Ablation of Vitamin D Signaling Compromises Cerebrovascular Adaptation to Carotid Artery Occlusion in Mice

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. In Vivo Experiments
2.3. Measurement of Cerebrocortical Blood Flow Using Laser-Speckle Imaging
2.4. Measurement of Carotid Artery Blood Flow Using a Transit-Time Ultrasonic Flowmeter
2.5. Evaluation of the Morphology of Pial Collaterals
2.6. Data Analysis
3. Results
3.1. Physiological Parameters
3.2. Effects of VDR Deficiency on the Regional CoBF Changes after CAO
3.3. Effects of VDR Deficiency on the Extracranial Collateral Circulation
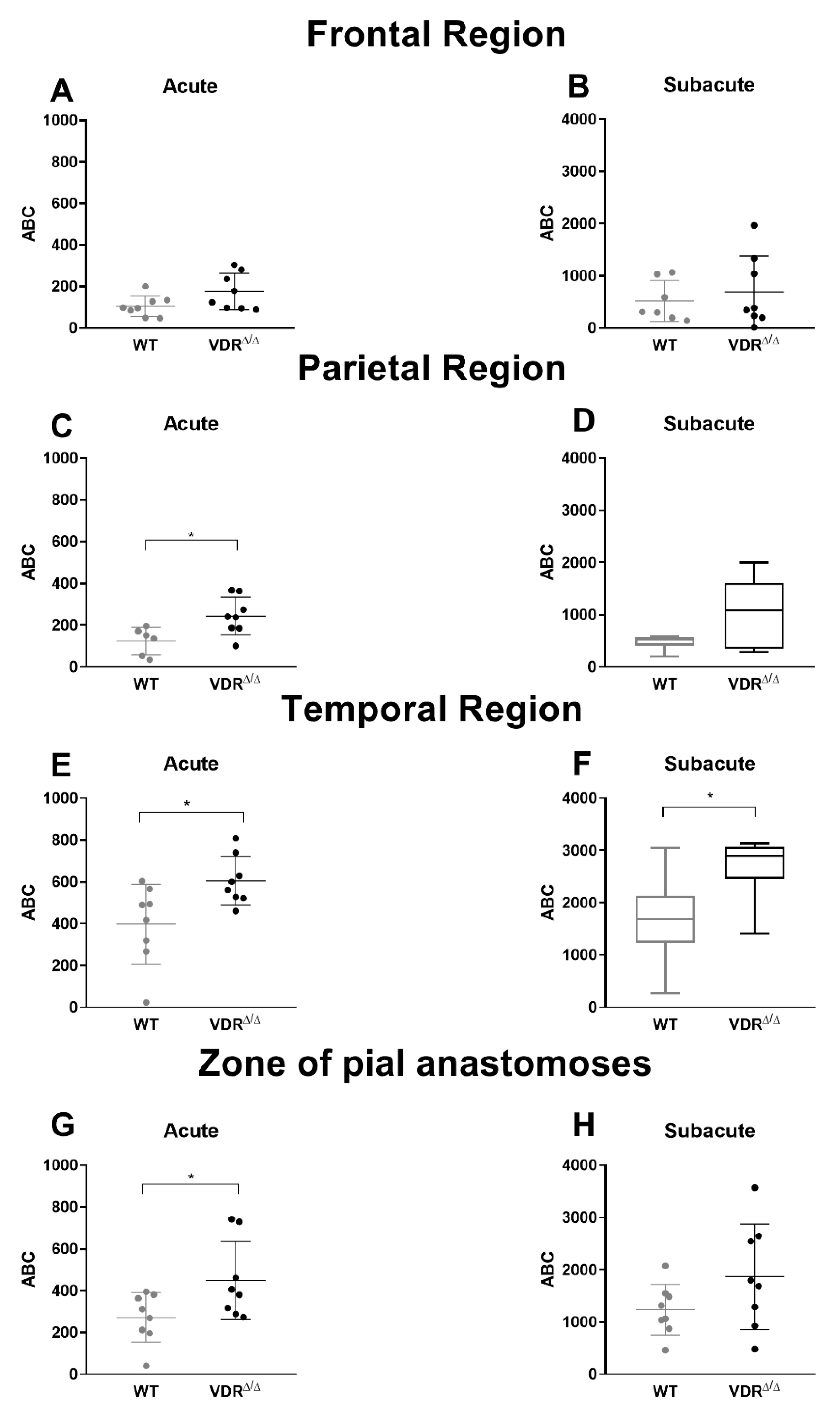
3.4. Effects of VDR Deficiency on the Intracranial Collateral Circulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Norman, P.E.; Powell, J.T. Vitamin D and Cardiovascular Disease. Circ. Res. 2014, 114, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Enkhjargal, B.; Malaguit, J.; Ho, W.M.; Jiang, W.; Wan, W.; Wang, G.; Tang, J.; Zhang, J.H. Vitamin D attenuates cerebral artery remodeling through VDR/AMPK/eNOS dimer phosphorylation pathway after subarachnoid hemorrhage in rats. Br. J. Pharmacol. 2017, 39, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Malloy, P.J.; Pike, J.W.; Feldman, D. The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D-resistant rickets. Endocr. Rev. 1999, 20, 156–188. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, T.; Afzal, S.; Yu, C.; Guo, Y.; Bian, Z.; Yang, L.; Millwood, I.Y.; Walters, R.G.; Chen, Y.; Chen, N.; et al. Vitamin D and cause-specific vascular disease and mortality: A Mendelian randomisation study involving 99,012 Chinese and 106,911 European adults. BMC Med. 2019, 17, 160. [Google Scholar] [CrossRef]
- Larsson, S.C.; Traylor, M.; Mishra, A.; Howson, J.M.M.; Michaelsson, K.; Markus, H.S. Serum 25-Hydroxyvitamin D Concentrations and Ischemic Stroke and Its Subtypes. Stroke 2018, 49, 2508–2511. [Google Scholar] [CrossRef]
- Leong, A.; Rehman, W.; Dastani, Z.; Greenwood, C.; Timpson, N.; Langsetmo, L.; Berger, C.; Fu, L.; Wong, B.Y.; Malik, S.; et al. The causal effect of vitamin D binding protein (DBP) levels on calcemic and cardiometabolic diseases: A Mendelian randomization study. PLoS Med. 2014, 11, e1001751. [Google Scholar] [CrossRef]
- Chowdhury, R.; Stevens, S.; Ward, H.; Chowdhury, S.; Sajjad, A.; Franco, O.H. Circulating vitamin D, calcium and risk of cerebrovascular disease: A systematic review and meta-analysis. Eur. J. Epidemiol. 2012, 27, 581–591. [Google Scholar] [CrossRef]
- Kojima, G.; Bell, C.; Abbott, R.D.; Launer, L.; Chen, R.; Motonaga, H.; Ross, G.W.; Curb, J.D.; Masaki, K. Low dietary vitamin D predicts 34-year incident stroke: The Honolulu Heart Program. Stroke 2012, 43, 2163–2167. [Google Scholar] [CrossRef]
- Sun, Q.; Pan, A.; Hu, F.B.; Manson, J.E.; Rexrode, K.M. 25-Hydroxyvitamin D levels and the risk of stroke: A prospective study and meta-analysis. Stroke 2012, 43, 1470–1477. [Google Scholar] [CrossRef]
- Zhou, R.; Wang, M.; Huang, H.; Li, W.; Hu, Y.; Wu, T. Lower Vitamin D Status Is Associated with an Increased Risk of Ischemic Stroke: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Turetsky, A.; Goddeau, R.P., Jr.; Henninger, N. Low Serum Vitamin D Is Independently Associated with Larger Lesion Volumes after Ischemic Stroke. J. Stroke Cereb. Dis. 2015, 24, 1555–1563. [Google Scholar] [CrossRef]
- Bajko, Z.; Balasa, R.; Motataianu, A.; Maier, S.; Chebut, O.C.; Szatmari, S. Common carotid artery occlusion: A case series. Isrn Neurol. 2013, 2013, 198595. [Google Scholar] [CrossRef] [PubMed]
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef]
- Shuaib, A.; Butcher, K.; Mohammad, A.A.; Saqqur, M.; Liebeskind, D.S. Collateral blood vessels in acute ischaemic stroke: A potential therapeutic target. Lancet Neurol. 2011, 10, 909–921. [Google Scholar] [CrossRef]
- Struys, T.; Govaerts, K.; Oosterlinck, W.; Casteels, C.; Bronckaers, A.; Koole, M.; Van Laere, K.; Herijgers, P.; Lambrichts, I.; Himmelreich, U.; et al. In vivo evidence for long-term vascular remodeling resulting from chronic cerebral hypoperfusion in mice. J. Cereb. Blood Flow Metab. 2017, 37, 726–739. [Google Scholar] [CrossRef]
- Polycarpou, A.; Hricisak, L.; Iring, A.; Safar, D.; Ruisanchez, E.; Horvath, B.; Sandor, P.; Benyo, Z. Adaptation of the cerebrocortical circulation to carotid artery occlusion involves blood flow redistribution between cortical regions and is independent of eNOS. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, 972–980. [Google Scholar] [CrossRef]
- Farkas, E.; Luiten, P.G.; Bari, F. Permanent, bilateral common carotid artery occlusion in the rat: A model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res. Rev. 2007, 54, 162–180. [Google Scholar] [CrossRef]
- Pal, E.; Hadjadj, L.; Fontanyi, Z.; Monori-Kiss, A.; Mezei, Z.; Lippai, N.; Magyar, A.; Heinzlmann, A.; Karvaly, G.; Monos, E.; et al. Vitamin D deficiency causes inward hypertrophic remodeling and alters vascular reactivity of rat cerebral arterioles. PLoS ONE 2018, 13, e0192480. [Google Scholar] [CrossRef]
- Pal, E.; Hadjadj, L.; Fontanyi, Z.; Monori-Kiss, A.; Lippai, N.; Horvath, E.M.; Magyar, A.; Horvath, E.; Monos, E.; Nadasy, G.L.; et al. Gender, hyperandrogenism and vitamin D deficiency related functional and morphological alterations of rat cerebral arteries. PLoS ONE 2019, 14, e0216951. [Google Scholar] [CrossRef]
- Erben, R.G.; Soegiarto, D.W.; Weber, K.; Zeitz, U.; Lieberherr, M.; Gniadecki, R.; Moller, G.; Adamski, J.; Balling, R. Deletion of deoxyribonucleic acid binding domain of the vitamin D receptor abrogates genomic and nongenomic functions of vitamin D. Mol. Endocrinol. 2002, 16, 1524–1537. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P.; Jokelainen, P.T. Dorsal cerebral arterial collaterals of the rat. Anat. Rec. 1982, 203, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.R.; Herz, J.; Hermann, D.M.; Doeppner, T.R. Visualization of macroscopic cerebral vessel anatomy—A new and reliable technique in mice. J. Neurosci. Methods 2012, 204, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Hata, R.; Bader, M.; Walther, T.; Hossmann, K.A. Larger anastomoses in angiotensinogen-knockout mice attenuate early metabolic disturbances after middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 1999, 19, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Liebeskind, D.S. Collateral circulation. Stroke 2003, 34, 2279–2284. [Google Scholar] [CrossRef]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef]
- Andrukhova, O.; Slavic, S.; Zeitz, U.; Riesen, S.C.; Heppelmann, M.S.; Ambrisko, T.D.; Markovic, M.; Kuebler, W.M.; Erben, R.G. Vitamin D is a regulator of endothelial nitric oxide synthase and arterial stiffness in mice. Mol. Endocrinol. 2014, 28, 53–64. [Google Scholar] [CrossRef]
- Li, Y.C.; Pirro, A.E.; Amling, M.; Delling, G.; Baron, R.; Bronson, R.; Demay, M.B. Targeted ablation of the vitamin D receptor: An animal model of vitamin D-dependent rickets type II with alopecia. Proc. Natl. Acad. Sci. USA 1997, 94, 9831–9835. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Handa, Y.; Uematsu, Y.; Takeda, S.; Sekine, K.; Yoshihara, Y.; Kawakami, T.; Arioka, K.; Sato, H.; Uchiyama, Y.; et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat. Genet. 1997, 16, 391–396. [Google Scholar] [CrossRef]
- Simpson, R.U.; Hershey, S.H.; Nibbelink, K.A. Characterization of heart size and blood pressure in the vitamin D receptor knockout mouse. J. Steroid Biochem. Mol. Biol. 2007, 103, 521–524. [Google Scholar] [CrossRef]
- Rahman, A.; Hershey, S.; Ahmed, S.; Nibbelink, K.; Simpson, R.U. Heart extracellular matrix gene expression profile in the vitamin D receptor knockout mice. J. Steroid Biochem. Mol. Biol. 2007, 103, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Kong, J.; Chen, S.; Cao, L.P.; Qiao, G.; Zheng, W.; Liu, W.; Li, X.; Gardner, D.G.; Li, Y.C. Cardiac hypertrophy in vitamin D receptor knockout mice: Role of the systemic and cardiac renin-angiotensin systems. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E125–E132. [Google Scholar] [CrossRef] [PubMed]
- Scremin, O.U.; Holschneider, D.P. The Mouse Nervous System-Vascular Supply; Watson, C., Paxinos, G., Puelles, L., Eds.; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Mozos, I.; Marginean, O. Links between Vitamin D Deficiency and Cardiovascular Diseases. Biomed. Res. Int. 2015, 2015, 109275. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Miguel, P.; Valdivielso, J.M.; Medrano-Andres, D.; Roman-Garcia, P.; Cano-Penalver, J.L.; Rodriguez-Puyol, M.; Rodriguez-Puyol, D.; Lopez-Ongil, S. The active form of vitamin D, calcitriol, induces a complex dual upregulation of endothelin and nitric oxide in cultured endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E1085–E1096. [Google Scholar] [CrossRef]
- Molinari, C.; Uberti, F.; Grossini, E.; Vacca, G.; Carda, S.; Invernizzi, M.; Cisari, C. 1alpha,25-dihydroxycholecalciferol induces nitric oxide production in cultured endothelial cells. Cell Physiol. Biochem. 2011, 27, 661–668. [Google Scholar] [CrossRef]
- Kovách, A.G.; Szabó, C.; Benyó, Z.; Csáki, C.; Greenberg, J.H.; Reivich, M. Effects of NG-nitro-L-arginine and L-arginine on regional cerebral blood flow in the cat. J. Physiol. 1992, 449, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wong, S.L.; Lau, C.W.; Lee, H.K.; Ng, C.F.; Zhang, L.; Yao, X.; Chen, Z.Y.; Vanhoutte, P.M.; Huang, Y. Calcitriol protects renovascular function in hypertension by down-regulating angiotensin II type 1 receptors and reducing oxidative stress. Eur. Heart J. 2012, 33, 2980–2990. [Google Scholar] [CrossRef]
- Zhong, W.; Gu, B.; Gu, Y.; Groome, L.J.; Sun, J.; Wang, Y. Activation of vitamin D receptor promotes VEGF and CuZn-SOD expression in endothelial cells. J. Steroid Biochem. Mol. Biol. 2014, 140, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Szabó, C.; Csáki, C.; Benyó, Z.; Marczis, J.; Reivich, M.; Kovách, A.G. Effect of superoxide dismutase on hemorrhagic hypotension and retransfusion-evoked middle cerebral artery endothelial dysfunction. Circ. Shock 1994, 44, 104–110. [Google Scholar]
- Zhang, H.; Prabhakar, P.; Sealock, R.; Faber, J.E. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J. Cereb. Blood Flow Metab. 2010, 30, 923–934. [Google Scholar] [CrossRef]
- Toriumi, H.; Tatarishvili, J.; Tomita, M.; Tomita, Y.; Unekawa, M.; Suzuki, N. Dually supplied T-junctions in arteriolo-arteriolar anastomosis in mice: Key to local hemodynamic homeostasis in normal and ischemic states? Stroke 2009, 40, 3378–3383. [Google Scholar] [CrossRef] [PubMed]
- Benyó, Z.; Ruisanchez, É.; Leszl-Ishiguro, M.; Sándor, P.; Pacher, P. Endocannabinoids in cerebrovascular regulation. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H785–H801. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.M. Morphology of cerebral arteries. Pharm. Ther. 1995, 66, 149–173. [Google Scholar] [CrossRef]
- Brozici, M.; van der Zwan, A.; Hillen, B. Anatomy and functionality of leptomeningeal anastomoses: A review. Stroke 2003, 34, 2750–2762. [Google Scholar] [CrossRef]
- Luo, C.; Liang, F.; Ren, H.; Yao, X.; Liu, Q.; Li, M.; Qin, D.; Yuan, T.F.; Pei, Z.; Su, H. Collateral blood flow in different cerebrovascular hierarchy provides endogenous protection in cerebral ischemia. Brain Pathol. 2017, 27, 809–821. [Google Scholar] [CrossRef]
- Hademenos, G.J.; Massoud, T.F. Biophysical mechanisms of stroke. Stroke 1997, 28, 2067–2077. [Google Scholar] [CrossRef]
- Maeda, K.; Hata, R.; Hossmann, K.A. Differences in the cerebrovascular anatomy of C57black/6 and SV129 mice. Neuroreport 1998, 9, 1317–1319. [Google Scholar] [CrossRef] [PubMed]
- Wu-Wong, J.R.; Nakane, M.; Ma, J.; Ruan, X.; Kroeger, P.E. VDR-mediated gene expression patterns in resting human coronary artery smooth muscle cells. J. Cell Biochem. 2007, 100, 1395–1405. [Google Scholar] [CrossRef]
- Clayton, J.A.; Chalothorn, D.; Faber, J.E. Vascular endothelial growth factor-A specifies formation of native collaterals and regulates collateral growth in ischemia. Circ. Res. 2008, 103, 1027–1036. [Google Scholar] [CrossRef]
- Dai, X.; Faber, J.E. Endothelial nitric oxide synthase deficiency causes collateral vessel rarefaction and impairs activation of a cell cycle gene network during arteriogenesis. Circ. Res. 2010, 106, 1870–1881. [Google Scholar] [CrossRef]
- Nishijima, Y.; Akamatsu, Y.; Weinstein, P.R.; Liu, J. Collaterals: Implications in cerebral ischemic diseases and therapeutic interventions. Brain Res. 2015, 1623, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Itoh, Y.; Toriumi, H.; Yamada, S.; Tomita, Y.; Hoshino, H.; Suzuki, N. Capillary remodeling and collateral growth without angiogenesis after unilateral common carotid artery occlusion in mice. Microcirculation 2011, 18, 221–227. [Google Scholar] [CrossRef]
- Hecht, N.; He, J.; Kremenetskaia, I.; Nieminen, M.; Vajkoczy, P.; Woitzik, J. Cerebral hemodynamic reserve and vascular remodeling in C57/BL6 mice are influenced by age. Stroke 2012, 43, 3052–3062. [Google Scholar] [CrossRef] [PubMed]
- Chalothorn, D.; Faber, J.E. Formation and maturation of the native cerebral collateral circulation. J. Mol. Cell Cardiol. 2010, 49, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Balden, R.; Selvamani, A.; Sohrabji, F. Vitamin D deficiency exacerbates experimental stroke injury and dysregulates ischemia-induced inflammation in adult rats. Endocrinology 2012, 153, 2420–2435. [Google Scholar] [CrossRef]
- Sayeed, I.; Turan, N.; Stein, D.G.; Wali, B. Vitamin D deficiency increases blood-brain barrier dysfunction after ischemic stroke in male rats. Exp. Neurol. 2019, 312, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.A.; Kim, H.A.; De Silva, T.; Arumugam, T.V.; Clarkson, A.N.; Drummond, G.R.; Zosky, G.R.; Broughton, B.R.; Sobey, C.G. Diet-induced vitamin D deficiency has no effect on acute post-stroke outcomes in young male mice. J. Cereb. Blood Flow Metab. 2017, 38, 1968–1978. [Google Scholar] [CrossRef]
- Gezmish, O.; Black, M.J. Vitamin D deficiency in early life and the potential programming of cardiovascular disease in adulthood. J. Cardiovasc. Transl. Res. 2013, 6, 588–603. [Google Scholar] [CrossRef]
- Hossein-nezhad, A.; Holick, M.F. Vitamin D for health: A global perspective. Mayo Clin. Proc. 2013, 88, 720–755. [Google Scholar] [CrossRef]
- Cui, X.; Gooch, H.; Petty, A.; McGrath, J.J.; Eyles, D. Vitamin D and the brain: Genomic and non-genomic actions. Mol. Cell Endocrinol. 2017, 453, 131–143. [Google Scholar] [CrossRef]
- Kesby, J.P.; Eyles, D.W.; Burne, T.H.; McGrath, J.J. The effects of vitamin D on brain development and adult brain function. Mol. Cell Endocrinol. 2011, 347, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Bivona, G.; Gambino, C.M.; Iacolino, G.; Ciaccio, M. Vitamin D and the nervous system. Neurol. Res. 2019, 41, 827–835. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | WT | VDRΔ/Δ |
|---|---|---|
| Body weight (g) | 30.0 ± 2.8 | 27.3 ± 2.4 ** |
| Tibial length (cm) | 1.80 (1.80–1.82) | 1.70 (1.60–1.70) **** |
| Heart weight (g) | 0.19 ± 0.03 | 0.17 ± 0.03 |
| Heart weight/body weight (%) | 0.60 ± 0.08 | 0.62 ± 0.11 |
| Left ventricle weight (g) | 0.12 (0.11–0.12) | 0.10 (0.09–0.11) |
| Brain weight (g) | 0.47 (0.44–0.48) | 0.45 (0.44–0.46) |
| Blood pressure (mmHg) | 84.09 ± 8.38 | 82.97 ± 9.60 |
| Heart rate (1/min) | 364.8 (310.3–423.3) | 351.6 (320.7–409.8) |
| Respiratory rate (1/min) | 59.56 (52.99–82.40) | 62.56 (54.42–78.91) |
| Hematocrit (%) | 42.63 ± 2.26 | 43.57 ± 1.51 |
| cNa+ (mmol/L) | 156.5 ± 3.67 | 156.7 ± 3.64 |
| cK+ (mmol/L) | 4.30 (3.67–4.46) | 4.02 (3.62–4.22) |
| cCa2+ (mmol/L) | 1.29 ± 0.09 | 1.24 ± 0.07 |
| cCl− (mmol/L) | 113.4 ± 2.50 | 115.5 ± 4.96 |
| Parameter | WT | VDRΔ/Δ |
|---|---|---|
| pH | 7.31 ± 0.10 | 7.29 ± 0.04 |
| pCO2 (mmHg) | 41.16 ± 9.13 | 42.51 ± 6.18 |
| pO2 (mmHg) | 93.5 (89.0–103.09) | 109.0 (85.5–112.8) |
| Standard base excess (mmol/L) | −5.08 ± 2.94 | −5.66 ± 2.81 |
| O2 saturation (%) | 96.10 (94.95–97.58) | 97.60 (93.40–97.98) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pál, É.; Hricisák, L.; Lékai, Á.; Nagy, D.; Fülöp, Á.; Erben, R.G.; Várbíró, S.; Sándor, P.; Benyó, Z. Ablation of Vitamin D Signaling Compromises Cerebrovascular Adaptation to Carotid Artery Occlusion in Mice. Cells 2020, 9, 1457. https://doi.org/10.3390/cells9061457
Pál É, Hricisák L, Lékai Á, Nagy D, Fülöp Á, Erben RG, Várbíró S, Sándor P, Benyó Z. Ablation of Vitamin D Signaling Compromises Cerebrovascular Adaptation to Carotid Artery Occlusion in Mice. Cells. 2020; 9(6):1457. https://doi.org/10.3390/cells9061457
Chicago/Turabian StylePál, Éva, László Hricisák, Ágnes Lékai, Dorina Nagy, Ágnes Fülöp, Reinhold G. Erben, Szabolcs Várbíró, Péter Sándor, and Zoltán Benyó. 2020. "Ablation of Vitamin D Signaling Compromises Cerebrovascular Adaptation to Carotid Artery Occlusion in Mice" Cells 9, no. 6: 1457. https://doi.org/10.3390/cells9061457
APA StylePál, É., Hricisák, L., Lékai, Á., Nagy, D., Fülöp, Á., Erben, R. G., Várbíró, S., Sándor, P., & Benyó, Z. (2020). Ablation of Vitamin D Signaling Compromises Cerebrovascular Adaptation to Carotid Artery Occlusion in Mice. Cells, 9(6), 1457. https://doi.org/10.3390/cells9061457