An Update on the Molecular Basis of Phosphoantigen Recognition by Vγ9Vδ2 T Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. T Cell Receptors and Antigens
2.1. αβ T Cells and γδ T Cells
2.2. Conventional vs. Unconventional T Cells
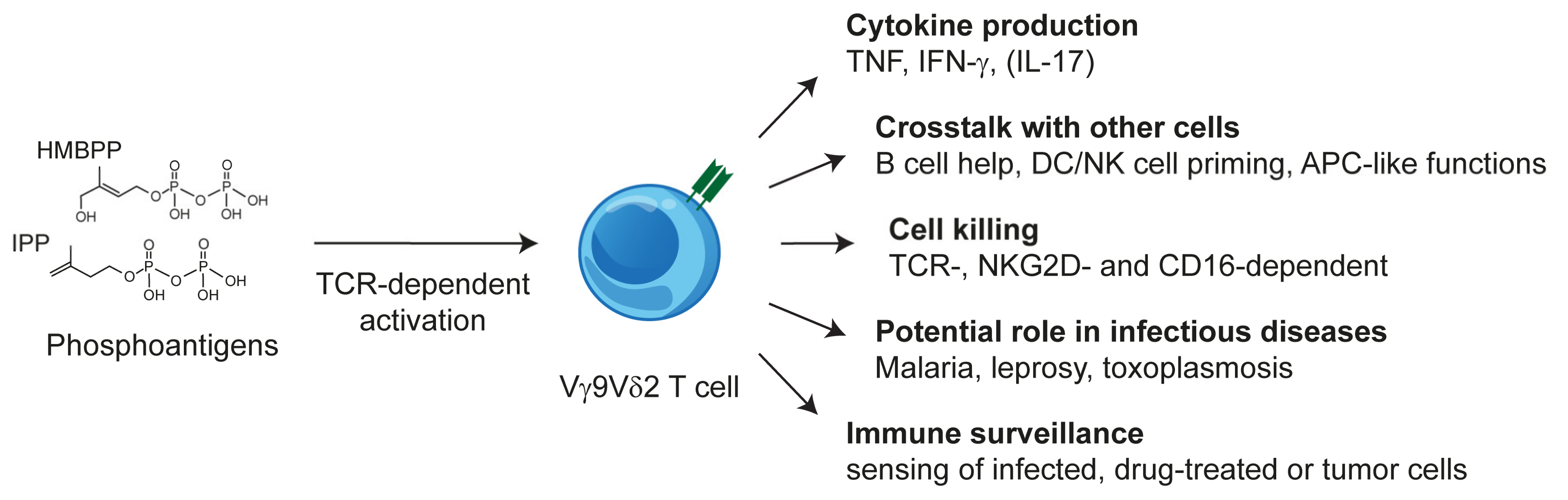
2.3. Vγ9Vδ2 T Cells: TCR and Phosphoantigen Reactivity
3. Butyrophilin 3 (BTN3) as PAg Sensor: Identification and Functional Analysis
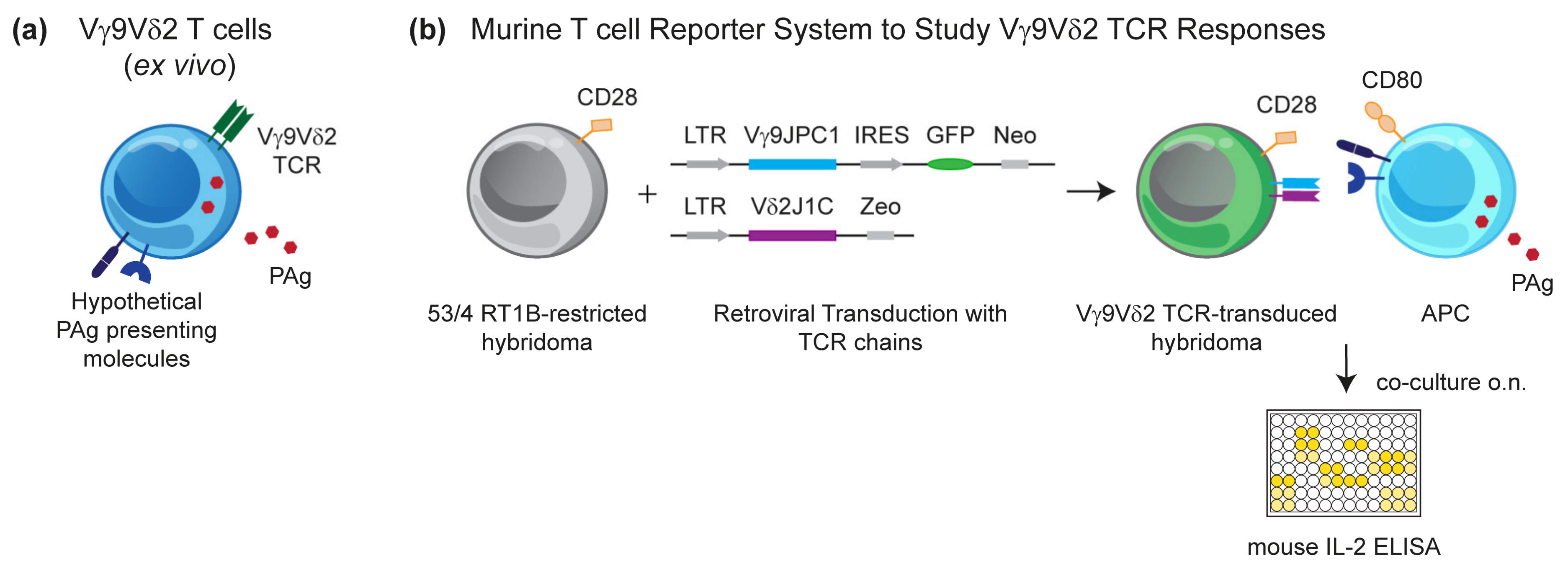
3.1. Murine Reporter Cells Identify the Crucial Role of Human Chromosome 6 (Chr:6) in Vγ9Vδ2 T Cell Activation by PAg
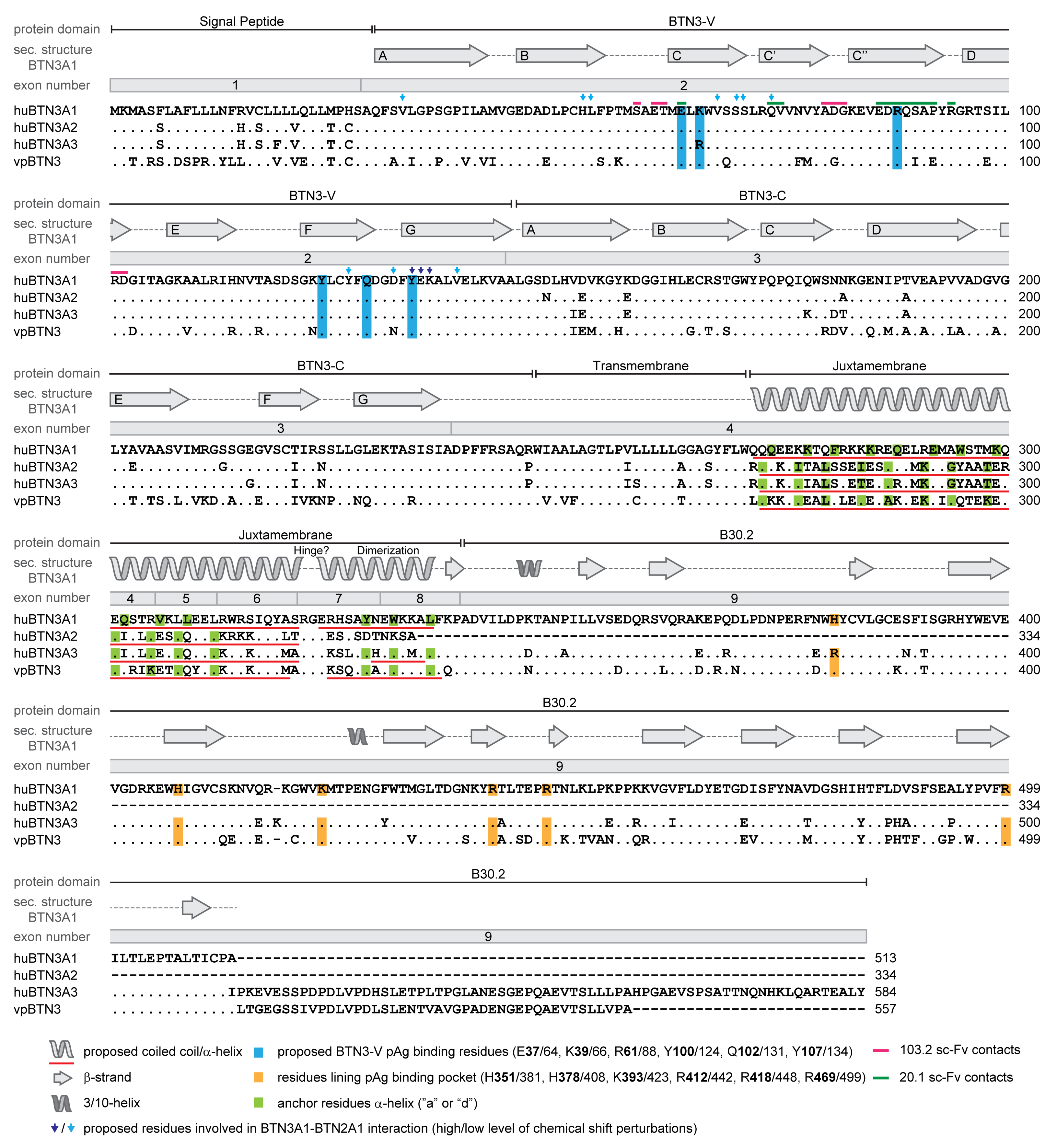
3.2. The Human Butyrophilin 3 (BTN3A) Family
3.3. BTN3A1 is Mandatory for Vγ9Vδ2 T Cell Activation
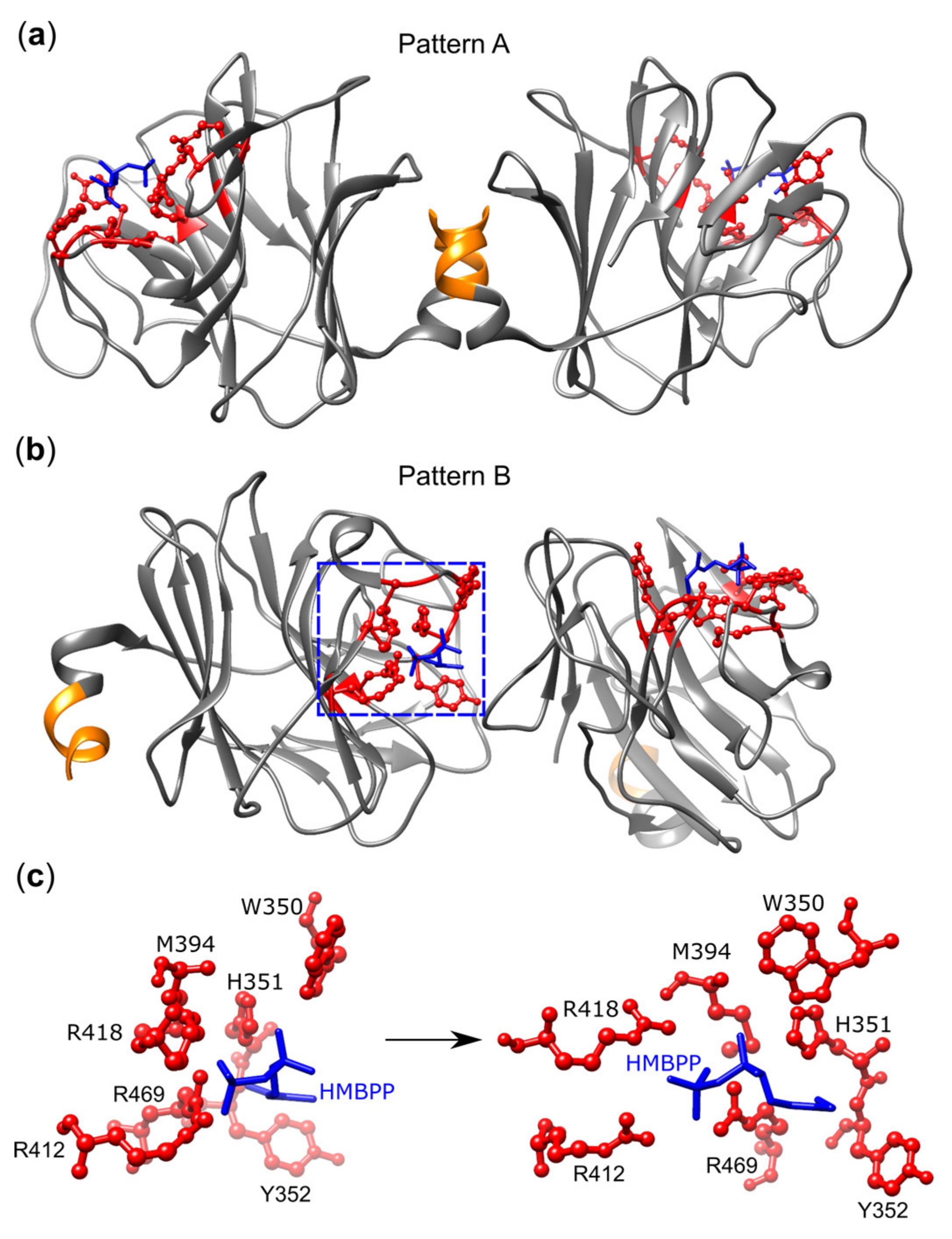
3.4. BTN3A1 Acts as a PAg Sensor
3.5. The Importance of the Juxtamembrane Domain
3.6. Cooperation of BTN3 Isoforms
3.7. The Role of TCR Clonotypes in the Response to mAb 20.1 and PAg
4. Lessons from Armadillo and Alpaca
4.1. Vγ9Vδ2 TCRs and BTN3s in Different Species
4.2. Armadillo: A Witness of Vγ9Vδ2 T Cell Evolution
4.3. Alpaca: The First Non-Primate Species with PAg-Reactive Cells
4.4. Alpaca BTN3: An All-in-One Solution
5. BTN2A1: A Missing Link Poses Questions
5.1. BTN2A1, a New Player in the Game: Identification of BTN2A1 as a Prerequisite of PAg Recognition
5.2. BTN2A1: Interaction with TCR and BTN3A1
5.3. A Composite Ligand Model of PAg Recognition
5.4. BTN3A1-Expressing Rodent Cells as Mediators of mAb 20.1-Induced Vγ9Vδ2 T Cell Activation
5.5. BTN3A–TCR Interaction: Yes, No or Yes and No?
6. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Fournie, J.J.; Bonneville, M. Stimulation of gamma delta T cells by phosphoantigens. Res. Immunol. 1996, 147, 338–347. [Google Scholar] [CrossRef]
- Rast, J.P.; Anderson, M.K.; Strong, S.J.; Luer, C.; Litman, R.T.; Litman, G.W. alpha, beta, gamma, and delta T cell antigen receptor genes arose early in vertebrate phylogeny. Immunity 1997, 6, 1–11. [Google Scholar] [CrossRef]
- Lefranc, M.P. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res. 2003, 31, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Flajnik, M.F.; Kasahara, M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat. Rev. Genet. 2010, 11, 47–59. [Google Scholar] [CrossRef]
- Chen, H.; Bernstein, H.; Ranganathan, P.; Schluter, S.F. Somatic hypermutation of TCR gamma V genes in the sandbar shark. Dev. Comp. Immunol. 2012, 37, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Ott, J.A.; Castro, C.D.; Deiss, T.C.; Ohta, Y.; Flajnik, M.F.; Criscitiello, M.F. Somatic hypermutation of T cell receptor alpha chain contributes to selection in nurse shark thymus. Elife 2018, 7. [Google Scholar] [CrossRef]
- Ciccarese, S.; Vaccarelli, G.; Lefranc, M.P.; Tasco, G.; Consiglio, A.; Casadio, R.; Linguiti, G.; Antonacci, R. Characteristics of the somatic hypermutation in the Camelus dromedarius T cell receptor gamma (TRG) and delta (TRD) variable domains. Dev. Comp. Immunol. 2014, 46, 300–313. [Google Scholar] [CrossRef]
- Antonacci, R.; Linguiti, G.; Burger, P.A.; Castelli, V.; Pala, A.; Fitak, R.; Massari, S.; Ciccarese, S. Comprehensive genomic analysis of the dromedary T cell receptor gamma (TRG) locus and identification of a functional TRGC5 cassette. Dev. Comp. Immunol. 2020, 106, 103614. [Google Scholar] [CrossRef]
- Hansen, V.L.; Miller, R.D. The Evolution and Structure of Atypical T Cell Receptors. Results Probl. Cell Differ. 2015, 57, 265–278. [Google Scholar] [CrossRef]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef]
- La Gruta, N.L.; Gras, S.; Daley, S.R.; Thomas, P.G.; Rossjohn, J. Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 2018, 18, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.I.; Koay, H.F.; McCluskey, J.; Gherardin, N.A. The biology and functional importance of MAIT cells. Nat. Immunol. 2019, 20, 1110–1128. [Google Scholar] [CrossRef] [PubMed]
- Legoux, F.; Salou, M.; Lantz, O. Unconventional or Preset alphabeta T Cells: Evolutionarily Conserved Tissue-Resident T Cells Recognizing Nonpeptidic Ligands. Annu. Rev. Cell Dev. Biol. 2017, 33, 511–535. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Kronenberg, M. Activation and Function of iNKT and MAIT Cells. Adv. Immunol. 2015, 127, 145–201. [Google Scholar] [CrossRef]
- Di Marco Barros, R.; Roberts, N.A.; Dart, R.J.; Vantourout, P.; Jandke, A.; Nussbaumer, O.; Deban, L.; Cipolat, S.; Hart, R.; Iannitto, M.L.; et al. Epithelia Use Butyrophilin-like Molecules to Shape Organ-Specific gammadelta T Cell Compartments. Cell 2016, 167, 203–218. [Google Scholar] [CrossRef]
- Boyden, L.M.; Lewis, J.M.; Barbee, S.D.; Bas, A.; Girardi, M.; Hayday, A.C.; Tigelaar, R.E.; Lifton, R.P. Skint1, the prototype of a newly identified immunoglobulin superfamily gene cluster, positively selects epidermal gammadelta T cells. Nat. Genet. 2008, 40, 656–662. [Google Scholar] [CrossRef]
- Melandri, D.; Zlatareva, I.; Chaleil, R.A.G.; Dart, R.J.; Chancellor, A.; Nussbaumer, O.; Polyakova, O.; Roberts, N.A.; Wesch, D.; Kabelitz, D.; et al. The gammadeltaTCR combines innate immunity with adaptive immunity by utilizing spatially distinct regions for agonist selection and antigen responsiveness. Nat. Immunol. 2018, 19, 1352–1365. [Google Scholar] [CrossRef]
- Willcox, C.R.; Vantourout, P.; Salim, M.; Zlatareva, I.; Melandri, D.; Zanardo, L.; George, R.; Kjaer, S.; Jeeves, M.; Mohammed, F.; et al. Butyrophilin-like 3 Directly Binds a Human Vgamma4(+) T Cell Receptor Using a Modality Distinct from Clonally-Restricted Antigen. Immunity 2019, 51, 813–825. [Google Scholar] [CrossRef]
- Rigau, M.; Ostrouska, S.; Fulford, T.S.; Johnson, D.N.; Woods, K.; Ruan, Z.; McWilliam, H.E.G.; Hudson, C.; Tutuka, C.; Wheatley, A.K.; et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by gammadelta T cells. Science 2020, 367. [Google Scholar] [CrossRef]
- Fichtner, A.S.; Karunakaran, M.M.; Gu, S.; Boughter, C.T.; Borowska, M.T.; Starick, L.; Nohren, A.; Gobel, T.W.; Adams, E.J.; Herrmann, T. Alpaca (Vicugna pacos), the first nonprimate species with a phosphoantigen-reactive Vgamma9Vdelta2 T cell subset. Proc. Natl. Acad. Sci. U S A 2020, 117, 6697–6707. [Google Scholar] [CrossRef]
- Hayday, A.C.; Vantourout, P. The Innate Biologies of Adaptive Receptors. Annu. Rev. Immunol. 2020, 38, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Pizzolato, G.; Kaminski, H.; Tosolini, M.; Franchini, D.M.; Pont, F.; Martins, F.; Valle, C.; Labourdette, D.; Cadot, S.; Quillet-Mary, A.; et al. Single-cell RNA sequencing unveils the shared and the distinct cytotoxic hallmarks of human TCRVdelta1 and TCRVdelta2 gammadelta T lymphocytes. Proc. Natl. Acad. Sci. U S A 2019, 116, 11906–11915. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, N.; La Mendola, C.; Orlando, V.; Meraviglia, S.; Todaro, M.; Stassi, G.; Sireci, G.; Fournie, J.J.; Dieli, F. Differentiation, phenotype, and function of interleukin-17-producing human Vgamma9Vdelta2 T cells. Blood 2011, 118, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D.; He, W. The multifunctionality of human Vgamma9Vdelta2 gammadelta T cells: clonal plasticity or distinct subsets? Scand. J. Immunol. 2012, 76, 213–222. [Google Scholar] [CrossRef]
- Moris, A.; Rothenfusser, S.; Meuer, E.; Hangretinger, R.; Fisch, P. Role of gammadelta T cells in tumor immunity and their control by NK receptors. Microbes Infect. 1999, 1, 227–234. [Google Scholar] [CrossRef]
- De Libero, G. Control of gammadelta T cells by NK receptors. Microbes Infect. 1999, 1, 263–267. [Google Scholar] [CrossRef]
- Rincon-Orozco, B.; Kunzmann, V.; Wrobel, P.; Kabelitz, D.; Steinle, A.; Herrmann, T. Activation of Vγ9Vδ2 T Cells by NKG2D. J. Immunol. 2005, 175, 2144–2151. [Google Scholar] [CrossRef]
- Tanaka, Y.; Morita, C.T.; Tanaka, Y.; Nieves, E.; Brenner, M.B.; Bloom, B.R. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature 1995, 375, 155–158. [Google Scholar] [CrossRef]
- Eberl, M.; Hintz, M.; Reichenberg, A.; Kollas, A.K.; Wiesner, J.; Jomaa, H. Microbial isoprenoid biosynthesis and human gammadelta T cell activation. FEBS Lett 2003, 544, 4–10. [Google Scholar] [CrossRef]
- Wiemer, A.J. Structure-activity relationships of butyrophilin 3 ligands. ChemMedChem 2020. [Google Scholar] [CrossRef]
- Kunzmann, V.; Bauer, E.; Feurle, J.; Tony, F.W.; Hans-Peter; Wilhelm, M. Stimulation of γδ T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood 2000, 96, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Gober, H.J.; Kistowska, M.; Angman, L.; Jeno, P.; Mori, L.; De Libero, G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef]
- Li, J.; Herold, M.J.; Kimmel, B.; Mueller, I.; Rincon-Orozco, B.; Kunzmann, V.; Herrmann, T. Reduced Expression of the Mevalonate Pathway Enzyme Farnesyl Pyrophosphate Synthase Unveils Recognition of Tumor Cells by V gamma 9V delta 2 T Cells. J. Immunol. 2009, 182, 8118–8124. [Google Scholar] [CrossRef] [PubMed]
- Moulin, M.; Alguacil, J.; Gu, S.; Mehtougui, A.; Adams, E.J.; Peyrottes, S.; Champagne, E. Vgamma9Vdelta2 T cell activation by strongly agonistic nucleotidic phosphoantigens. Cell Mol. Life Sci. 2017, 74, 4353–4367. [Google Scholar] [CrossRef] [PubMed]
- Fisch, P.; Oettel, K.; Fudim, N.; Surfus, J.E.; Malkovsky, M.; Sondel, P.M. MHC-unrestricted cytotoxic and proliferative responses of two distinct human gamma/delta T cell subsets to Daudi cells. J. Immunol. 1992, 148, 2315–2323. [Google Scholar]
- Scotet, E.; Martinez, L.O.; Grant, E.; Barbaras, R.; Jeno, P.; Guiraud, M.; Monsarrat, B.; Saulquin, X.; Maillet, S.; Esteve, J.P.; et al. Tumor recognition following Vgamma9Vdelta2 T cell receptor interactions with a surface F1-ATPase-related structure and apolipoprotein A-I. Immunity 2005, 22, 71–80. [Google Scholar] [CrossRef]
- Kong, Y.; Cao, W.; Xi, X.; Ma, C.; Cui, L.; He, W. The NKG2D ligand ULBP4 binds to TCRγ9/δ2 and induces cytotoxicity to tumor cells through both TCRγδ and NKG2D. Blood 2009, 114, 310–317. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, H.; Mo, C.; Cui, L.; He, W. Ectopically expressed human tumor biomarker MutS homologue 2 is a novel endogenous ligand that is recognized by human gammadelta T cells to induce innate anti-tumor/virus immunity. J. Biol. Chem. 2012, 287, 16812–16819. [Google Scholar] [CrossRef]
- Fisch, P.; Malkovsky, M.; Kovats, S.; Sturm, E.; Braakman, E.; Klein, B.S.; Voss, S.D.; Morrissey, L.W.; DeMars, R.; Welch, W.J.; et al. Recognition by human V gamma 9/V delta 2 T cells of a GroEL homolog on Daudi Burkitt’s lymphoma cells. Science 1990, 250, 1269–1273. [Google Scholar] [CrossRef]
- Kistowska, M.; Rossy, E.; Sansano, S.; Gober, H.J.; Landmann, R.; Mori, L.; De Libero, G. Dysregulation of the host mevalonate pathway during early bacterial infection activates human TCR gamma delta cells. Eur. J. Immunol. 2008, 38, 2200–2209. [Google Scholar] [CrossRef]
- Hoeres, T.; Smetak, M.; Pretscher, D.; Wilhelm, M. Improving the Efficiency of Vgamma9Vdelta2 T-Cell Immunotherapy in Cancer. Front. Immunol. 2018, 9, 800. [Google Scholar] [CrossRef] [PubMed]
- Kunkele, K.P.; Wesch, D.; Oberg, H.H.; Aichinger, M.; Supper, V.; Baumann, C. Vgamma9Vdelta2 T Cells: Can We Re-Purpose a Potent Anti-Infection Mechanism for Cancer Therapy? Cells 2020, 9, 829. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.W. Multifunctional immune responses of HMBPP-specific Vgamma2Vdelta2 T cells in M. tuberculosis and other infections. Cell Mol. Immunol. 2013, 10, 58–64. [Google Scholar] [CrossRef]
- Chen, Z.W. Protective immune responses of major Vgamma2Vdelta2 T-cell subset in M. tuberculosis infection. Curr. Opin. Immunol. 2016, 42, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Belmant, C.; Espinosa, E.; Halary, F.; Tang, Y.; Peyrat, M.A.; Sicard, H.; Kozikowski, A.; Buelow, R.; Poupot, R.; Bonneville, M.; et al. A chemical basis for selective recognition of nonpeptide antigens by human delta T cells. FASEB J. 2000, 14, 1669–1670. [Google Scholar] [CrossRef]
- Allison, T.J.; Winter, C.C.; Fournie, J.J.; Bonneville, M.; Garboczi, D.N. Structure of a human gammadelta T-cell antigen receptor. Nature 2001, 411, 820–824. [Google Scholar] [CrossRef]
- Morita, C.T.; Beckman, E.M.; Bukowski, J.F.; Tanaka, Y.; Band, H.; Bloom, B.R.; Golan, D.E.; Brenner, M.B. Direct presentation of nonpeptide prenyl pyrophosphate antigens to human gamma delta T cells. Immunity 1995, 3, 495–507. [Google Scholar] [CrossRef]
- Kato, Y.; Tanaka, Y.; Tanaka, H.; Yamashita, S.; Minato, N. Requirement of species-specific interactions for the activation of human gamma delta T cells by pamidronate. J. Immunol. 2003, 170, 3608–3613. [Google Scholar] [CrossRef]
- Green, A.E.; Lissina, A.; Hutchinson, S.L.; Hewitt, R.E.; Temple, B.; James, D.; Boulter, J.M.; Price, D.A.; Sewell, A.K. Recognition of nonpeptide antigens by human V gamma 9V delta 2 T cells requires contact with cells of human origin. Clin. Exp. Immunol. 2004, 136, 472–482. [Google Scholar] [CrossRef]
- Morita, C.T.; Jin, C.; Sarikonda, G.; Wang, H. Nonpeptide antigens, presentation mechanisms, and immunological memory of human Vgamma2Vdelta2 T cells: discriminating friend from foe through the recognition of prenyl pyrophosphate antigens. Immunol. Rev. 2007, 215, 59–76. [Google Scholar] [CrossRef]
- Li, J.-Q. Modulating the expression of enzymes of isoprenoid synthesis: effects on Vgamma9Vdelta2 T cell activation and tumor cell growth. Ph.D. Thesis, Julius-Maximilians-University Wuerzburg, Wuerzburg, Germany, 2010. [Google Scholar]
- Kreiss, M. T-Zellrezeptorbindung und Modulation der T-Zellaktivierung durch Autoantigene der Ratte. Available online: https://opus.bibliothek.uni-wuerzburg.de/opus4-wuerzburg/frontdoor/deliver/index/docId/903/file/Developing_Emotional_Intelligence.pdf (accessed on 5 June 2020).
- Harly, C.; Guillaume, Y.; Nedellec, S.; Peigne, C.M.; Monkkonen, H.; Monkkonen, J.; Li, J.; Kuball, J.; Adams, E.J.; Netzer, S.; et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human gammadelta T-cell subset. Blood 2012, 120, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Starick, L.; Riano, F.; Karunakaran, M.M.; Kunzmann, V.; Li, J.; Kreiss, M.; Amslinger, S.; Scotet, E.; Olive, D.; De Libero, G.; et al. Butyrophilin 3A (BTN3A, CD277)-specific antibody 20.1 differentially activates Vgamma9Vdelta2 TCR clonotypes and interferes with phosphoantigen activation. Eur. J. Immunol. 2017, 47, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.A.; Stammers, M.; Malcherek, G.; Beck, S.; Trowsdale, J. The cluster of BTN genes in the extended major histocompatibility complex. Genomics 2001, 71, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Compte, E.; Pontarotti, P.; Collette, Y.; Lopez, M.; Olive, D. Frontline: Characterization of BT3 molecules belonging to the B7 family expressed on immune cells. Eur. J. Immunol. 2004, 34, 2089–2099. [Google Scholar] [CrossRef]
- Palakodeti, A.; Sandstrom, A.; Sundaresan, L.; Harly, C.; Nedellec, S.; Olive, D.; Scotet, E.; Bonneville, M.; Adams, E.J. The molecular basis for modulation of human Vgamma9Vdelta2 T cell responses by CD277/butyrophilin-3 (BTN3A)-specific antibodies. J. Biol. Chem. 2012, 287, 32780–32790. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Reith, W.; Trowsdale, J. Regulation of Immunity by Butyrophilins. Annu. Rev. Immunol. 2016, 34, 151–172. [Google Scholar] [CrossRef]
- Afrache, H.; Pontarotti, P.; Abi-Rached, L.; Olive, D. Evolutionary and polymorphism analyses reveal the central role of BTN3A2 in the concerted evolution of the BTN3 gene family. Immunogenetics 2017, 69, 379–390. [Google Scholar] [CrossRef]
- Rhodes, D.A.; de Bono, B.; Trowsdale, J. Relationship between SPRY and B30.2 protein domains. Evolution of a component of immune defence? Immunology 2005, 116, 411–417. [Google Scholar] [CrossRef]
- Yamashiro, H.; Yoshizaki, S.; Tadaki, T.; Egawa, K.; Seo, N. Stimulation of human butyrophilin 3 molecules results in negative regulation of cellular immunity. J. Leukoc. Biol. 2010, 88, 757–767. [Google Scholar] [CrossRef]
- Simone, R.; Barbarat, B.; Rabellino, A.; Icardi, G.; Bagnasco, M.; Pesce, G.; Olive, D.; Saverino, D. Ligation of the BT3 molecules, members of the B7 family, enhance the proinflammatory responses of human monocytes and monocyte-derived dendritic cells. Mol. Immunol. 2010, 48, 109–118. [Google Scholar] [CrossRef]
- Messal, N.; Mamessier, E.; Sylvain, A.; Celis-Gutierrez, J.; Thibult, M.L.; Chetaille, B.; Firaguay, G.; Pastor, S.; Guillaume, Y.; Wang, Q.; et al. Differential role for CD277 as a co-regulator of the immune signal in T and NK cells. Eur. J. Immunol. 2011, 41, 3443–3454. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Li, J.; Puthenveetil, R.; Lin, X.; Poe, M.M.; Hsiao, C.C.; Vinogradova, O.; Wiemer, A.J. The butyrophilin 3A1 intracellular domain undergoes a conformational change involving the juxtamembrane region. FASEB J. 2017, 31, 4697–4706. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Nada, M.H.; Tanaka, Y.; Sakuraba, S.; Morita, C.T. Critical Roles for Coiled-Coil Dimers of Butyrophilin 3A1 in the Sensing of Prenyl Pyrophosphates by Human Vgamma2Vdelta2 T Cells. J. Immunol. 2019, 203, 607–626. [Google Scholar] [CrossRef] [PubMed]
- Vavassori, S.; Kumar, A.; Wan, G.S.; Ramanjaneyulu, G.S.; Cavallari, M.; El Daker, S.; Beddoe, T.; Theodossis, A.; Williams, N.K.; Gostick, E.; et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human gammadelta T cells. Nat. Immunol. 2013, 14, 908–916. [Google Scholar] [CrossRef]
- Sandstrom, A.; Peigne, C.M.; Leger, A.; Crooks, J.E.; Konczak, F.; Gesnel, M.C.; Breathnach, R.; Bonneville, M.; Scotet, E.; Adams, E.J. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vgamma9Vdelta2 T cells. Immunity 2014, 40, 490–500. [Google Scholar] [CrossRef]
- Karunakaran, M.M.; Willcox, C.R.; Salim, M.; Paletta, D.; Fichtner, A.S.; Noll, A.; Starick, L.; Nohren, A.; Begley, C.R.; Berwick, K.A.; et al. Butyrophilin-2A1 Directly Binds Germline-Encoded Regions of the Vgamma9Vdelta2 TCR and Is Essential for Phosphoantigen Sensing. Immunity 2020, 52, 487–498. [Google Scholar] [CrossRef]
- Wang, H.; Henry, O.; Distefano, M.D.; Wang, Y.C.; Raikkonen, J.; Monkkonen, J.; Tanaka, Y.; Morita, C.T. Butyrophilin 3A1 plays an essential role in prenyl pyrophosphate stimulation of human Vgamma2Vdelta2 T cells. J. Immunol. 2013, 191, 1029–1042. [Google Scholar] [CrossRef]
- Wang, H.; Morita, C.T. Sensor Function for Butyrophilin 3A1 in Prenyl Pyrophosphate Stimulation of Human Vgamma2Vdelta2 T Cells. J. Immunol. 2015, 195, 4583–4594. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Chen, H.C.; Price, A.J.; Keeble, A.H.; Davey, M.S.; James, L.C.; Eberl, M.; Trowsdale, J. Activation of human gammadelta T cells by cytosolic interactions of BTN3A1 with soluble phosphoantigens and the cytoskeletal adaptor periplakin. J. Immunol. 2015, 194, 2390–2398. [Google Scholar] [CrossRef]
- Vantourout, P.; Laing, A.; Woodward, M.J.; Zlatareva, I.; Apolonia, L.; Jones, A.W.; Snijders, A.P.; Malim, M.H.; Hayday, A.C. Heteromeric interactions regulate butyrophilin (BTN) and BTN-like molecules governing γδ T cell biology. Proc. Natl. Acad. Sci. U S A 2018. [Google Scholar] [CrossRef]
- Gu, S.; Sachleben, J.R.; Boughter, C.T.; Nawrocka, W.I.; Borowska, M.T.; Tarrasch, J.T.; Skiniotis, G.; Roux, B.; Adams, E.J. Phosphoantigen-induced conformational change of butyrophilin 3A1 (BTN3A1) and its implication on Vgamma9Vdelta2 T cell activation. Proc. Natl. Acad. Sci. U S A 2017, 114, E7311–E7320. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, L.; Yuan, L.; Zhou, X.; Duan, J.; Xiao, H.; Cai, N.; Han, S.; Ma, X.; Liu, W.; et al. A Structural Change in Butyrophilin upon Phosphoantigen Binding Underlies Phosphoantigen-Mediated Vgamma9Vdelta2 T Cell Activation. Immunity 2019, 50, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; Scotet, E.; Olive, D. An X-ray Vision for Phosphoantigen Recognition. Immunity 2019, 50, 1026–1028. [Google Scholar] [CrossRef]
- Hsiao, C.H.; Lin, X.; Barney, R.J.; Shippy, R.R.; Li, J.; Vinogradova, O.; Wiemer, D.F.; Wiemer, A.J. Synthesis of a phosphoantigen prodrug that potently activates Vgamma9Vdelta2 T-lymphocytes. Chem. Biol. 2014, 21, 945–954. [Google Scholar] [CrossRef]
- Salim, M.; Knowles, T.J.; Baker, A.T.; Davey, M.S.; Jeeves, M.; Sridhar, P.; Wilkie, J.; Willcox, C.R.; Kadri, H.; Taher, T.E.; et al. BTN3A1 Discriminates gammadelta T Cell Phosphoantigens from Nonantigenic Small Molecules via a Conformational Sensor in Its B30.2 Domain. ACS Chem Biol 2017, 12, 2631–2643. [Google Scholar] [CrossRef]
- Poe, M.M.; Agabiti, S.S.; Liu, C.; Li, V.; Teske, K.A.; Hsiao, C.C.; Wiemer, A.J. Probing the Ligand-Binding Pocket of BTN3A1. J. Med. Chem. 2019, 62, 6814–6823. [Google Scholar] [CrossRef]
- Sebestyen, Z.; Scheper, W.; Vyborova, A.; Gu, S.; Rychnavska, Z.; Schiffler, M.; Cleven, A.; Cheneau, C.; van Noorden, M.; Peigne, C.M.; et al. RhoB Mediates Phosphoantigen Recognition by Vgamma9Vdelta2 T Cell Receptor. Cell Rep. 2016, 15, 1973–1985. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Chen, H.C.; Williamson, J.C.; Hill, A.; Yuan, J.; Smith, S.; Rhodes, H.; Trowsdale, J.; Lehner, P.J.; Herrmann, T.; et al. Regulation of Human gammadelta T Cells by BTN3A1 Protein Stability and ATP-Binding Cassette Transporters. Front. Immunol. 2018, 9, 662. [Google Scholar] [CrossRef]
- Seo, M.; Lee, S.-O.; Kim, J.-H.; Hong, Y.; Kim, S.; Kim, Y.; Min, D.-H.; Kong, Y.-Y.; Shin, J.; Ahn, K. MAP4-regulated dynein-dependent trafficking of BTN3A1 controls the TBK1–IRF3 signaling axis. Proc. Natl. Acad. Sci. USA 2016, 113, 14390–14395. [Google Scholar] [CrossRef]
- Benyamine, A.; Le Roy, A.; Mamessier, E.; Gertner-Dardenne, J.; Castanier, C.; Orlanducci, F.; Pouyet, L.; Goubard, A.; Collette, Y.; Vey, N.; et al. BTN3A molecules considerably improve Vgamma9Vdelta2T cells-based immunotherapy in acute myeloid leukemia. Oncoimmunology 2016, 5, e1146843. [Google Scholar] [CrossRef]
- Benyamine, A.; Loncle, C.; Foucher, E.; Blazquez, J.L.; Castanier, C.; Chretien, A.S.; Modesti, M.; Secq, V.; Chouaib, S.; Gironella, M.; et al. BTN3A is a prognosis marker and a promising target for Vgamma9Vdelta2 T cells based-immunotherapy in pancreatic ductal adenocarcinoma (PDAC). Oncoimmunology 2017, 7, e1372080. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, M.M. The Evolution of Vγ9Vδ2 T cells. Ph.D. Thesis, Julius-Maximilians-University Wuerzburg, Wuerzburg, Germany, 2014. [Google Scholar]
- Karunakaran, M.M.; Goebel, T.W.; Starick, L.; Walter, L.; Herrmann, T. V gamma 9 and V delta 2 T cell antigen receptor genes and butyrophilin 3 (BTN3) emerged with placental mammals and are concomitantly preserved in selected species like alpaca (Vicugna pacos). Immunogenetics 2014, 66, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, M.M.; Herrmann, T. The Vγ9Vδ2 T Cell Antigen Receptor and Butyrophilin-3 A1: Models of Interaction, the Possibility of Co-Evolution, and the Case of Dendritic Epidermal T Cells. Front. Immunol. 2014, 5, 648. [Google Scholar] [CrossRef]
- Balamayooran, G.; Pena, M.; Sharma, R.; Truman, R.W. The armadillo as an animal model and reservoir host for Mycobacterium leprae. Clin. Dermatol. 2015, 33, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Ohmen, J.D.; Barnes, P.F.; Uyemura, K.; Lu, S.Z.; Grisso, C.L.; Modlin, R.L. The T cell receptors of human gamma delta T cells reactive to Mycobacterium tuberculosis are encoded by specific V genes but diverse V-J junctions. J. Immunol. 1991, 147, 3353–3359. [Google Scholar]
- Lathrop, G.; Scollard, D.M.; Dietrich, M. Reactivity of a population of armadillo lymphocytes with an antibody to human gamma, delta T-cells. Clin. Immunol. Immunopathol. 1997, 82, 68–72. [Google Scholar] [CrossRef]
- Fichtner, A.S.; Karunakaran, M.M.; Starick, L.; Truman, R.W.; Herrmann, T. The Armadillo (Dasypus novemcinctus): A Witness but Not a Functional Example for the Emergence of the Butyrophilin 3/Vγ9Vδ2 System in Placental Mammals. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Linguiti, G.; Antonacci, R.; Tasco, G.; Grande, F.; Casadio, R.; Massari, S.; Castelli, V.; Consiglio, A.; Lefranc, M.P.; Ciccarese, S. Genomic and expression analyses of Tursiops truncatus T cell receptor gamma (TRG) and alpha/delta (TRA/TRD) loci reveal a similar basic public gammadelta repertoire in dolphin and human. BMC Genomics 2016, 17, 634. [Google Scholar] [CrossRef]
- Fichtner, A.S.; Bubke, A.; Rampoldi, F.; Wilharm, A.; Tan, L.; Steinbruck, L.; Schultze-Florey, C.; von Kaisenberg, C.; Prinz, I.; Herrmann, T.; et al. TCR repertoire analysis reveals phosphoantigen-induced polyclonal proliferation of Vgamma9Vdelta2 T cells in neonates and adults. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef]
- Fichtner, A.S.; Ravens, S.; Prinz, I. Human gammadelta TCR Repertoires in Health and Disease. Cells 2020, 9, 800. [Google Scholar] [CrossRef]
- Fichtner, A.S. Alpaca, armadillo and cotton rat as new animal models for nonconventional T cells: Identification of cell populations and analysis of antigen receptors and ligands. Ph.D. Thesis, Julius-Maximilians-University, Würzburg, Germany, 2018. [Google Scholar]
- Riano, F.; Karunakaran, M.M.; Starick, L.; Li, J.Q.; Scholz, C.J.; Kunzmann, V.; Olive, D.; Amslinger, S.; Herrmann, T. V gamma 9V delta 2 TCR-activation by phosphorylated antigens requires butyrophilin 3 A1 (BTN3A1) and additional genes on human chromosome 6. European Journal of Immunology 2014, 44, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Riaño-Arias, R.F. BTN3A1 in the immune response of Vγ9Vδ2 T cells. Ph.D. Thesis, Julius-Maximilans-Universität Würzburg, Würzburg, Germany, 2016. [Google Scholar]
- Ross, S.R. Commentary: phenotypic screening of radiation hybrid panels. Mamm. Genome 2001, 12, 879–881. [Google Scholar] [CrossRef] [PubMed]
- Goss, S.J.; Harris, H. New method for mapping genes in human chromosomes. Nature 1975, 255, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Chaudhri, A.; Xiao, Y.; Klee, A.N.; Wang, X.; Zhu, B.; Freeman, G.J. PD-L1 Binds to B7-1 Only In Cis on the Same Cell Surface. Cancer Immunol. Res. 2018, 6, 921–929. [Google Scholar] [CrossRef]
- Wang, H.; Fang, Z.; Morita, C.T. Vgamma2Vdelta2 T Cell Receptor recognition of prenyl pyrophosphates is dependent on all CDRs. J. Immunol. 2010, 184, 6209–6222. [Google Scholar] [CrossRef]
- Grunder, C.; van Dorp, S.; Hol, S.; Drent, E.; Straetemans, T.; Heijhuurs, S.; Scholten, K.; Scheper, W.; Sebestyen, Z.; Martens, A.; et al. gamma9 and delta2CDR3 domains regulate functional avidity of T cells harboring gamma9delta2TCRs. Blood 2012, 120, 5153–5162. [Google Scholar] [CrossRef]
- Willcox, C.R.; Pitard, V.; Netzer, S.; Couzi, L.; Salim, M.; Silberzahn, T.; Moreau, J.-F.; Hayday, A.C.; Willcox, B.E.; Déchanet-Merville, J. Cytomegalovirus and tumor stress surveillance by binding of a human γδ T cell antigen receptor to endothelial protein C receptor. Nat. Immunol. 2012, 13, 872. [Google Scholar] [CrossRef]
- Afrache, H.; Gouret, P.; Ainouche, S.; Pontarotti, P.; Olive, D. The butyrophilin (BTN) gene family: From milk fat to the regulation of the immune response. Immunogenetics 2012, 64, 781–794. [Google Scholar] [CrossRef]
- Sarter, K.; Leimgruber, E.; Gobet, F.; Agrawal, V.; Dunand-Sauthier, I.; Barras, E.; Mastelic-Gavillet, B.; Kamath, A.; Fontannaz, P.; Guery, L.; et al. Btn2a2, a T cell immunomodulatory molecule coregulated with MHC class II genes. J. Exp. Med. 2016, 213, 177–187. [Google Scholar] [CrossRef]
- Abeler-Dorner, L.; Swamy, M.; Williams, G.; Hayday, A.C.; Bas, A. Butyrophilins: An emerging family of immune regulators. Trends Immunol. 2012, 33, 34–41. [Google Scholar] [CrossRef]
- Kistowska, M. Antigen recognition and thymic maturation of human TCR Vgamma9-Vdelta2 cells. Ph.D. Thesis, University of Basel, Basel, Switzerland, 2007. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrmann, T.; Fichtner, A.S.; Karunakaran, M.M. An Update on the Molecular Basis of Phosphoantigen Recognition by Vγ9Vδ2 T Cells. Cells 2020, 9, 1433. https://doi.org/10.3390/cells9061433
Herrmann T, Fichtner AS, Karunakaran MM. An Update on the Molecular Basis of Phosphoantigen Recognition by Vγ9Vδ2 T Cells. Cells. 2020; 9(6):1433. https://doi.org/10.3390/cells9061433
Chicago/Turabian StyleHerrmann, Thomas, Alina Suzann Fichtner, and Mohindar Murugesh Karunakaran. 2020. "An Update on the Molecular Basis of Phosphoantigen Recognition by Vγ9Vδ2 T Cells" Cells 9, no. 6: 1433. https://doi.org/10.3390/cells9061433
APA StyleHerrmann, T., Fichtner, A. S., & Karunakaran, M. M. (2020). An Update on the Molecular Basis of Phosphoantigen Recognition by Vγ9Vδ2 T Cells. Cells, 9(6), 1433. https://doi.org/10.3390/cells9061433