Heme | Decomposition of hydrogen peroxide |
| Lipoxygenase | Amino acids | HPETE and leukotriene synthesis |
| Cyclooxygenase | Heme and Amino acids | Prostaglandin and thromboxane synthesis |
| Adrenodoxin | 2Fe-2S | Electron transport. Oxidation/reduction |
| Aconitase | 4Fe–4S | Tricarboxylic acid cycle |
| Succinate dehydrogenase | 2Fe-2S, 4Fe–4S, 3Fe-4S | Tricarboxylic acid cycle |
| NADH dehydrogenase | Fe–S Clusters | Electron transport. Respiration |
| Xanthine oxidase | 4x (2Fe-2S) | Conversion of xanthine to uric acid |
| Aldehyde oxidase | 2x (2Fe-2S) | Metabolism of aldehydes |
| Transferrin | Amino Acids | Iron transport in plasma |
| Lactoferrin | Amino Acids | Iron binding in milk and secretions |
| Ferritin | Oxyhydroxide, phosphate Fe | Iron storage |
| Hemosiderin | Oxyhydroxide, phosphate Fe | Iron storage |
| Hephaestin | Not carrying or containing Fe | Ferroxidase and influx transmembrane iron transport |
| Ferroportin | Not carrying or containing Fe | Efflux transmembrane iron transporter in cells |
| Hepcidin | Not carrying or containing Fe | Regulatory protein affecting iron uptake and release |
| Phosphates | Pyridoxal phosphate, thiamine pyrophosphate, ribonucleoside and deoxyribonucleoside phosphates, phytic acid (IP6), Pyrophosphate, ATP, ADP, AMP, etc. |
| Amino acids | Aspartic acid, glutamic acid, histidine, cysteine, tyrosine, etc. |
| Carboxylic acids | Citric acid, aconitic acid, oxaloacetic acid, etc. |
| Mono- and di- saccharides | Fructose, glucose, lactose, etc. |
| Vitamins | Ascorbic acid, lipoic acid, riboflavin. |
| Fatty acids and phosphoglycerides | Oleic acid, linoleic acid, phosphatidic acid. |
| Other naturally occurring chelators | Catecholamines, pteridines, purines, spermine, spermidine. Glutathione. Folic acid. |
| Dietary molecules | In addition to food components containing the above molecules, there are also many plant products including most polyphenols and other phytochelators with iron chelating properties such as: gallic acid, caffeic acid, quercetin, ellagic acid, curcumin, catechin, maltol, etc. |
| Ion | EDTA | DTPA | Deferoxamine | Deferiprone | Deferasirox |
|---|---|---|---|---|---|
| Fe3+ | 25.1 | 28.6 | 30.6 | 35.0 | 27.0 |
| Cu2+ | 18.8 | 21.0 | 14.0 | 19.6 | – |
| Zn2+ | 16.5 | 18.4 | 11.1 | 13.5 | – |
| Charge | |||||
| (pH 7) | −ve | −ve | +ve | neutral | −ve |
| MWt | 292 | 393 | 561 | 139 | 373 |
| Iron oxidation | Oxidation of Fe (II) to Fe (III) by L1, DFO or transferrin at pH 7.4 Oxidation of hemoglobin to methemoglobin by DFO Oxidation of cytochrome c by 2,3-dihydroxybenzoic acid |
| Iron reduction | Heme Fe (IV) to Fe (III) in myoglobin and hemoglobin by DFO and L1 |
| Allosteric interactions | L1 and hemoglobin. Hydroxyurea and ribonucleotide reductase. |
| Competition with other metals | Order of stability constants of L1, DFO with metals: Fe>Al> Zn>Mg |
| Lipid / water partition coefficients (Kpar: n-octanol/water) | Order of hydrophilicity: DTPA and EDTA >DFO>L1>DFRA Order of lipophilicity: 8-hydroxyquinoline >tropolone>maltol |
| Inhibition or increase of iron induced free radical damage | L1 and DFO inhibit iron induced free radical damage to the DNA sugar deoxyribose. EDTA causes an increase in the iron induced free radical damage to deoxyribose. |
| Inhibition of iron-containing enzymes by iron chelating drugs | Lipoxygenase and cyclooxygenase inhibition by L1 and DFO. Catechol-O-methyltransferase, tyrosine and tryptophan hydroxylase inhibition by L1. |
| Promotion and inhibition of cell growth by iron binding and transport to cells | Maltol promotes cell growth. L1 and DFO inhibit cell growth. |
| Iron donors to transferrin | Ascorbate, citrate and L1 bound iron. DFO bound iron is not available to transferrin. |
| Iron mobilization from diferric transferrin and lactoferrin | L1 mobilizes iron preferentially from the C-terminal site and mimosine preferentially from the N-terminal site of transferrin. DFO and DFRA are not effective in transferrin or lactoferrin iron mobilization. |
| Differential rate of mobilization of iron species and forms by L1 | Mononuclear> oligonuclear> polynuclear. Transferrin, lactoferrin > ferritin, hemosiderin. |
| Increase in iron excretion and route of elimination in iron loaded patients | L1: Urinary iron. DFRA: Fecal iron. DFO: Urinary and fecal iron. |
| Differential iron removal from various organs. Efficacy is dose related. | L1 preferential iron removal from the heart and DFRA from the liver. DFO from the liver and to lesser extent from the heart. L1 iron removal from focal iron deposits in the brain of patients with neurodegenerative diseases. |
| Iron removal from diferric transferrin in iron loaded patients | About 40% at L1 concentrations > 0.1 mM, but not by DFO or DFRA. |
| Iron redistribution | DFO and especially L1 redistribute iron from the reticuloendothelial system to the erythron in anemic rheumatoid arthritis patients. DFO in cell studies. DFRA may cause redistribution of iron from the liver to other organs in thalassemia and other iron loaded patients. |
| Increase excretion of metals other than iron, e.g., zinc (Zn) and aluminum (Al). | DTPA > L1 > DFO. (Order of increased Zn excretion in iron loaded patients). DFO and L1 cause increase Al excretion in renal dialysis patients. DFRA causes Al and other xenobiotic metal absorption. |
| Iron mobilization and excretion of chelator metabolite iron complexes | Several DFO metabolites have iron chelation potential and cause increase in iron excretion. No increase in iron excretion by the L1 glucuronide and DFRA glucuronide metabolites. |
| Combination chelation therapy | L1 and DFO or L1 and DFRA or other chelator combinations are likely to be more effective than monotherapy. |
| Chelating drug synergism with reducing agents | Ascorbic acid acts synergistically with DFO, but not with L1 or DFRA for increasing iron excretion. |
| Effects on iron absorption by lipophilic and hydrophilic chelators | Increase of iron absorption by maltol, 8-hydroxyquinoline and DFRA. Decrease of iron absorption by DFO, DTPA, EDTA and L1. |
| Chelating drugs minimizing toxicity of other drugs | L1 and ICRF187 (Dexrazoxane), but not DFRA, inhibit doxorubicin induced cardiotoxicity. |
| Chelator prodrugs | ICRF 187 (Dexrazoxane) is converted in vivo to an EDTA like chelator. |
| Chelators with enterohepatic circulation | DFRA and cholyl hydroxamic acid. |
| Deferiprone (L1) | Deferasirox (DFRA) | |
|---|---|---|
| Molecular Differences | ||
| Molecular weight of chelators | 139 | 373 |
| Molecular weight of iron complexes | 470 | 798 |
| Charge of chelators at pH 7.4 | Neutral | Negative |
| Charge of iron complexes at pH 7.4 | Neutral | Negative |
| Partition coefficient of chelators (Kpar: n-octanol/water) | 0.19 | 6.3 |
| Partition coefficient of iron complexes (Kpar: n-octanol/water) | 0.05 | Not reported |
| Stability constant (Log K) of chelator iron complexes– (Transferrin: 36 ) | 35 | 27 |
| Metabolic and Pharmacokinetic Differences | ||
| Metabolite(s) | Glucuronide conjugate, which is cleared through the urine and have no iron chelation properties | Glucuronide conjugate cleared through the fecal route |
| T1/2 absorption | 0.7–32 min | estimated within 1 h |
| T max of the chelator | Mostly within 1 h | 1–3 h |
| T1/2 elimination of chelator | 47–134 min at 35–71 mg/kg | 19 +/− 6.5 h at 20 and 40 mg/kg |
| T1/2 elimination of the iron complex | Estimated within 47–134 min | 17.2 +/− 7.8 h at 20 mg/kg and 17.7 +/− 5.1 h at 40 mg/kg |
| T max of the iron complex | Estimated within 1 h | at 20 mg/kg 1–6 h and at 40 mg/kg 4–8 h |
| T max of the metabolite | glucuronide: 1–3 h | glucuronide: Not known |
| Route of elimination of chelator and its iron complex | urine | Almost exclusively in feces and less than 0.1% of the administered dose in urine |
| Enterohepatic re-circulation | L1 and iron complex not shown or suspected | DFRA and iron complex suspected from pharmacokinetic data |
| Clinical Use and Dose Ranges | ||
| Longest period of treatment | 33 years | 11 years |
| Time of experience of clinical use | 33 years | 16 years |
| Maximum dose in humans in 24 h | 250 mg/kg | 80 mg/kg |
| Maximum iron excretion in 24 h | 325 mg | 55 mg (estimated from the reported iron balance studies using 40 mg/kg ) |
| Dose in current use in 24 h | 75–110 mg/kg in divided doses | 20–40 mg/kg single dose |
| Effective dose for iron balance in most thalassemia patients | >80 mg/kg/day | >40 mg/kg |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kontoghiorghes, G.J.; Kontoghiorghe, C.N. Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases. Cells 2020, 9, 1456. https://doi.org/10.3390/cells9061456
Kontoghiorghes GJ, Kontoghiorghe CN. Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases. Cells. 2020; 9(6):1456. https://doi.org/10.3390/cells9061456
Chicago/Turabian StyleKontoghiorghes, George J., and Christina N. Kontoghiorghe. 2020. "Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases" Cells 9, no. 6: 1456. https://doi.org/10.3390/cells9061456
APA StyleKontoghiorghes, G. J., & Kontoghiorghe, C. N. (2020). Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases. Cells, 9(6), 1456. https://doi.org/10.3390/cells9061456