Cancer Stem Cells in Soft-Tissue Sarcomas

 , ,
, ,  , and
, and 
Abstract
1. Introduction
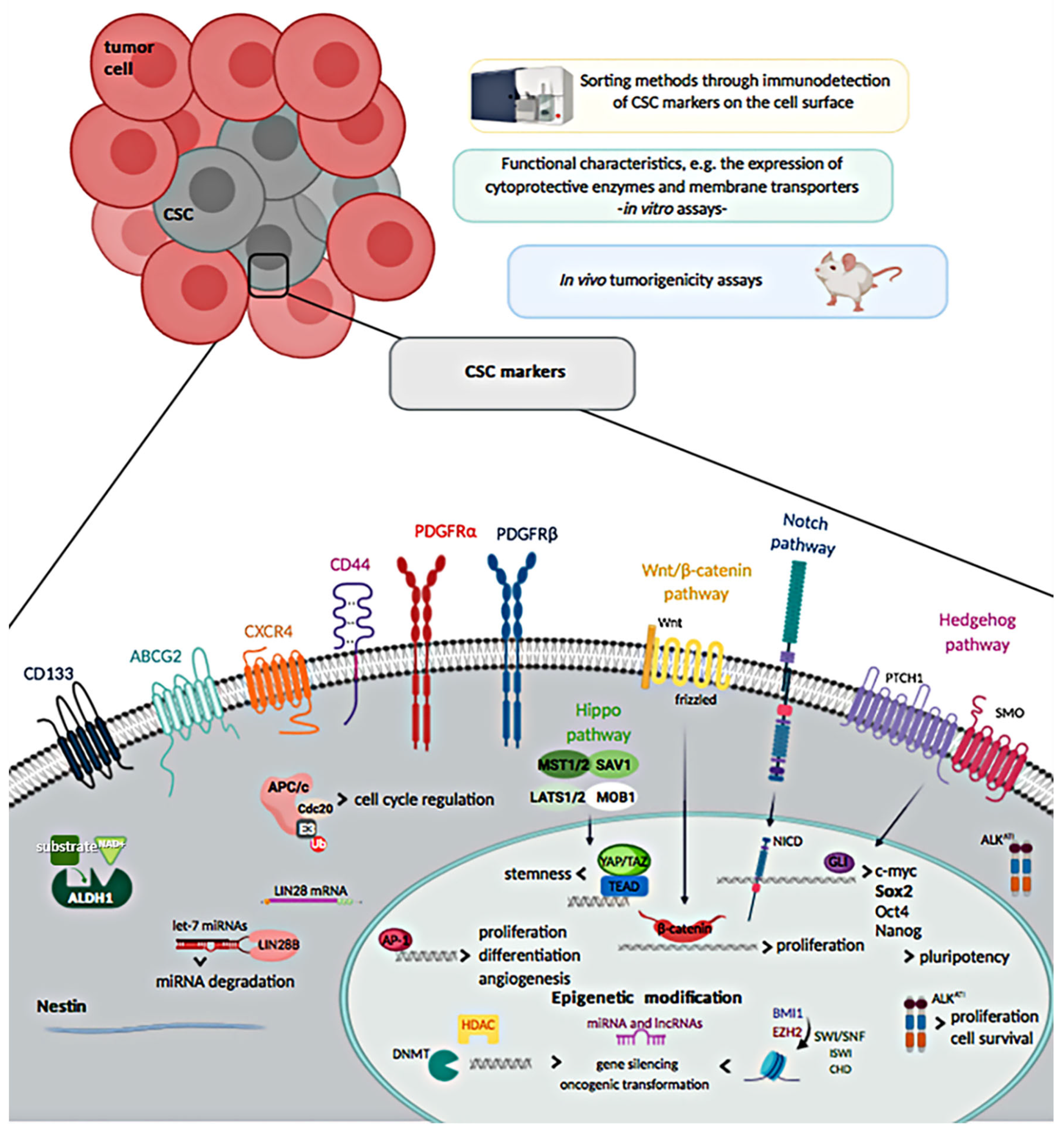
2. Isolation and Molecular Characterization of Cancer Stem Cells (CSCs) in Soft Tissue Sarcomas (STS)
2.1. Cell Surface Markers
2.1.1. CD133
2.1.2. Chemokine Receptor Type-4
2.1.3. CD44
2.1.4. Nestin
2.1.5. ALDH1
2.1.6. PDGFRα and PDGFRβ
2.1.7. ATP-Binding Cassette (ABC) Efflux Pumps
2.2. Side Population
2.3. Stemness Markers
2.4. Epigenetic Alterations
3. New Therapeutic Approaches against CSCs in STS and Future Directions
3.1. Inhibition of CSC-Dependent Pathways and Surface Markers
3.1.1. ABC Efflux Pumps
3.1.2. JAK–STAT Signaling Pathway
3.1.3. DNA Damage Repair Pathways
3.1.4. mTOR Pathway
3.1.5. Wnt Pathway
3.1.6. ALKATI-Targeted Therapy
3.2. Targeting the Tumor Microenvironment
3.3. Differentiation Therapy
3.4. Directed Immunotherapy
3.5. Epigenetic Modulation
4. Conclusions
Conflicts of Interest
References
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Moghaddam, S.H.Z. Cancer stem cells: A review from origin to therapeutic implications. J. Cell Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Abarrategi, A.; Tornin, J.; Martinez-Cruzado, L.; Hamilton, A.; Martinez-Campos, E.; Rodrigo, J.P.; González, M.V.; Baldini, N.; Garcia-Castro, J.; Rodriguez, R. Osteosarcoma: Cells-of-Origin, Cancer Stem Cells, and Targeted Therapies. Stem Cells Int. 2016, 2016, 3631764. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Birbrair, A. Stem Cells Heterogeneity. Adv. Exp. Med. Biol. 2019, 1123, 1–3. [Google Scholar] [CrossRef]
- Skoda, J.; Veselska, R. Cancer stem cells in sarcomas: Getting to the stemness core. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2134–2139. [Google Scholar] [CrossRef]
- Honoki, K. Do stem-like cells play a role in drug resistance of sarcomas? Expert Rev. Anticancer Ther. 2010, 10, 261–270. [Google Scholar] [CrossRef]
- Tap, W.D.; Wagner, A.J.; Schoffski, P.; Martin-Broto, J.; Krarup-Hansen, A.; Ganjoo, K.N.; Yen, C.C.; Abdul Razak, A.R.; Spira, A.; Kawai, A.; et al. Effect of Doxorubicin Plus Olaratumab vs Doxorubicin Plus Placebo on Survival in Patients With Advanced Soft Tissue Sarcomas: The ANNOUNCE Randomized Clinical Trial. JAMA 2020, 323, 1266–1276. [Google Scholar] [CrossRef]
- Tap, W.D.; Papai, Z.; Van Tine, B.A.; Attia, S.; Ganjoo, K.N.; Jones, R.L.; Schuetze, S.; Reed, D.; Chawla, S.P.; Riedel, R.F.; et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): An international, multicentre, open-label, randomised phase 3 trial. The Lancet. Oncology 2017, 18, 1089–1103. [Google Scholar] [CrossRef]
- Ryan, C.W.; Merimsky, O.; Agulnik, M.; Blay, J.Y.; Schuetze, S.M.; Van Tine, B.A.; Jones, R.L.; Elias, A.D.; Choy, E.; Alcindor, T.; et al. PICASSO III: A Phase III, Placebo-Controlled Study of Doxorubicin With or Without Palifosfamide in Patients With Metastatic Soft Tissue Sarcoma. J. Clin. Oncol. 2016, 34, 3898–3905. [Google Scholar] [CrossRef]
- Potter, J.W.; Jones, K.B.; Barrott, J.J. Sarcoma–The standard-bearer in cancer discovery. Crit. Rev. Oncol. Hematol. 2018, 126, 1–5. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Karsten, U.; Goletz, S. What makes cancer stem cell markers different? Springerplus 2013, 2, 301. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nature Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduc. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Alison, M.R.; Lim, S.M.; Nicholson, L.J. Cancer stem cells: Problems for therapy? J. Pathol. 2011, 223, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Genadry, K.C.; Pietrobono, S.; Rota, R.; Linardic, C.M. Soft Tissue Sarcoma Cancer Stem Cells: An Overview. Front. Oncol. 2018, 8, 475. [Google Scholar] [CrossRef]
- Suvà, M.L.; Riggi, N.; Stehle, J.C.; Baumer, K.; Tercier, S.; Joseph, J.M.; Suvà, D.; Clément, V.; Provero, P.; Cironi, L.; et al. Identification of cancer stem cells in Ewing’s sarcoma. Cancer Res. 2009, 69, 1776–1781. [Google Scholar] [CrossRef]
- Sana, J.; Zambo, I.; Skoda, J.; Neradil, J.; Chlapek, P.; Hermanova, M.; Mudry, P.; Vasikova, A.; Zitterbart, K.; Hampl, A.; et al. CD133 expression and identification of CD133/nestin positive cells in rhabdomyosarcomas and rhabdomyosarcoma cell lines. Anal. Cell. Pathol. (Amst.) 2011, 34, 303–318. [Google Scholar] [CrossRef][Green Version]
- Walter, D.; Satheesha, S.; Albrecht, P.; Bornhauser, B.C.; D’Alessandro, V.; Oesch, S.M.; Rehrauer, H.; Leuschner, I.; Koscielniak, E.; Gengler, C.; et al. CD133 positive embryonal rhabdomyosarcoma stem-like cell population is enriched in rhabdospheres. PLoS ONE 2011, 6, e19506. [Google Scholar] [CrossRef]
- Pressey, J.G.; Haas, M.C.; Pressey, C.S.; Kelly, V.M.; Parker, J.N.; Gillespie, G.Y.; Friedman, G.K. CD133 marks a myogenically primitive subpopulation in rhabdomyosarcoma cell lines that are relatively chemoresistant but sensitive to mutant HSV. Pediatr. Blood Cancer 2013, 60, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.H.; Liu, A.G.; Gu, W.G.; Deng, L.; Cheng, X.G.; Tong, T.J.; Zhang, H.Z. CD133+ subpopulation of the HT1080 human fibrosarcoma cell line exhibits cancer stem-like characteristics. Oncol. Rep. 2013, 30, 815–823. [Google Scholar] [CrossRef]
- Liu, A.; Feng, B.; Gu, W.; Cheng, X.; Tong, T.; Zhang, H.; Hu, Y. The CD133+ subpopulation of the SW982 human synovial sarcoma cell line exhibits cancer stem-like characteristics. Int. J. Oncol. 2013, 42, 1399–1407. [Google Scholar] [CrossRef]
- Liu, W.D.; Zhang, T.; Wang, C.L.; Meng, H.M.; Song, Y.W.; Zhao, Z.; Li, Z.M.; Liu, J.K.; Pan, S.H.; Wang, W.B. Sphere-forming tumor cells possess stem-like properties in human fibrosarcoma primary tumors and cell lines. Oncol. Lett. 2012, 4, 1315–1320. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Terry, J.; Nielsen, T. Expression of CD133 in synovial sarcoma. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Skoda, J.; Nunukova, A.; Loja, T.; Zambo, I.; Neradil, J.; Mudry, P.; Zitterbart, K.; Hermanova, M.; Hampl, A.; Sterba, J.; et al. Cancer stem cell markers in pediatric sarcomas: Sox2 is associated with tumorigenicity in immunodeficient mice. Tumour Biol. 2016, 37, 9535–9548. [Google Scholar] [CrossRef] [PubMed]
- Zambo, I.; Hermanova, M.; Zapletalova, D.; Skoda, J.; Mudry, P.; Kyr, M.; Zitterbart, K.; Sterba, J.; Veselska, R. Expression of nestin, CD133 and ABCG2 in relation to the clinical outcome in pediatric sarcomas. Cancer Biomark. 2016, 17, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Wang, L.; Tabu, K.; Tsuda, M.; Tanino, M.; Maekawa, A.; Nishihara, H.; Hiraga, H.; Taga, T.; Oda, Y.; et al. Identification and analysis of CXCR4-positive synovial sarcoma-initiating cells. Oncogene 2016, 35, 3932–3943. [Google Scholar] [CrossRef]
- Pazzaglia, L.; Pollino, S.; Vitale, M.; Bientinesi, E.; Benini, S.; Ferrari, C.; Palmerini, E.; Gambarotti, M.; Picci, P.; Benassi, M.S. miR494.3p expression in synovial sarcoma: Role of CXCR4 as a potential target gene. Int. J. Oncol. 2019, 54, 361–369. [Google Scholar] [CrossRef]
- Palmerini, E.; Benassi, M.S.; Quattrini, I.; Pazzaglia, L.; Donati, D.; Benini, S.; Gamberi, G.; Gambarotti, M.; Picci, P.; Ferrari, S. Prognostic and predictive role of CXCR4, IGF-1R and Ezrin expression in localized synovial sarcoma: Is chemotaxis important to tumor response? Orphanet J. Rare Dis. 2015, 10, 6. [Google Scholar] [CrossRef]
- Skubitz, K.M.; Wilson, J.D.; Cheng, E.Y.; Lindgren, B.R.; Boylan, K.L.M.; Skubitz, A.P.N. Effect of chemotherapy on cancer stem cells and tumor-associated macrophages in a prospective study of preoperative chemotherapy in soft tissue sarcoma. J. Transl. Med. 2019, 17, 130. [Google Scholar] [CrossRef]
- Henderson, T.; Chen, M.; Darrow, M.A.; Li, C.S.; Chiu, C.L.; Monjazeb, A.M.; Murphy, W.J.; Canter, R.J. Alterations in cancer stem-cell marker CD44 expression predict oncologic outcome in soft-tissue sarcomas. J. Surg. Res. 2018, 223, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Matsuda, Y.; Naito, Z. Nestin in gastrointestinal and other cancers: Effects on cells and tumor angiogenesis. World J. Gastroenterol. 2011, 17, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Neradil, J.; Veselska, R. Nestin as a marker of cancer stem cells. Cancer Sci. 2015, 106, 803–811. [Google Scholar] [CrossRef]
- Murphy, A.J.; Viero, S.; Ho, M.; Thorner, P.S. Diagnostic utility of nestin expression in pediatric tumors in the region of the kidney. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Spyra, M.; Kluwe, L.; Hagel, C.; Nguyen, R.; Panse, J.; Kurtz, A.; Mautner, V.F.; Rabkin, S.D.; Demestre, M. Cancer stem cell-like cells derived from malignant peripheral nerve sheath tumors. PLoS ONE 2011, 6, e21099. [Google Scholar] [CrossRef]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef]
- Awad, O.; Yustein, J.T.; Shah, P.; Gul, N.; Katuri, V.; O’Neill, A.; Kong, Y.; Brown, M.L.; Toretsky, J.A.; Loeb, D.M. High ALDH activity identifies chemotherapy-resistant Ewing’s sarcoma stem cells that retain sensitivity to EWS-FLI1 inhibition. PLoS ONE 2010, 5, e13943. [Google Scholar] [CrossRef]
- Nakahata, K.; Uehara, S.; Nishikawa, S.; Kawatsu, M.; Zenitani, M.; Oue, T.; Okuyama, H. Aldehyde Dehydrogenase 1 (ALDH1) Is a Potential Marker for Cancer Stem Cells in Embryonal Rhabdomyosarcoma. PLoS ONE 2015, 10, e0125454. [Google Scholar] [CrossRef]
- Lohberger, B.; Rinner, B.; Stuendl, N.; Absenger, M.; Liegl-Atzwanger, B.; Walzer, S.M.; Windhager, R.; Leithner, A. Aldehyde dehydrogenase 1, a potential marker for cancer stem cells in human sarcoma. PLoS ONE 2012, 7, e43664. [Google Scholar] [CrossRef]
- Stratford, E.W.; Castro, R.; Wennerstrom, A.; Holm, R.; Munthe, E.; Lauvrak, S.; Bjerkehagen, B.; Myklebost, O. Liposarcoma Cells with Aldefluor and CD133 Activity have a Cancer Stem Cell Potential. Clin. Sarcoma Res. 2011, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; Feng, F.E.; Wang, Q.M.; Zhu, X.L.; Fu, H.X.; Xu, L.P.; Liu, K.Y.; Huang, X.J.; Zhang, X.H. Platelet-Derived Growth Factor-BB Protects Mesenchymal Stem Cells (MSCs) Derived From Immune Thrombocytopenia Patients Against Apoptosis and Senescence and Maintains MSC-Mediated Immunosuppression. Stem Cells Transl. Med. 2016, 5, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.; Boucher, S.; Koh, S.; Sastry, K.S.; Chase, L.; Lakshmipathy, U.; Choong, C.; Yang, Z.; Vemuri, M.C.; Rao, M.S.; et al. PDGF, TGF-beta, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): Transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood 2008, 112, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.K.; Yoon, C.; Yi, B.C.; Tap, W.D.; Simon, M.C.; Yoon, S.S. Platelet-derived growth factor receptor-α and -β promote cancer stem cell phenotypes in sarcomas. Oncogenesis 2018, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Pedeutour, F.; Simon, M.P.; Minoletti, F.; Barcelo, G.; Terrier-Lacombe, M.J.; Combemale, P.; Sozzi, G.; Ayraud, N.; Turc-Carel, C. Translocation, t(17;22)(q22;q13), in dermatofibrosarcoma protuberans: A new tumor-associated chromosome rearrangement. Cytogen. Cell Genet. 1996, 72, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, P.; Van Glabbeke, M.; Rankin, C.J.; Ruka, W.; Rubin, B.P.; Debiec-Rychter, M.; Lazar, A.; Gelderblom, H.; Sciot, R.; Lopez-Terrada, D.; et al. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: Pooled analysis of two phase II clinical trials. J. Clin. Oncol. 2010, 28, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Noujaim, J.; Thway, K.; Fisher, C.; Jones, R.L. Dermatofibrosarcoma protuberans: From translocation to targeted therapy. Cancer Biol. Med. 2015, 12, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Moitra, K. Overcoming Multidrug Resistance in Cancer Stem Cells. Biomed. Res. Int. 2015, 2015, 635745. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Gutierrez, A.M.; Ramos, R.F.; Lopez-Guerrero, J.A.; Ferrari, S.; Stacchiotti, S.; Picci, P.; Calabuig, S.; Collini, P.; Gambarotti, M.; et al. MRP1 overexpression determines poor prognosis in prospectively treated patients with localized high-risk soft tissue sarcoma of limbs and trunk wall: An ISG/GEIS study. Mol. Cancer Ther. 2014, 13, 249–259. [Google Scholar] [CrossRef]
- Deng, L.; Li, D.; Gu, W.; Liu, A.; Cheng, X. Formation of spherical cancer stem-like cell colonies with resistance to chemotherapy drugs in the human malignant fibrous histiocytoma NMFH-1 cell line. Oncol. Lett. 2015, 10, 3323–3331. [Google Scholar] [CrossRef][Green Version]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef]
- Christgen, M.; Ballmaier, M.; Lehmann, U.; Kreipe, H. Detection of putative cancer stem cells of the side population phenotype in human tumor cell cultures. Methods Mol. Biol. 2012, 878, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.P.; Nishijo, K.; Chen, H.I.; Yi, X.; Schuetze, D.P.; Pal, R.; Prajapati, S.I.; Abraham, J.; Arenkiel, B.R.; Chen, Q.R.; et al. Evidence for an unanticipated relationship between undifferentiated pleomorphic sarcoma and embryonal rhabdomyosarcoma. Cancer Cell 2011, 19, 177–191. [Google Scholar] [CrossRef]
- Hotfilder, M.; Mallela, N.; Seggewiss, J.; Dirksen, U.; Korsching, E. Defining a Characteristic Gene Expression Set Responsible for Cancer Stem Cell-Like Features in a Sub-Population of Ewing Sarcoma Cells CADO-ES1. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wei, Q.; Utomo, V.; Nadesan, P.; Whetstone, H.; Kandel, R.; Wunder, J.S.; Alman, B.A. Side Population Cells Isolated from Mesenchymal Neoplasms Have Tumor Initiating Potential. Cancer Res. 2007, 67, 8216–8222. [Google Scholar] [CrossRef] [PubMed]
- Hatina, J.; Kripnerova, M.; Houfkova, K.; Pesta, M.; Kuncova, J.; Sana, J.; Slaby, O.; Rodríguez, R. Sarcoma Stem Cell Heterogeneity. Adv. Exp. Med. Biol. 2019, 1123, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Roma, J.; Almazán-Moga, A.; Sánchez de Toledo, J.; Gallego, S. Notch, wnt, and hedgehog pathways in rhabdomyosarcoma: From single pathways to an integrated network. Sarcoma 2012, 2012, 695603. [Google Scholar] [CrossRef]
- Satheesha, S.; Manzella, G.; Bovay, A.; Casanova, E.A.; Bode, P.K.; Belle, R.; Feuchtgruber, S.; Jaaks, P.; Dogan, N.; Koscielniak, E.; et al. Targeting hedgehog signaling reduces self-renewal in embryonal rhabdomyosarcoma. Oncogene 2016, 35, 2020–2030. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wei, Q.; Han, I.; Sato, S.; Ghanbari-Azarnier, R.; Whetstone, H.; Poon, R.; Hu, J.; Zheng, F.; Zhang, P.; et al. Hedgehog and Notch signaling regulate self-renewal of undifferentiated pleomorphic sarcomas. Cancer Res. 2012, 72, 1013–1022. [Google Scholar] [CrossRef]
- Ignatius, M.S.; Hayes, M.N.; Lobbardi, R.; Chen, E.Y.; McCarthy, K.M.; Sreenivas, P.; Motala, Z.; Durbin, A.D.; Molodtsov, A.; Reeder, S.; et al. The NOTCH1/SNAIL1/MEF2C Pathway Regulates Growth and Self-Renewal in Embryonal Rhabdomyosarcoma. Cell Rep. 2017, 19, 2304–2318. [Google Scholar] [CrossRef]
- Slemmons, K.K.; Crose, L.E.S.; Riedel, S.; Sushnitha, M.; Belyea, B.; Linardic, C.M. A Novel Notch-YAP Circuit Drives Stemness and Tumorigenesis in Embryonal Rhabdomyosarcoma. Mol. Cancer Res. 2017, 15, 1777–1791. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338.e315–354.e315. [Google Scholar] [CrossRef]
- Riggi, N.; Suvà, M.L.; De Vito, C.; Provero, P.; Stehle, J.C.; Baumer, K.; Cironi, L.; Janiszewska, M.; Petricevic, T.; Suvà, D.; et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010, 24, 916–932. [Google Scholar] [CrossRef]
- Mizushima, E.; Tsukahara, T.; Emori, M.; Murata, K.; Akamatsu, A.; Shibayama, Y.; Hamada, S.; Watanabe, Y.; Kaya, M.; Hirohashi, Y.; et al. Osteosarcoma-initiating cells show high aerobic glycolysis and attenuation of oxidative phosphorylation mediated by LIN28B. Cancer Sci. 2020, 111, 36–46. [Google Scholar] [CrossRef]
- Vishnubalaji, R.; Elango, R.; Al-Toub, M.; Manikandan, M.; Al-Rikabi, A.; Harkness, L.; Ditzel, N.; Atteya, M.; Hamam, R.; Alfayez, M.; et al. Neoplastic Transformation of Human Mesenchymal Stromal Cells Mediated via LIN28B. Sci. Rep. 2019, 9, 8101. [Google Scholar] [CrossRef] [PubMed]
- Mazzu, Y.Z.; Hu, Y.; Soni, R.K.; Mojica, K.M.; Qin, L.X.; Agius, P.; Waxman, Z.M.; Mihailovic, A.; Socci, N.D.; Hendrickson, R.C.; et al. miR-193b-Regulated Signaling Networks Serve as Tumor Suppressors in Liposarcoma and Promote Adipogenesis in Adipose-Derived Stem Cells. Cancer Res. 2017, 77, 5728–5740. [Google Scholar] [CrossRef]
- Megiorni, F.; Camero, S.; Ceccarelli, S.; McDowell, H.P.; Mannarino, O.; Marampon, F.; Pizer, B.; Shukla, R.; Pizzuti, A.; Marchese, C.; et al. DNMT3B in vitro knocking-down is able to reverse embryonal rhabdomyosarcoma cell phenotype through inhibition of proliferation and induction of myogenic differentiation. Oncotarget 2016, 7, 79342–79356. [Google Scholar] [CrossRef] [PubMed]
- Ciarapica, R.; Carcarino, E.; Adesso, L.; De Salvo, M.; Bracaglia, G.; Leoncini, P.P.; Dall’agnese, A.; Verginelli, F.; Milano, G.M.; Boldrini, R.; et al. Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC Cancer 2014, 14, 139. [Google Scholar] [CrossRef]
- Shen, J.K.; Cote, G.M.; Gao, Y.; Choy, E.; Mankin, H.J.; Hornicek, F.J.; Duan, Z. Targeting EZH2-mediated methylation of H3K27 inhibits proliferation and migration of Synovial Sarcoma in vitro. Sci. Rep. 2016, 6, 25239. [Google Scholar] [CrossRef]
- Changchien, Y.C.; Tátrai, P.; Papp, G.; Sápi, J.; Fónyad, L.; Szendrői, M.; Pápai, Z.; Sápi, Z. Poorly differentiated synovial sarcoma is associated with high expression of enhancer of zeste homologue 2 (EZH2). J. Transl. Med. 2012, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Grassian, A.R.; Tsuda, M.; Knutson, S.K.; Warholic, N.M.; Kuznetsov, G.; Xu, S.; Xiao, Y.; Pollock, R.M.; Smith, J.J.; et al. Correction: Preclinical Evidence of Anti-Tumor Activity Induced by EZH2 Inhibition in Human Models of Synovial Sarcoma. PLoS ONE 2017, 12, e0170539. [Google Scholar] [CrossRef]
- Karlsson, J.; Valind, A.; Jansson, C.; O’Sullivan, M.J.; Holmquist Mengelbier, L.; Gisselsson, D. Aberrant epigenetic regulation in clear cell sarcoma of the kidney featuring distinct DNA hypermethylation and EZH2 overexpression. Oncotarget 2016, 7, 11127–11136. [Google Scholar] [CrossRef]
- Dolatabadi, S.; Jonasson, E.; Lindén, M.; Fereydouni, B.; Bäcksten, K.; Nilsson, M.; Martner, A.; Forootan, A.; Fagman, H.; Landberg, G.; et al. JAK-STAT signalling controls cancer stem cell properties including chemotherapy resistance in myxoid liposarcoma. Int. J. Cancer 2019, 145, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Zayed, H.; Petersen, I. Stem cell transcription factor SOX2 in synovial sarcoma and other soft tissue tumors. Pathol. Res. Pract. 2018, 214, 1000–1007. [Google Scholar] [CrossRef]
- Taulli, R.; Foglizzo, V.; Morena, D.; Coda, D.M.; Ala, U.; Bersani, F.; Maestro, N.; Ponzetto, C. Failure to downregulate the BAF53a subunit of the SWI/SNF chromatin remodeling complex contributes to the differentiation block in rhabdomyosarcoma. Oncogene 2014, 33, 2354–2362. [Google Scholar] [CrossRef] [PubMed]
- Barham, W.; Frump, A.L.; Sherrill, T.P.; Garcia, C.B.; Saito-Diaz, K.; VanSaun, M.N.; Fingleton, B.; Gleaves, L.; Orton, D.; Capecchi, M.R.; et al. Targeting the Wnt pathway in synovial sarcoma models. Cancer Discov. 2013, 3, 1286–1301. [Google Scholar] [CrossRef] [PubMed]
- Nooter, K.; Stoter, G. Molecular mechanisms of multidrug resistance in cancer chemotherapy. Pathol. Res. Pract. 1996, 192, 768–780. [Google Scholar] [CrossRef]
- Martinez-Cruzado, L.; Tornin, J.; Rodriguez, A.; Santos, L.; Allonca, E.; Fernandez-Garcia, M.T.; Astudillo, A.; Garcia-Pedrero, J.M.; Rodriguez, R. Trabectedin and Campthotecin Synergistically Eliminate Cancer Stem Cells in Cell-of-Origin Sarcoma Models. Neoplasia 2017, 19, 460–470. [Google Scholar] [CrossRef]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genomics 2009, 3, 281–290. [Google Scholar] [CrossRef]
- Villar, V.H.; Vögler, O.; Barceló, F.; Gómez-Florit, M.; Martínez-Serra, J.; Obrador-Hevia, A.; Martín-Broto, J.; Ruiz-Gutiérrez, V.; Alemany, R. Oleanolic and maslinic acid sensitize soft tissue sarcoma cells to doxorubicin by inhibiting the multidrug resistance protein MRP-1, but not P-glycoprotein. J. Nutr. Biochem. 2014, 25, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Villar, V.H.; Vögler, O.; Martínez-Serra, J.; Ramos, R.; Calabuig-Fariñas, S.; Gutiérrez, A.; Barceló, F.; Martín-Broto, J.; Alemany, R. Nilotinib counteracts P-glycoprotein-mediated multidrug resistance and synergizes the antitumoral effect of doxorubicin in soft tissue sarcomas. PLoS ONE 2012, 7, e37735. [Google Scholar] [CrossRef] [PubMed]
- Aggerholm-Pedersen, N.; Demuth, C.; Safwat, A.; Meldgaard, P.; Kassem, M.; Sandahl Sorensen, B. Dasatinib and Doxorubicin Treatment of Sarcoma Initiating Cells: A Possible New Treatment Strategy. Stem Cells Int. 2016, 2016, 9601493. [Google Scholar] [CrossRef] [PubMed]
- Kerklaan, B.M.; Lolkema, M.P.; Devriese, L.A.; Voest, E.E.; Nol-Boekel, A.; Mergui-Roelvink, M.; Langenberg, M.; Mykulowycz, K.; Stoebenau, J.; Lane, S.; et al. Phase I and pharmacological study of pazopanib in combination with oral topotecan in patients with advanced solid tumours. Br. J. Cancer 2015, 113, 706–715. [Google Scholar] [CrossRef]
- Alemany, R.; Moura, D.S.; Redondo, A.; Martinez-Trufero, J.; Calabuig, S.; Saus, C.; Obrador-Hevia, A.; Ramos, R.; Villar, V.H.; Valverde, C.; et al. Nilotinib as Coadjuvant Treatment with Doxorubicin in Patients with Sarcomas: A Phase I Trial of the Spanish Group for Research on Sarcoma. Clin. Cancer Res. 2018, 24, 5239–5249. [Google Scholar] [CrossRef]
- Fujii, H.; Honoki, K.; Tsujiuchi, T.; Kido, A.; Yoshitani, K.; Takakura, Y. Sphere-forming stem-like cell populations with drug resistance in human sarcoma cell lines. Int. J. Oncol. 2009, 34, 1381–1386. [Google Scholar]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Okuno, S.; Bailey, H.; Mahoney, M.R.; Adkins, D.; Maples, W.; Fitch, T.; Ettinger, D.; Erlichman, C.; Sarkaria, J.N. A phase 2 study of temsirolimus (CCI-779) in patients with soft tissue sarcomas: A study of the Mayo phase 2 consortium (P2C). Cancer 2011, 117, 3468–3475. [Google Scholar] [CrossRef]
- Chawla, S.P.; Staddon, A.P.; Baker, L.H.; Schuetze, S.M.; Tolcher, A.W.; D’Amato, G.Z.; Blay, J.Y.; Mita, M.M.; Sankhala, K.K.; Berk, L.; et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J. Clin. Oncol. 2012, 30, 78–84. [Google Scholar] [CrossRef]
- Martin-Liberal, J.; Lopez-Pousa, A.; Martinez-Trufero, J.; Martin-Broto, J.; Cubedo, R.; Lavernia, J.; Redondo, A.; Lopez-Martin, J.A.; Mulet-Margalef, N.; Sanjuan, X.; et al. Phase II Study of Gemcitabine Plus Sirolimus in Previously Treated Patients with Advanced Soft-Tissue Sarcoma: A Spanish Group for Research on Sarcomas (GEIS) Study. Target Oncol. 2018, 13, 81–87. [Google Scholar] [CrossRef]
- Trucco, M.M.; Meyer, C.F.; Thornton, K.A.; Shah, P.; Chen, A.R.; Wilky, B.A.; Carrera-Haro, M.A.; Boyer, L.C.; Ferreira, M.F.; Shafique, U.; et al. A phase II study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcomas. Clin. Sarcoma Res. 2018, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Thornton, K.A.; Chen, A.R.; Trucco, M.M.; Shah, P.; Wilky, B.A.; Gul, N.; Carrera-Haro, M.A.; Ferreira, M.F.; Shafique, U.; Powell, J.D.; et al. A dose-finding study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcoma. Int. J. Cancer 2013, 133, 997–1005. [Google Scholar] [CrossRef]
- De Sanctis, R.; Bertuzzi, A.; Basso, U.; Comandone, A.; Marchetti, S.; Marrari, A.; Colombo, P.; Lutman, R.F.; Giordano, L.; Santoro, A. Non-pegylated liposomal doxorubicin plus ifosfamide in metastatic soft tissue sarcoma: Results from a phase-II trial. Anticancer Res. 2015, 35, 543–547. [Google Scholar] [PubMed]
- Poveda, A.; Lopez-Pousa, A.; Martin, J.; Del Muro, J.G.; Bernabe, R.; Casado, A.; Balana, C.; Sanmartin, O.; Menendez, M.D.; Escudero, P.; et al. Phase II Clinical Trial With Pegylated Liposomal Doxorubicin (CAELYX(R)/Doxil(R)) and Quality of Life Evaluation (EORTC QLQ-C30) in Adult Patients With Advanced Soft Tissue Sarcomas: A study of the Spanish Group for Research in Sarcomas (GEIS). Sarcoma 2005, 9, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.W.; Palle, K. Aldehyde dehydrogenases in cancer stem cells: Potential as therapeutic targets. Ann. Transl. Med. 2016, 4, 518. [Google Scholar] [CrossRef] [PubMed]
- Honoki, K.; Tsujiuchi, T. Senescence bypass in mesenchymal stem cells: A potential pathogenesis and implications of pro-senescence therapy in sarcomas. Expert Rev. Anticancer Ther. 2013, 13, 983–996. [Google Scholar] [CrossRef]
- Matushansky, I.; Hernando, E.; Socci, N.D.; Mills, J.E.; Matos, T.A.; Edgar, M.A.; Singer, S.; Maki, R.G.; Cordon-Cardo, C. Derivation of sarcomas from mesenchymal stem cells via inactivation of the Wnt pathway. J. Clin. Invest. 2007, 117, 3248–3257. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Junttila, M.R.; Mao, W.; Wang, X.; Wang, B.E.; Pham, T.; Flygare, J.; Yu, S.F.; Yee, S.; Goldenberg, D.; Fields, C.; et al. Targeting LGR5+ cells with an antibody-drug conjugate for the treatment of colon cancer. Sci. Transl. Med. 2015, 7, 314ra186. [Google Scholar] [CrossRef]
- Gong, X.; Azhdarinia, A.; Ghosh, S.C.; Xiong, W.; An, Z.; Liu, Q.; Carmon, K.S. LGR5-Targeted Antibody-Drug Conjugate Eradicates Gastrointestinal Tumors and Prevents Recurrence. Mol. Cancer Ther. 2016, 15, 1580–1590. [Google Scholar] [CrossRef]
- Nile, A.H.; Hannoush, R.N. Fatty acylation of Wnt proteins. Nat. Chem. Biol. 2016, 12, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, S.; Liu, G.; Rus, I.A.; Yao, S.; Chen, Y.; Akiri, G.; Grumolato, L.; Aaronson, S.A. High-frequency canonical Wnt activation in multiple sarcoma subtypes drives proliferation through a TCF/beta-catenin target gene, CDC25A. Cancer Cell 2011, 19, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Font, E.; Felipe-Abrio, I.; Calabuig-Farinas, S.; Ramos, R.; Terrasa, J.; Vogler, O.; Alemany, R.; Martin-Broto, J.; Obrador-Hevia, A. Disruption of TCF/beta-Catenin Binding Impairs Wnt Signaling and Induces Apoptosis in Soft Tissue Sarcoma Cells. Mol. Cancer Ther. 2017, 16, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.S.; Chen, H.Y.; Que, Y.; Xiao, W.; Zeng, M.S.; Zhang, X. ALK(ATI) interacts with c-Myc and promotes cancer stem cell-like properties in sarcoma. Oncogene 2020, 39, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.N.; Lv, Y.F.; Guo, Q.N. Advances in osteosarcoma stem cell research and opportunities for novel therapeutic targets. Cancer Lett. 2016, 370, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Avnet, S.; Cortini, M. Role of Pericellular Matrix in the Regulation of Cancer Stemness. Stem Cell Rev. Rep. 2016, 12, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef]
- Pollack, S.M.; Ingham, M.; Spraker, M.B.; Schwartz, G.K. Emerging Targeted and Immune-Based Therapies in Sarcoma. J. Clin. Oncol. 2018, 36, 125–135. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Cruz, J.; Penel, N.; Le Cesne, A.; Hindi, N.; Luna, P.; Moura, D.S.; Bernabeu, D.; de Alava, E.; Lopez-Guerrero, J.A.; et al. Pazopanib for treatment of typical solitary fibrous tumours: A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 456–466. [Google Scholar] [CrossRef]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology 2019, 8, 1596004. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Saito, T.; Nagai, M.; Ladanyi, M. SYT-SSX1 and SYT-SSX2 interfere with repression of E-cadherin by snail and slug: A potential mechanism for aberrant mesenchymal to epithelial transition in human synovial sarcoma. Cancer Res. 2006, 66, 6919–6927. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Oda, Y.; Kawaguchi, K.; Sugimachi, K.; Yamamoto, H.; Tateishi, N.; Tanaka, K.; Matsuda, S.; Iwamoto, Y.; Ladanyi, M.; et al. E-cadherin mutation and Snail overexpression as alternative mechanisms of E-cadherin inactivation in synovial sarcoma. Oncogene 2004, 23, 8629–8638. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Joseph, J.V.; Kruyt, F.A.E. EMT- and MET-related processes in nonepithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol. Oncol. 2017, 11, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Eddy, J.A.; Pan, Y.; Hategan, A.; Tabus, I.; Wang, Y.; Cogdell, D.; Price, N.D.; Pollock, R.E.; Lazar, A.J.; et al. Integrated proteomics and genomics analysis reveals a novel mesenchymal to epithelial reverting transition in leiomyosarcoma through regulation of slug. Mol. Cell Proteomics 2010, 9, 2405–2413. [Google Scholar] [CrossRef]
- Chang, C.C.; Campoli, M.; Ferrone, S. Classical and nonclassical HLA class I antigen and NK Cell-activating ligand changes in malignant cells: Current challenges and future directions. Adv. Cancer Res. 2005, 93, 189–234. [Google Scholar] [CrossRef]
- Han, D.; Rodriguez-Bravo, V.; Charytonowicz, E.; Demicco, E.; Domingo-Domenech, J.; Maki, R.G.; Cordon-Cardo, C. Targeting sarcoma tumor-initiating cells through differentiation therapy. Stem Cell Res. 2017, 21, 117–123. [Google Scholar] [CrossRef]
- Takahashi, N.; Nobusue, H.; Shimizu, T.; Sugihara, E.; Yamaguchi-Iwai, S.; Onishi, N.; Kunitomi, H.; Kuroda, T.; Saya, H. ROCK Inhibition Induces Terminal Adipocyte Differentiation and Suppresses Tumorigenesis in Chemoresistant Osteosarcoma Cells. Cancer Res. 2019, 79, 3088–3099. [Google Scholar] [CrossRef] [PubMed]
- Giraudo, L.; Gammaitoni, L.; Cangemi, M.; Rotolo, R.; Aglietta, M.; Sangiolo, D. Cytokine-induced killer cells as immunotherapy for solid tumors: Current evidence and perspectives. Immunotherapy 2015, 7, 999–1010. [Google Scholar] [CrossRef]
- Sangiolo, D.; Mesiano, G.; Gammaitoni, L.; Leuci, V.; Todorovic, M.; Giraudo, L.; Cammarata, C.; Dell’Aglio, C.; D’Ambrosio, L.; Pisacane, A.; et al. Cytokine-induced killer cells eradicate bone and soft-tissue sarcomas. Cancer Res. 2014, 74, 119–129. [Google Scholar] [CrossRef]
- Mesiano, G.; Grignani, G.; Fiorino, E.; Leuci, V.; Rotolo, R.; D’Ambrosio, L.; Salfi, C.; Gammaitoni, L.; Giraudo, L.; Pisacane, A.; et al. Cytokine Induced Killer cells are effective against sarcoma cancer stem cells spared by chemotherapy and target therapy. Oncoimmunology 2018, 7, e1465161. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Takahashi, S. Precision Medicine in Soft Tissue Sarcoma Treatment. Cancers 2020, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Souza, B.K.; da Costa Lopez, P.L.; Menegotto, P.R.; Vieira, I.A.; Kersting, N.; Abujamra, A.L.; Brunetto, A.T.; Brunetto, A.L.; Gregianin, L.; de Farias, C.B.; et al. Targeting Histone Deacetylase Activity to Arrest Cell Growth and Promote Neural Differentiation in Ewing Sarcoma. Mol. Neurobiol. 2018, 55, 7242–7258. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Choy, E.; Tu, C.; Hornicek, F.; Duan, Z. Therapeutic applications of histone deacetylase inhibitors in sarcoma. Cancer Treat. Rev. 2017, 59, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Di Pompo, G.; Salerno, M.; Rotili, D.; Valente, S.; Zwergel, C.; Avnet, S.; Lattanzi, G.; Baldini, N.; Mai, A. Novel histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in sarcoma cancer stem cells. J. Med. Chem. 2015, 58, 4073–4079. [Google Scholar] [CrossRef]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Lahiri, P.; Zatloukal, K. Epigenetic silencing of apoptosis-inducing gene expression can be efficiently overcome by combined SAHA and TRAIL treatment in uterine sarcoma cells. PLoS ONE 2014, 9, e91558. [Google Scholar] [CrossRef]
- Vitfell-Rasmussen, J.; Judson, I.; Safwat, A.; Jones, R.L.; Rossen, P.B.; Lind-Hansen, M.; Knoblauch, P.; Krarup-Hansen, A. A Phase I/II Clinical Trial of Belinostat (PXD101) in Combination with Doxorubicin in Patients with Soft Tissue Sarcomas. Sarcoma 2016, 2016, 2090271. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Fourneaux, B.; Bourdon, A.; Dadone, B.; Lucchesi, C.; Daigle, S.R.; Richard, E.; Laroche-Clary, A.; Le Loarer, F.; Italiano, A. Identifying and targeting cancer stem cells in leiomyosarcoma: Prognostic impact and role to overcome secondary resistance to PI3K/mTOR inhibition. J. Hematol. Oncol. 2019, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Leslie, M. First EZH2 Inhibitor Approved-for Rare Sarcoma. Cancer Discov. 2020, 10, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Tazemetostat: First Approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef]
- Cole, A.J.; Fayomi, A.P.; Anyaeche, V.I.; Bai, S.; Buckanovich, R.J. An evolving paradigm of cancer stem cell hierarchies: Therapeutic implications. Theranostics 2020, 10, 3083–3098. [Google Scholar] [CrossRef]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Ann. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef]


{kind=link}
| STS Subtype | CSC Marker/Feature | Evidence |
|---|---|---|
| Clear Cell Sarcoma | EZH2 [73] | Patient samples |
| Fibrosarcoma | ALDH [40] | In vitro |
| CD133 [22] | In vitro | |
| PDGFRα and PDGFRβ [44] | In vitro and in vivo | |
| Embryonic stem cell transcription factors [24] | In vitro and in vivo | |
| Leiomyosarcoma | PDGFRα and PDGFRβ [44] | In vitro and in vivo |
| Liposarcoma | ALDH [41] | In vivo and patient samples |
| CD133 [41] | In vivo and patient samples | |
| DNMT1 [67] | In vitro and in vivo | |
| PDGFRα and PDGFRβ [44] | In vitro and in vivo | |
| Myxoid Liposarcoma | SWI/ SNF, ISWI and CHD [74] | In vitro |
| MPNST 1 | Nestin [36] | In vitro and in vivo |
| Rhabdomyosarcoma | ALDH [39] | In vitro and in vivo |
| CD133 [19,20,21] | In vitro, in vivo and patient samples | |
| DNMT3B [68] | In vitro | |
| EZH2 [69] | In vitro and in vivo | |
| Nestin [19,27,33,34,35] | In vitro, in vivo and patient samples | |
| NOTCH-YAP1-Sox2 [62] | In vitro, in vivo and patient samples | |
| Side Population [54] | In vitro, in vivo and patient samples | |
| Embryonic stem cell transcription factors [64] | In vitro, in vivo and patient samples | |
| SWI/ SNF, ISWI and CHD [76] | In vitro, in vivo and patient samples | |
| Synovial Sarcoma | ALDH [40] | In vitro |
| Active Wnt pathway [77] | In vitro and in vivo | |
| BMI1 [23] | In vitro | |
| CD133 [23,25] | In vitro and patient samples | |
| CXCR4 [28] | In vitro, in vivo and patient samples | |
| EZH2 [70,71,72] | In vitro, in vivo and patient samples | |
| SWI/ SNF, ISWI and CHD [75] | Patient samples | |
| UPS 2 | Side Population [54] | In vitro, in vivo and patient samples |
| ABC transporters [51] | In vitro | |
| CD133 [51] | In vitro | |
| Embryonic stem cell transcription factors [51] | In vitro |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Delgado, P.; Lacerenza, S.; Obrador-Hevia, A.; Lopez-Alvarez, M.; Mondaza-Hernandez, J.L.; Blanco-Alcaina, E.; Sanchez-Bustos, P.; Hindi, N.; S. Moura, D.; Martin-Broto, J. Cancer Stem Cells in Soft-Tissue Sarcomas. Cells 2020, 9, 1449. https://doi.org/10.3390/cells9061449
Martínez-Delgado P, Lacerenza S, Obrador-Hevia A, Lopez-Alvarez M, Mondaza-Hernandez JL, Blanco-Alcaina E, Sanchez-Bustos P, Hindi N, S. Moura D, Martin-Broto J. Cancer Stem Cells in Soft-Tissue Sarcomas. Cells. 2020; 9(6):1449. https://doi.org/10.3390/cells9061449
Chicago/Turabian StyleMartínez-Delgado, Paula, Serena Lacerenza, Antonia Obrador-Hevia, Maria Lopez-Alvarez, José L. Mondaza-Hernandez, Elena Blanco-Alcaina, Paloma Sanchez-Bustos, Nadia Hindi, David S. Moura, and Javier Martin-Broto. 2020. "Cancer Stem Cells in Soft-Tissue Sarcomas" Cells 9, no. 6: 1449. https://doi.org/10.3390/cells9061449
APA StyleMartínez-Delgado, P., Lacerenza, S., Obrador-Hevia, A., Lopez-Alvarez, M., Mondaza-Hernandez, J. L., Blanco-Alcaina, E., Sanchez-Bustos, P., Hindi, N., S. Moura, D., & Martin-Broto, J. (2020). Cancer Stem Cells in Soft-Tissue Sarcomas. Cells, 9(6), 1449. https://doi.org/10.3390/cells9061449