Breast Cancer Chemotherapeutic Options: A General Overview on the Preclinical Validation of a Multi-Target Ruthenium(III) Complex Lodged in Nucleolipid Nanosystems

 ,
,  ,
, 
 ,
,  ,
,  ,
,  ,
, 
Abstract
1. Breast Cancer Clinical Classification
2. Outline on the Therapeutic Approaches for BC Treatment
3. Dysregulated Mechanisms Controlling Apoptosis and Cell Death/Cell Survival in Breast Cancer Cells (BCC): Prospective Targeted Therapies
4. Autophagy in BCC: Towards Novel Targets in Anticancer Strategies
5. An Outlook on Ru-Based Complexes in the Landscape of Anticancer Metallodrugs in Clinical Trials
6. Ru-Based Drugs Upgrading for Cancer Treatments: Advancements and Prospective Nanostructured Materials
7. The Idea of Biocompatible Ru(III)-Based Nucleolipid Nanosystems
8. Preclinical Validation of Nucleolipid Ru-Based Nanoformulations in Models In Vitro
9. Selectivity and Efficacy of Nucleolipid Ru-Based Nanoformulations in BCC Models
10. Biological Responses to Nucleolipid Ru-Based Nanoformulations in BCC Models
11. Conclusive Remarks and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Woolston, C. Breast cancer. Nature 2015, 527, S101. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A. Breast Cancer Statistics: Recent Trends. Adv. Exp. Med. Biol. 2019, 1152, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Leon-Ferre, R.A.; Giridhar, K.; Hieken, T.J.; Mutter, R.W.; Couch, F.J.; Jimenez, R.E.; Hawse, J.R.; Boughey, J.C.; Ruddy, K.J. A contemporary review of male breast cancer: Current evidence and unanswered questions. Cancer Metastasis Rev. 2018, 37, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadi, Z.; Lianos, G.D.; Ignatiadou, E.; Harissis, H.V.; Mitsis, M. Breast cancer in young women: An overview. Updates Surg. 2017, 69, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Roulot, A.; Héquet, D.; Guinebretière, J.-M.; Vincent-Salomon, A.; Lerebours, F.; Dubot, C.; Rouzier, R. Tumoral heterogeneity of breast cancer. Ann. Biol. Clin. (Paris) 2016, 74, 653–660. [Google Scholar] [CrossRef]
- Pandya, S.; Moore, R.G. Breast Development and Anatomy. Clin. Obstet. Gynecol. 2011, 54, 91–95. [Google Scholar] [CrossRef]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Holm, J.; Eriksson, L.; Ploner, A.; Eriksson, M.; Rantalainen, M.; Li, J.; Hall, P.; Czene, K. Assessment of Breast Cancer Risk Factors Reveals Subtype Heterogeneity. Cancer Res. 2017, 77, 3708–3717. [Google Scholar] [CrossRef]
- Kalinowski, L.; Sanus, J.M.; Reed, A.E.M.; Lakhani, S.R. Breast Cancer Heterogeneity in Primary and Metastatic Disease. Adv. Exp. Med. Biol. 2019, 1152, 75–104. [Google Scholar] [CrossRef]
- Gao, J.J.; Swain, S. Luminal A Breast Cancer and Molecular Assays: A Review. Oncology 2018, 23, 556–565. [Google Scholar] [CrossRef]
- Shah, A.N.; Metzger, O.; Bartlett, C.H.; Liu, Y.; Huang, X.; Cristofanilli, M. Hormone Receptor–Positive/Human Epidermal Growth Receptor 2–Negative Metastatic Breast Cancer in Young Women: Emerging Data in the Era of Molecularly Targeted Agents. Oncology 2020, 2019-0729. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-H.; Hu, P.-H.; Tu, J.-H.; Yu, N.-S. Luminal B breast cancer: Patterns of recurrence and clinical outcome. Oncotarget 2016, 7, 65024–65033. [Google Scholar] [CrossRef] [PubMed]
- Asif, H.M.; Sultana, S.; Ahmed, S.; Akhtar, N.; Tariq, M. HER-2 Positive Breast Cancer - a Mini-Review. Asian Pac. J. Cancer Prev. 2016, 17, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.L.; Nunes, N.C.C.; Izetti, P.; De Mesquita, G.G.; De Melo, A.C. Triple negative breast cancer: A thorough review of biomarkers. Crit. Rev. Oncol. 2020, 145, 102855. [Google Scholar] [CrossRef]
- Eskiler, G.G.; Cecener, G.; Egeli, U.; Tunca, B. Triple negative breast cancer: New therapeutic approaches andBRCAstatus. APMIS 2018, 126, 371–379. [Google Scholar] [CrossRef]
- Jitariu, A.-A.; Cîmpean, A.M.; Ribatti, D.; Raica, M. Triple negative breast cancer: The kiss of death. Oncotarget 2017, 8, 46652–46662. [Google Scholar] [CrossRef]
- Russnes, H.G.; Lingjærde, O.C.; Borresen-Dale, A.-L.; Caldas, C. Breast Cancer Molecular Stratification. Am. J. Pathol. 2017, 187, 2152–2162. [Google Scholar] [CrossRef]
- Peart, O. Breast intervention and breast cancer treatment options. Radiol. Technol. 2015, 86, 535–558. [Google Scholar]
- Tray, N.; Taff, J.; Adams, S. Therapeutic landscape of metaplastic breast cancer. Cancer Treat. Rev. 2019, 79, 101888. [Google Scholar] [CrossRef]
- Savard, M.-F.; Khan, O.; Hunt, K.K.; Verma, S. Redrawing the Lines: The Next Generation of Treatment in Metastatic Breast Cancer. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, e8–e21. [Google Scholar] [CrossRef]
- Le Du, F.; Perrin, C.; Brunot, A.; Crouzet, L.; Rouge, T.D.L.M.; Lefeuvre-Plesse, C.; Diéras, V. Therapeutic innovations in breast cancer. La Presse Médicale 2019, 48, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Samadi, P.; Saki, S.; Dermani, F.K.; Pourjafar, M.; Saidijam, M. Emerging ways to treat breast cancer: Will promises be met? Cell. Oncol. 2018, 41, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Abderrahman, B.; Jordan, V.C. Telling details of breast-cancer recurrence. Nature 2018, 553, 155. [Google Scholar] [CrossRef] [PubMed]
- Szostakowska-Rodzos, M.; Trebinska-Stryjewska, A.; Grzybowska, E.A.; Fabisiewicz, A. Resistance to endocrine therapy in breast cancer: Molecular mechanisms and future goals. Breast Cancer Res. Treat. 2018, 173, 489–497. [Google Scholar] [CrossRef]
- Richman, J.; Dowsett, M. Beyond 5 years: Eenduring risk of recurrence in oestrogen receptor-positive breast cancer. Nat. Rev. Clin. Oncol. 2018, 16, 296–311. [Google Scholar] [CrossRef]
- Loibl, S.; Gianni, L. HER2-positive breast cancer. Lancet 2017, 389, 2415–2429. [Google Scholar] [CrossRef]
- Cameron, D.; Piccart-Gebhart, M.J.; Gelber, R.D.; Procter, M.; Goldhirsch, A.; De Azambuja, E.; Castro, G.; Untch, M.; Smith, I.; Gianni, L.; et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: Final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 2017, 389, 1195–1205. [Google Scholar] [CrossRef]
- Shagufta; Ahmad, I. Tamoxifen a pioneering drug: An update on the therapeutic potential of tamoxifen derivatives. Eur. J. Med. Chem. 2018, 143, 515–531. [Google Scholar] [CrossRef]
- Solinas, C.; Aiello, M.; Migliori, E.; Willard-Gallo, K.; Emens, L.A. Breast cancer vaccines: Heeding the lessons of the past to guide a path forward. Cancer Treat. Rev. 2020, 84, 101947. [Google Scholar] [CrossRef]
- Marra, A.; Viale, G.; Curigliano, G. Recent advances in triple negative breast cancer: The immunotherapy era. BMC Med. 2019, 17, 90. [Google Scholar] [CrossRef]
- Abotaleb, M.; Kubatka, P.; Caprnda, M.; Varghese, E.; Zolakova, B.; Zubor, P.; Opatřilová, R.; Kruzliak, P.; Stefanicka, P.; Büsselberg, D. Chemotherapeutic agents for the treatment of metastatic breast cancer: An update. Biomed. Pharmacother. 2018, 101, 458–477. [Google Scholar] [CrossRef] [PubMed]
- Pathak, M.; Dwivedi, S.N.; Deo, S.V.S.; Thakur, B.; Sreenivas, V.; Rath, G.K. Neoadjuvant chemotherapy regimens in treatment of breast cancer: A systematic review and network meta-analysis protocol. Syst. Rev. 2018, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Biersack, B.; Schobert, R. Current State of Platinum Complexes for the Treatment of Advanced and Drug-Resistant Breast Cancers. Adv. Exp. Med. Biol. 2019, 1152, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Wahid, M.; Mandal, R.; Dar, S.A.; Jawed, A.; Lohani, M.; Areeshi, M.Y.; Akhter, N.; Haque, S.; Areeshi, M.Y. Therapeutic potential and critical analysis of trastuzumab and bevacizumab in combination with different chemotherapeutic agents against metastatic breast/colorectal cancer affecting various endpoints. Crit. Rev. Oncol. 2016, 104, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Lu, Y.; Tian, H.; Meng, X.; Wei, M.; Cho, W.C. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed. Pharmacother. 2019, 114, 108800. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.W.S.; Wu, M.; Cho, W.C.S.; To, K.K. Recent Advances in the Treatment of Breast Cancer. Front. Oncol. 2018, 8, 227. [Google Scholar] [CrossRef]
- Majidinia, M.; Yousefi, B. DNA repair and damage pathways in breast cancer development and therapy. DNA Repair 2017, 54, 22–29. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Burgio, E.; Piscitelli, P.; Colao, A. Environmental Carcinogenesis and Transgenerational Transmission of Carcinogenic Risk: From Genetics to Epigenetics. Int. J. Environ. Res. Public Health 2018, 15, 1791. [Google Scholar] [CrossRef]
- Matsuura, K.; Canfield, K.; Feng, W.; Kurokawa, M. Metabolic Regulation of Apoptosis in Cancer. Int. Rev. Cell Mol. Boil. 2016, 327, 43–87. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genome Res. 2019, 33, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Longo, F.; Giuliano, M.; Sabbatino, F.; Favia, G.; Ionna, F.; Addeo, R.; Scarpati, G.D.V.; Di Lorenzo, G.; Pisconti, S.; et al. Epigenetic control of gene expression: Potential implications for cancer treatment. Crit. Rev. Oncol. 2017, 111, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Kaserer, T.; Blagg, J. Combining Mutational Signatures, Clonal Fitness, and Drug Affinity to Define Drug-Specific Resistance Mutations in Cancer. Cell Chem. Boil. 2018, 25, 1359–1371.e2. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ferrell, J.E. Apoptosis propagates through the cytoplasm as trigger waves. Science 2018, 361, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Ferrari, P.; Diodati, L.; Carpi, A. Recent Advances in Comprehending the Signaling Pathways Involved in the Progression of Breast Cancer. Int. J. Mol. Sci. 2017, 18, 2321. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Kønig, S.M.; Rissler, V.; Terkelsen, T.; Lambrughi, M.; Papaleo, E. Alterations of the interactome of Bcl-2 proteins in breast cancer at the transcriptional, mutational and structural level. PLoS Comput. Boil. 2019, 15, e1007485. [Google Scholar] [CrossRef]
- Williams, M.M.; Cook, R.S. Bcl-2 family proteins in breast development and cancer: Could Mcl-1 targeting overcome therapeutic resistance? Oncotarget 2015, 6, 3519–3530. [Google Scholar] [CrossRef]
- Moody, S.E.; Schinzel, A.C.; Singh, S.; Izzo, F.; Strickland, M.R.; Luo, L.; Thomas, S.R.; Boehm, J.S.; Kim, S.Y.; Wang, Z.C.; et al. PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling. Oncogene 2014, 34, 2061–2071. [Google Scholar] [CrossRef]
- Honma, N.; Horii, R.; Ito, Y.; Saji, S.; Younes, M.; Iwase, T.; Akiyama, F. Differences in clinical importance of Bcl-2 in breast cancer according to hormone receptors status or adjuvant endocrine therapy. BMC Cancer 2015, 15, 698. [Google Scholar] [CrossRef]
- Mohamed, Z.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and Molecular Targeting Therapy in Cancer. Biomed. Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.D.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. A BH3 Mimetic for Killing Cancer Cells. Cell 2016, 165, 1560. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2017, 25, 27–36. [Google Scholar] [CrossRef]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure–activity relationships for ruthenium and osmium anticancer agents – towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef]
- Abid, M.; Shamsi, F.; Azam, A. Ruthenium Complexes: An Emerging Ground to the Development of Metallopharmaceuticals for Cancer Therapy. Mini-Rev. Med. Chem. 2016, 16, 772–786. [Google Scholar] [CrossRef]
- Irace, C.; Misso, G.; Capuozzo, A.; Piccolo, M.; Riccardi, C.; Luchini, A.; Caraglia, M.; Paduano, L.; Montesarchio, D.; Santamaria, R. Antiproliferative effects of ruthenium-based nucleolipidic nanoaggregates in human models of breast cancer in vitro: Insights into their mode of action. Sci. Rep. 2017, 7, 45236. [Google Scholar] [CrossRef]
- Zheng, K.; Wu, Q. Ruthenium(II) Complexes as Potential Apoptosis Inducers in Chemotherapy. Anticancer Agents Med. Chem. 2017, 17, 29–39. [Google Scholar]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- White, E.; Mehnert, J.M. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Fan, S.; Qin, T.; Yang, J.; Sun, Y.; Lu, Y.; Mao, J.; Li, L. Role of autophagy in breast cancer and breast cancer stem cells (Review). Int. J. Oncol. 2018, 52, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed]
- Lisiak, N.; Toton, E.; Rybczynska, M. Autophagy as a Potential Therapeutic Target in Breast Cancer Treatment. Curr. Cancer Drug Targets 2018, 18, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.E.; Tee, A.R. Exploiting cancer vulnerabilities: mTOR, autophagy, and homeostatic imbalance. Essays Biochem. 2017, 61, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Misso, G.; Zarone, M.R.; Lombardi, A.; Grimaldi, A.; Cossu, A.M.; Ferri, C.; Russo, M.; Vuoso, D.C.; Luce, A.; Kawasaki, H.; et al. miR-125b Upregulates miR-34a and Sequentially Activates Stress Adaption and Cell Death Mechanisms in Multiple Myeloma. Mol. Ther. Nucleic Acids 2019, 16, 391–406. [Google Scholar] [CrossRef]
- Shen, P.; Chen, M.; He, M.; Chen, L.; Song, Y.; Xiao, P.; Wan, X.; Dai, F.; Pan, T.; Wang, Q. Inhibition of ERα/ERK/P62 cascades induces “autophagic switch” in the estrogen receptor-positive breast cancer cells exposed to gemcitabine. Oncotarget 2016, 7, 48501–48516. [Google Scholar] [CrossRef]
- Das, C.K.; Parekh, A.; Parida, P.K.; Bhutia, S.K.; Mandal, M. Lactate dehydrogenase A regulates autophagy and tamoxifen resistance in breast cancer. Biochim. Biophys. Acta 2019, 1866, 1004–1018. [Google Scholar] [CrossRef]
- Ronghe, A.; Chatterjee, A. Tamoxifen synergizes with 4-(E)-{(4-hydroxyphenylimino)-methylbenzene, 1,2-diol} and 4-(E)-{(p-tolylimino)-methylbenzene-1,2-diol}, novel azaresveratrol analogs, in inhibiting the proliferation of breast cancer cells. Oncotarget 2016, 7, 51747–51762. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Liu, X. How does estrogen work on autophagy? Autophagy. 2019, 15, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Janser, F.A.; Tschan, M.P.; Langer, R.; A Janser, F. The role of autophagy in HER2-targeted therapy. Swiss Med. Wkly. 2019, 149, w20138. [Google Scholar] [CrossRef] [PubMed]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Garbar, C.; Mascaux, C.; Giustiniani, J.; Merrouche, Y.; Bensussan, A. Chemotherapy treatment induces an increase of autophagy in the luminal breast cancer cell MCF7, but not in the triple-negative MDA-MB231. Sci. Rep. 2017, 7, 7201. [Google Scholar] [CrossRef]
- Chen, M.; He, M.; Song, Y.; Chen, L.; Xiao, P.; Wan, X.; Dai, F.; Shen, P. The cytoprotective role of gemcitabine-induced autophagy associated with apoptosis inhibition in triple-negative MDA-MB-231 breast cancer cells. Int. J. Mol. Med. 2014, 34, 276–282. [Google Scholar] [CrossRef]
- Tekedereli, I.; Alpay, S.N.; Akar, U.; Yuca, E.; Ayugo-Rodriguez, C.; Han, H.-D.; Sood, A.K.; Lopez-Berestein, G.; Ozpolat, B. Therapeutic Silencing of Bcl-2 by Systemically Administered siRNA Nanotherapeutics Inhibits Tumor Growth by Autophagy and Apoptosis and Enhances the Efficacy of Chemotherapy in Orthotopic Xenograft Models of ER (-) and ER (+) Breast Cancer. Mol. Ther. Nucleic Acids 2013, 2, e121. [Google Scholar] [CrossRef]
- Piccolo, M.; Misso, G.; Ferraro, M.G.; Riccardi, C.; Capuozzo, A.; Zarone, M.R.; Maione, F.; Trifuoggi, M.; Stiuso, P.; D’Errico, G.; et al. Exploring cellular uptake, accumulation and mechanism of action of a cationic Ru-based nanosystem in human preclinical models of breast cancer. Sci. Rep. 2019, 9, 7006. [Google Scholar] [CrossRef]
- Koceva-Chyła, A.; Matczak, K.; Hikisz, P.; Durka, M.K.; Kochel, M.K.; Süss-Fink, G.; Furrer, J.; Kowalski, K. Insights into the in vitro Anticancer Effects of Diruthenium-1. ChemMedChem 2016, 11, 2171–2187. [Google Scholar] [CrossRef]
- Yuan, J.; Lei, Z.; Wang, X.; Zhu, F.; Chen, D. Ruthenium complex ?-WH0402 induces hepatocellular carcinoma LM6 (HCCLM6) cell death by triggering the Beclin-1-dependent autophagy pathway. Metallomics 2015, 7, 896–907. [Google Scholar] [CrossRef]
- Song, X.; Lee, D.H. Crosstalk Between Apoptosis and Autophagy Is Regulated by the Arginylated BiP/Beclin-1/p62 Complex. Mol. Cancer Res. 2018, 16, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Shamseddine, A.I.; Farhat, F.S. Platinum-Based Compounds for the Treatment of Metastatic Breast Cancer. Chemotherapy 2011, 57, 468–487. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhang, W.; Ji, W.; Yang, F.; Guan, X. Predictive biomarkers for triple negative breast cancer treated with platinum-based chemotherapy. Cancer Boil. Ther. 2017, 18, 369–378. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Simpson, P.; Desai, N.M.; Casari, I.; Massi, M.; Falasca, M. Metal-based antitumor compounds: Beyond cisplatin. Future Med. Chem. 2019, 11, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Zhao, Z.-Z.; Bo, H.-B.; Hao, X.; Wang, J.-Q. Applications of Ruthenium Complex in Tumor Diagnosis and Therapy. Front. Pharmacol. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Coverdale, J.P.C.; Laroiya-McCarron, T.; Canelon, I.R. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D.; Trifuoggi, M.; Irace, C.; Paduano, L.; Montesarchio, D. Anticancer Ruthenium(III) Complexes and Ru(III)-Containing Nanoformulations: An Update on the Mechanism of Action and Biological Activity. Pharmacy 2019, 12, 146. [Google Scholar] [CrossRef]
- Pal, M.; Nandi, U.; Mukherjee, D. Detailed account on activation mechanisms of ruthenium coordination complexes and their role as antineoplastic agents. Eur. J. Med. Chem. 2018, 150, 419–445. [Google Scholar] [CrossRef]
- Thota, S.; Rodrigues, D.A.; Crans, D.C.; Barreiro, E.J. Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.-S. The development of anticancer ruthenium(ii) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef]
- Thangavel, P.; Viswanath, B.; Kim, S. Recent developments in the nanostructured materials functionalized with ruthenium complexes for targeted drug delivery to tumors. Int. J. Nanomed. 2017, 12, 2749–2758. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E.; Messori, L. NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry. Molecules 2019, 24, 1995. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E.; Messori, L. The DECEPTIVELY SIMILAR RUTHENIUM(III) DRUG CANDIDATES KP1019 AND NAMI-A HAVE DIFFERENT ACTIONS. WHAT DID WE LEARN IN THE PAST 30 YEARS? Met. Drugs Dev. Action Anticancer Agents 2018, 141–170. [Google Scholar] [CrossRef]
- Casini, A.; Gabbiani, C.; Sorrentino, F.; Rigobello, M.P.; Bindoli, A.; Geldbach, T.J.; Marrone, A.; Re, N.; Hartinger, C.G.; Dyson, P.J.; et al. Emerging Protein Targets for Anticancer Metallodrugs: Inhibition of Thioredoxin Reductase and Cathepsin B by Antitumor Ruthenium(II)−Arene Compounds. J. Med. Chem. 2008, 51, 6773–6781. [Google Scholar] [CrossRef]
- Babak, M.V.; Meier-Menches, S.M.; Huber, K.; Reynisson, J.; Legin, A.A.; Jakupec, M.A.; Roller, A.; Stukalov, A.; Gridling, M.; Bennett, K.L.; et al. Target profiling of an antimetastatic RAPTA agent by chemical proteomics: Relevance to the mode of action. Chem. Sci. 2015, 6, 2449–2456. [Google Scholar] [CrossRef]
- Licona, C.; Spaety, M.-E.; Capuozzo, A.; Ali, M.; Santamaria, R.; Armant, O.; Delalande, F.; Van Dorsselaer, A.; Cianferani, S.; Spencer, J.; et al. A ruthenium anticancer compound interacts with histones and impacts differently on epigenetic and death pathways compared to cisplatin. Oncotarget 2016, 8, 2568–2584. [Google Scholar] [CrossRef]
- Parveen, S.; Arjmand, F.; Tabassum, S. Development and future prospects of selective organometallic compounds as anticancer drug candidates exhibiting novel modes of action. Eur. J. Med. Chem. 2019, 175, 269–286. [Google Scholar] [CrossRef]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef]
- Clarke, M.; Bitler, S.; Rennert, D.; Buchbinder, M.; Kelman, A. Reduction and Subsequent Binding of Ruthenium Ions Catalyzed by Subcellular Components. J. Inorg. Biochem. 1980, 12, 79–87. [Google Scholar] [CrossRef]
- Keppler, B.K.; Rupp, W. Antitumor activity of imidazolium-bisimidazole-tetrachlororuthenate (III). A representative of a new class of inorganic antitumor agents. J. Cancer Res. Clin. Oncol. 1986, 111, 166–168. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside – preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Pacor, S.; Luxich, E.; Ceschia, V.; Sava, G.; Alessio, E.; Mestroni, G. Effects of trans-RuCl2(DMSO)4 on B16 melanoma in mice. Pharmacol. Res. 1989, 21, 127–128. [Google Scholar] [CrossRef]
- Sava, G.; Pacor, S.; Mestroni, G.; Alessio, E. Na[trans-RuCl4(DMSO)Im], a metal complex of ruthenium with antimetastatic properties. Clin. Exp. Metastasis 1992, 10, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Mestroni, G.; Alessio, E.; Sava, G.; Pacor, S.; Coluccia, M.; Boccarelli, A. Water-Soluble Ruthenium(III)-Dimethyl Sulfoxide Complexes: Chemical Behaviour and Pharmaceutical Properties. Met. Drugs 1994, 1, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Sava, G.; Capozzi, I.; Clerici, K.; Gagliardi, G.; Alessio, E.; Mestroni, G. Pharmacological control of lung metastases of solid tumours by a novel ruthenium complex. Clin. Exp. Metastasis 1998, 16, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Sava, G.; Clerici, K.; Capozzi, I.; Cocchietto, M.; Gagliardi, R.; Alessio, E.; Mestroni, G.; Perbellini, A. Reduction of lung metastasis by ImH[trans-RuCl4(DMSO)Im]: Mechanism of the selective action investigated on mouse tumors. Anti-Cancer Drugs 1999, 10, 129–138. [Google Scholar] [CrossRef]
- Bergamo, A.; Gagliardi, R.; Scarcia, V.; Furlani, A.; Alessio, E.; Mestroni, G.; Sava, G. In vitro cell cycle arrest, in vivo action on solid metastasizing tumors, and host toxicity of the antimetastatic drug NAMI-A and cisplatin. J. Pharmacol. Exp. Ther. 1999, 289, 559–564. [Google Scholar]
- Alessio, E.; Mestroni, G.; Bergamo, A.; Sava, G. Bentham Science Publisher Enzo Alessio; Bentham Science Publisher Giovanni Mestroni; Bentham Science Publisher Alberta Bergamo; Bentham Science Publisher Gianni Sava Ruthenium Antimetastatic Agents. Curr. Top. Med. Chem. 2004, 4, 1525–1535. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; Bongard, D.V.D.; Pluim, D.; Beijnen, J.H.; Schellens, J. A Phase I and Pharmacological Study with Imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a Novel Ruthenium Anticancer Agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; Van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Investig. New Drugs 2014, 33, 201–214. [Google Scholar] [CrossRef]
- Wernitznig, D.; Kiakos, K. First-in-class ruthenium anticancer drug (KP1339/IT-139) induces an immunogenic cell death signature in colorectal spheroids in vitro. Metallomics 2019, 11, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Heffeter, P.; Böck, K.; Atil, B.; Hoda, M.A.R.; Körner, W.; Bartel, C.; Jungwirth, U.; Keppler, B.K.; Micksche, M.; Berger, W.; et al. Intracellular protein binding patterns of the anticancer ruthenium drugs KP1019 and KP1339. JBIC J. Boil. Inorg. Chem. 2010, 15, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Dömötör, O.; Hartinger, C.G.; Bytzek, A.K.; Kiss, T.; Keppler, B.K.; Enyedy, É.A. Characterization of the binding sites of the anticancer ruthenium(III) complexes KP1019 and KP1339 on human serum albumin via competition studies. JBIC J. Boil. Inorg. Chem. 2012, 18, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef]
- Flocke, L.S.; Trondl, R.; Jakupec, M.A.; Keppler, B.K. Molecular mode of action of NKP-1339 - a clinically investigated ruthenium-based drug - involves ER- and ROS-related effects in colon carcinoma cell lines. Investig. New Drugs 2016, 34, 261–268. [Google Scholar] [CrossRef]
- Schoenhacker-Alte, B.; Mohr, T. Sensitivity towards the GRP78 inhibitor KP1339/IT-139 is characterized by apoptosis induction via caspase 8 upon disruption of ER homeostasis. Cancer Lett. 2017, 404, 79–88. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A New Redox-Active Anticancer Agent - Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef]
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open 2017, 1, e000154. [Google Scholar] [CrossRef]
- Bytzek, A.K.; Koellensperger, G. Biodistribution of the novel anticancer drug sodium trans-[tetrachloridobis(1H-indazole)ruthenate(III)] KP-1339/IT139 in nude BALB/c mice and implications on its mode of action. J. Inorg. Biochem. 2016, 160, 250–255. [Google Scholar] [CrossRef]
- Monro, S.; Colón, K.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2018, 119, 797–828. [Google Scholar] [CrossRef]
- Bergamo, A.; Sava, G. Ruthenium anticancer compounds: Myths and realities of the emerging metal-based drugs. Dalton Trans. 2011, 40, 7817. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E. Thirty Years of the Drug Candidate NAMI-A and the Myths in the Field of Ruthenium Anticancer Compounds: A Personal Perspective. Eur. J. Inorg. Chem. 2016, 2017, 1549–1560. [Google Scholar] [CrossRef]
- Capper, M.S.; Packman, H.; Rehkämper, M. Rhenium-Based Complexes and in Vivo Testing: A Brief History. ChemBioChem 2020. [Google Scholar] [CrossRef] [PubMed]
- Golbaghi, G.; Castonguay, A. Rationally Designed Ruthenium Complexes for Breast Cancer Therapy. Molecules 2020, 25, 265. [Google Scholar] [CrossRef]
- Liu, J.; Lai, H.; Xiong, Z.; Chen, B.; Chen, T. Functionalization and cancer-targeting design of ruthenium complexes for precise cancer therapy. Chem. Commun. 2019, 55, 9904–9914. [Google Scholar] [CrossRef]
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Co-Ord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Webb, M.I.; Chard, R.A.; Al-Jobory, Y.M.; Jones, M.R.; Wong, E.W.Y.; Walsby, C.J. Pyridine Analogues of the Antimetastatic Ru(III) Complex NAMI-A Targeting Non-Covalent Interactions with Albumin. Inorg. Chem. 2011, 51, 954–966. [Google Scholar] [CrossRef]
- Vaccaro, M.; Del Litto, R.; Mangiapia, G.; Carnerup, A.M.; D’Errico, G.; Ruffo, F.; Paduano, L. Lipid based nanovectors containing ruthenium complexes: A potential route in cancer therapy. Chem. Commun. 2009, 11, 1404. [Google Scholar] [CrossRef]
- Mangiapia, G.; D’Errico, G.; Simeone, L.; Irace, C.; Radulescu, A.; Di Pascale, A.; Colonna, A.; Montesarchio, D.; Paduano, L. Ruthenium-based complex nanocarriers for cancer therapy. Biomaterials 2012, 33, 3770–3782. [Google Scholar] [CrossRef]
- Mangiapia, G.; Vitiello, G.; Irace, C.; Santamaria, R.; Colonna, A.; Angelico, R.; Radulescu, A.; D’Errico, G.; Montesarchio, D.; Paduano, L. Anticancer Cationic Ruthenium Nanovectors: From Rational Molecular Design to Cellular Uptake and Bioactivity. Biomacromolecules 2013, 14, 2549–2560. [Google Scholar] [CrossRef][Green Version]
- Ringhieri, P.; Morelli, G.; Accardo, A. Supramolecular Delivery Systems for Non-Platinum Metal-Based Anticancer Drugs. Crit. Rev. Ther. Drug Carr. Syst. 2017, 34, 149–183. [Google Scholar] [CrossRef] [PubMed]
- Sava, G.; Bergamo, A.; Zorzet, S.; Gava, B.; Casarsa, C.; Cocchietto, M.; Furlani, A.; Scarcia, V.; Serli, B.; Iengo, E.; et al. Influence of chemical stability on the activity of the antimetastasis ruthenium compound NAMI-A. Eur. J. Cancer 2002, 38, 427–435. [Google Scholar] [CrossRef]
- Bouma, M.; Nuijen, B.; Jansen, M.T.; Sava, G.; Flaibani, A.; Bult, A.; Beijnen, J.H. A kinetic study of the chemical stability of the antimetastatic ruthenium complex NAMI-A. Int. J. Pharm. 2002, 248, 239–246. [Google Scholar] [CrossRef]
- Baillet, J.; Desvergnes, V.; Hamoud, A.; Latxague, L.; Barthelemy, P. Lipid and Nucleic Acid Chemistries: Combining the Best of Both Worlds to Construct Advanced Biomaterials. Adv. Mater. 2018, 30, 1705078. [Google Scholar] [CrossRef] [PubMed]
- Rosemeyer, H. Nucleolipids: Natural Occurrence, Synthesis, Molecular Recognition, and Supramolecular Assemblies as Potential Precursors of Life and Bioorganic Materials. Chem. Biodivers. 2005, 2, 977–1063. [Google Scholar] [CrossRef] [PubMed]
- Allain, V.; Bourgaux, C.; Couvreur, P. Self-assembled nucleolipids: From supramolecular structure to soft nucleic acid and drug delivery devices. Nucleic Acids Res. 2011, 40, 1891–1903. [Google Scholar] [CrossRef]
- Simeone, L.; Mangiapia, G.; Irace, C.; Di Pascale, A.; Colonna, A.; Ortona, O.; De Napoli, L.; Montesarchio, D.; Paduano, L. Nucleolipid nanovectors as molecular carriers for potential applications in drug delivery. Mol. Biosyst. 2011, 7, 3075. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D. RuIII Complexes for Anticancer Therapy: The Importance of Being Nucleolipidic. Eur. J. Org. Chem. 2017, 1100–1119. [Google Scholar] [CrossRef]
- Simeone, L.; Mangiapia, G.; Vitiello, G.; Irace, C.; Colonna, A.; Ortona, O.; Montesarchio, D.; Paduano, L. Cholesterol-Based Nucleolipid-Ruthenium Complex Stabilized by Lipid Aggregates for Antineoplastic Therapy. Bioconjugate Chem. 2012, 23, 758–770. [Google Scholar] [CrossRef]
- Vitiello, G.; Luchini, A.; D’Errico, G.; Santamaria, R.; Capuozzo, A.; Irace, C.; Montesarchio, D.; Paduano, L. Cationic liposomes as efficient nanocarriers for the drug delivery of an anticancer cholesterol-based ruthenium complex. J. Mater. Chem. B 2015, 3, 3011–3023. [Google Scholar] [CrossRef]
- Montesarchio, D.; Mangiapia, G.; Vitiello, G.; Musumeci, D.; Irace, C.; Santamaria, R.; D’Errico, G.; Paduano, L. A new design for nucleolipid-based Ru(iii) complexes as anticancer agents. Dalton Trans. 2013, 42, 16697. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Reyes-Reyes, E.; Malik, M.T.; Murphy, E.M.; O’Toole, M.G.; Trent, J.O. G-quadruplex oligonucleotide AS1411 as a cancer-targeting agent: Uses and mechanisms. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1414–1428. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Musumeci, D.; Krauss, I.R.; Piccolo, M.; Irace, C.; Paduano, L.; Montesarchio, D. Exploring the conformational behaviour and aggregation properties of lipid-conjugated AS1411 aptamers. Int. J. Boil. Macromol. 2018, 118, 1384–1399. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Comşa, Ş.; Cîmpean, A.M.; Raica, M. The Story of MCF-7 Breast Cancer Cell Line: 40 years of Experience in Research. Anticancer. Res. 2015, 35, 3147–3154. [Google Scholar]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef]
- Chen, Z.; Han, L.; Xu, M.; Xu, Y.; Qian, X. Rationally designed multitarget anticancer agents. Curr. Med. Chem. 2013, 20, 1694–1714. [Google Scholar] [CrossRef]
- Zheng, W.; Zhao, Y.; Luo, Q.; Zhang, Y.; Wu, K.; Wang, F. Multi-Targeted Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 3084–3098. [Google Scholar] [CrossRef]
- Geromichalos, G.D.; Alifieris, K.; Geromichalou, E.G.; Trafalis, D.T. Overview on the current status of virtual high-throughput screening and combinatorial chemistry approaches in multi-target anticancer drug discovery; Part I. Off. J. Balk. Union Oncol. 2016, 21, 764–779. [Google Scholar]
- Vitali, F.; Cohen, L.D.; DeMartini, A.; Amato, A.; Eterno, V.; Zambelli, A.; Bellazzi, R. A Network-Based Data Integration Approach to Support Drug Repurposing and Multi-Target Therapies in Triple Negative Breast Cancer. PLoS ONE 2016, 11, e0162407. [Google Scholar] [CrossRef]
- Motadi, P.M.A.L.R. Apoptotic Molecular Advances in Breast Cancer Management. In Cell Death—Autophagy, Apoptosis and Necrosis; Intech Open Science: London, UK, 2015. [Google Scholar]
- Jena, M.K.; Jaswal, S.; Kumar, S.; Mohanty, A.K. Molecular mechanism of mammary gland involution: An update. Dev. Boil. 2019, 445, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Boise, L.H.; Shanmugam, M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers 2019, 11, 1144. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Magistrato, A.; Riedel, T.; Von Erlach, T.; Davey, C.A.; Dyson, P.J.; Rothlisberger, U. Fighting Cancer with Transition Metal Complexes: From Naked DNA to Protein and Chromatin Targeting Strategies. ChemMedChem 2015, 11, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Rozza, L.; Merlino, A.; Paduano, L.; Marzo, T.; Massai, L.; Messori, L.; Montesarchio, D. Interaction of anticancer Ru(iii) complexes with single stranded and duplex DNA model systems. Dalton Trans. 2015, 44, 13914–13925. [Google Scholar] [CrossRef]
- Che, C.; Siu, F.-M. Metal complexes in medicine with a focus on enzyme inhibition. Curr. Opin. Chem. Boil. 2010, 14, 255–261. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial performance in apoptosis. FEBS J. 2017, 285, 416–431. [Google Scholar] [CrossRef]
- Wang, S.; He, M.; Li, L.; Liang, Z.; Zou, Z.; Tao, A. Cell-in-Cell Death Is Not Restricted by Caspase-3 Deficiency in MCF-7 Cells. J. Breast Cancer 2016, 19, 231–241. [Google Scholar] [CrossRef]
- Chung, C. Restoring the switch for cancer cell death: Targeting the apoptosis signaling pathway. Am. J. Health Pharm. 2018, 75, 945–952. [Google Scholar] [CrossRef]
- Walensky, L.D. Targeting BAX to drug death directly. Nat. Methods 2019, 15, 657–665. [Google Scholar] [CrossRef]
- Deng, Z.; Gao, P.; Yu, L.; Ma, B.; You, Y.; Chan, L.; Mei, C.; Chen, T. Ruthenium complexes with phenylterpyridine derivatives target cell membrane and trigger death receptors-mediated apoptosis in cancer cells. Biomaterilas 2017, 129, 111–126. [Google Scholar] [CrossRef]
- Kocaturk, N.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Russo, G.L. Autophagy inducers in cancer. Biochem. Pharmacol. 2018, 153, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Ojha, R.; Ishaq, M. Caspase-mediated crosstalk between autophagy and apoptosis: Mutual adjustment or matter of dominance. J. Cancer Res. Ther. 2015, 11, 514. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Tavallai, S.; Hamed, H.A.; Cruickshanks, N.; Dent, P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell. Signal. 2013, 26, 549–555. [Google Scholar] [CrossRef]
- Tang, B.; Wan, D.; Lai, S.-H.; Yang, H.-H.; Zhang, C.; Wang, X.-Z.; Zeng, C.-C.; Liu, Y.-J. Design, synthesis and evaluation of anticancer activity of ruthenium (II) polypyridyl complexes. J. Inorg. Biochem. 2017, 173, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-C.; Wu, A.-G.; Huang, Y.-Z.; Shao, G.-L.; Ji, S.-F.; Wang, R.-W.; Yuan, H.-J.; Fan, X.-L.; Zheng, L.-H.; Jiao, Q.-L. Autophagic regulation of cell growth by altered expression of Beclin 1 in triple-negative breast cancer. Int. J. Clin. Exp. Med. 2015, 8, 7049–7058. [Google Scholar]
- Vega-Rubín-De-Celis, S. The Role of Beclin 1-Dependent Autophagy in Cancer. Boilogy 2019, 9, 4. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Marquez, R.T.; Xu, L. Bcl-2: Beclin 1 complex: Multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am. J. Cancer Res. 2012, 2, 214–221. [Google Scholar]
- Riccardi, C.; Musumeci, D.; Capuozzo, A.; Irace, C.; King, S.; Krauss, I.R.; Paduano, L.; Montesarchio, D. “Dressing up” an Old Drug: An Aminoacyl Lipid for the Functionalization of Ru(III)-Based Anticancer Agents. Acs Biomater. Sci. Eng. 2017, 4, 163–174. [Google Scholar] [CrossRef]
- Acampora, F.; Marzaioli, A.M.; Capuozzo, A.; Appavou, M.-S.; Campanella, A.; D’Errico, G.; Irace, C.; Montesarchio, D.; Musumeci, D.; Szekely, N.K.; et al. Lipooligosaccharides as Amphiphiles to Build Liposomes for Effective Drug Delivery: The Case of Anticancer Ruthenium Complex-Based Aggregates. ChemistrySelect 2016, 1, 2129–2139. [Google Scholar] [CrossRef]
- Grimaldi, A.; Zarone, M.R.; Irace, C.; Zappavigna, S.; Lombardi, A.; Kawasaki, H.; Caraglia, M.; Misso, G. Non-coding RNAs as a new dawn in tumor diagnosis. Semin. Cell Dev. Boil. 2018, 78, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Boil. 2020, 98, 139–153. [Google Scholar] [CrossRef]
- Vock, C.A.; Ang, W.H.; Scolaro, C.; Phillips, A.D.; Lagopoulos, L.; Juillerat-Jeanneret, L.; Sava, G.; Scopelliti, R.; Dyson, P.J. Development of Ruthenium Antitumor Drugs that Overcome Multidrug Resistance Mechanisms. J. Med. Chem. 2007, 50, 2166–2175. [Google Scholar] [CrossRef] [PubMed]






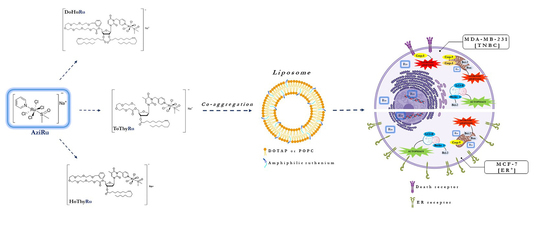
{kind=link}
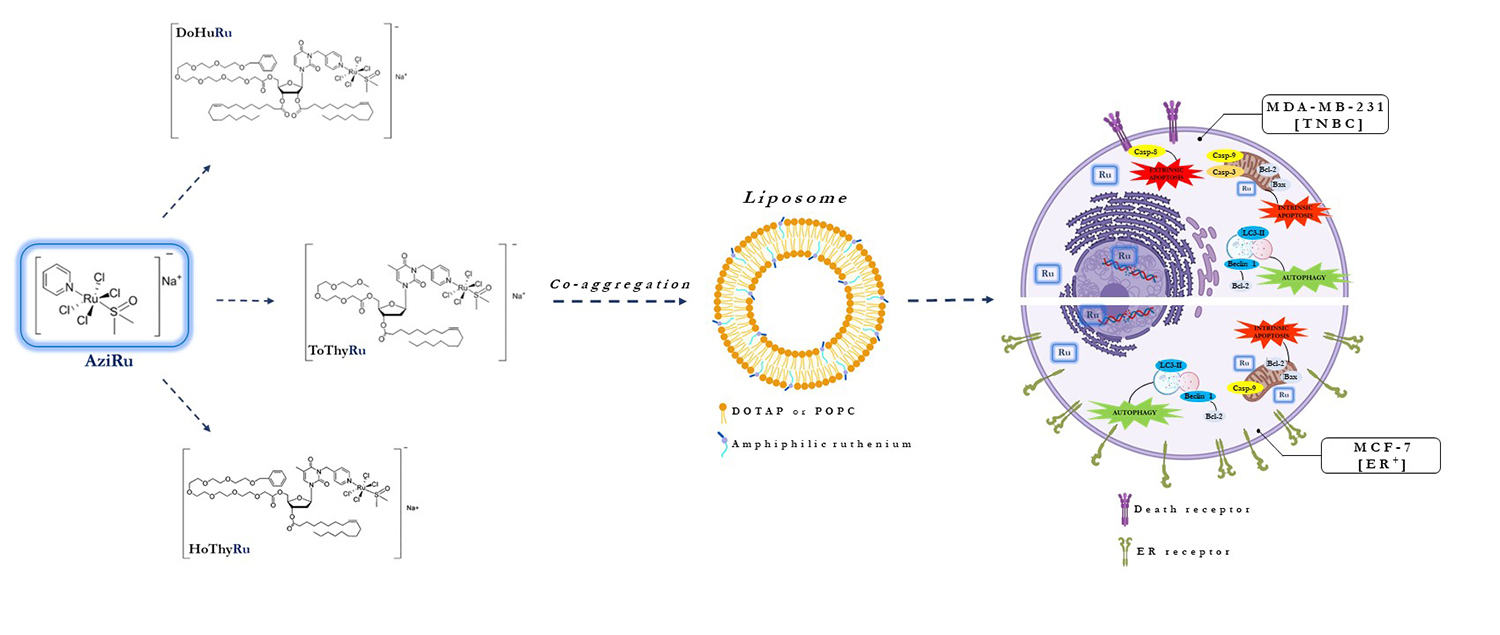
{kind=link}
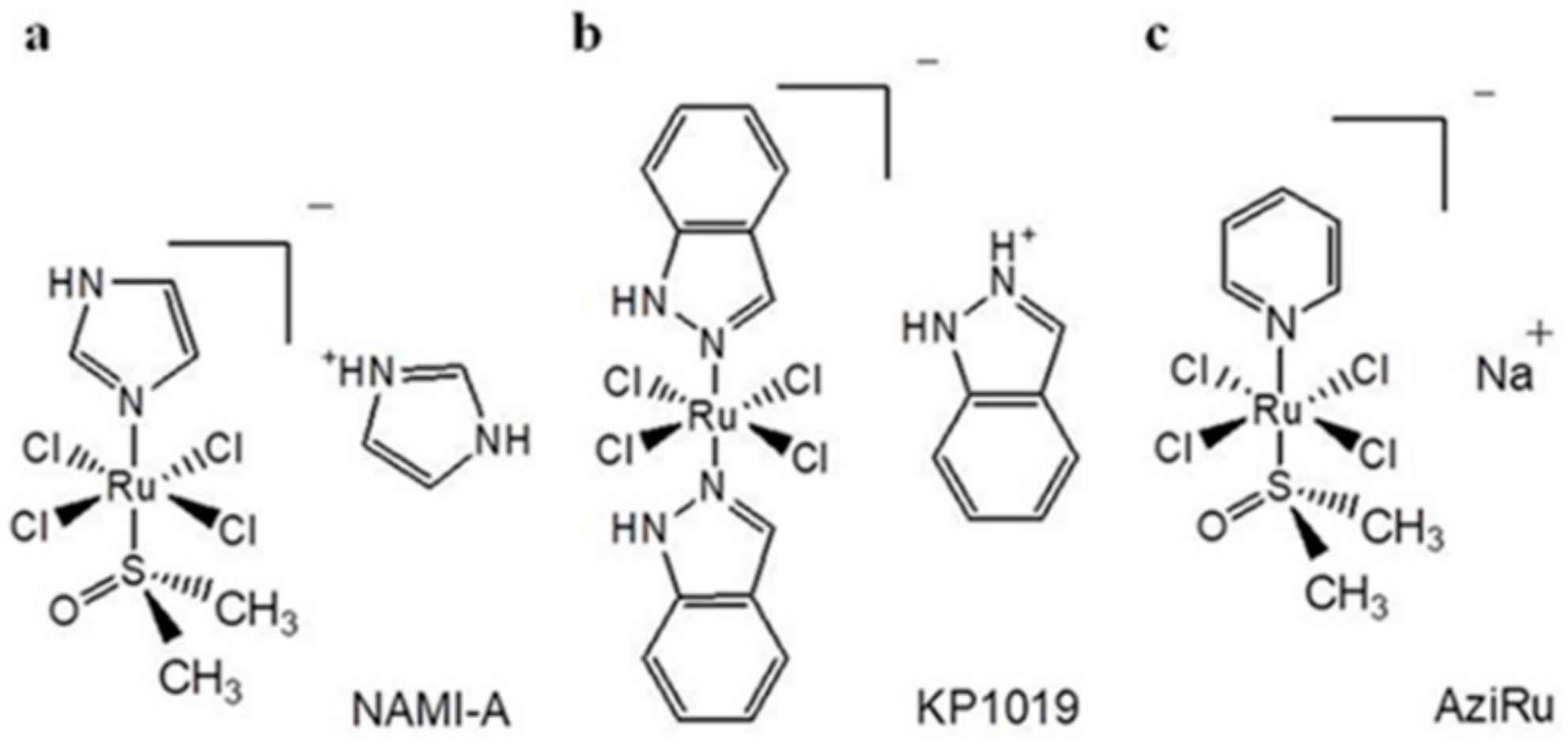
{kind=link}
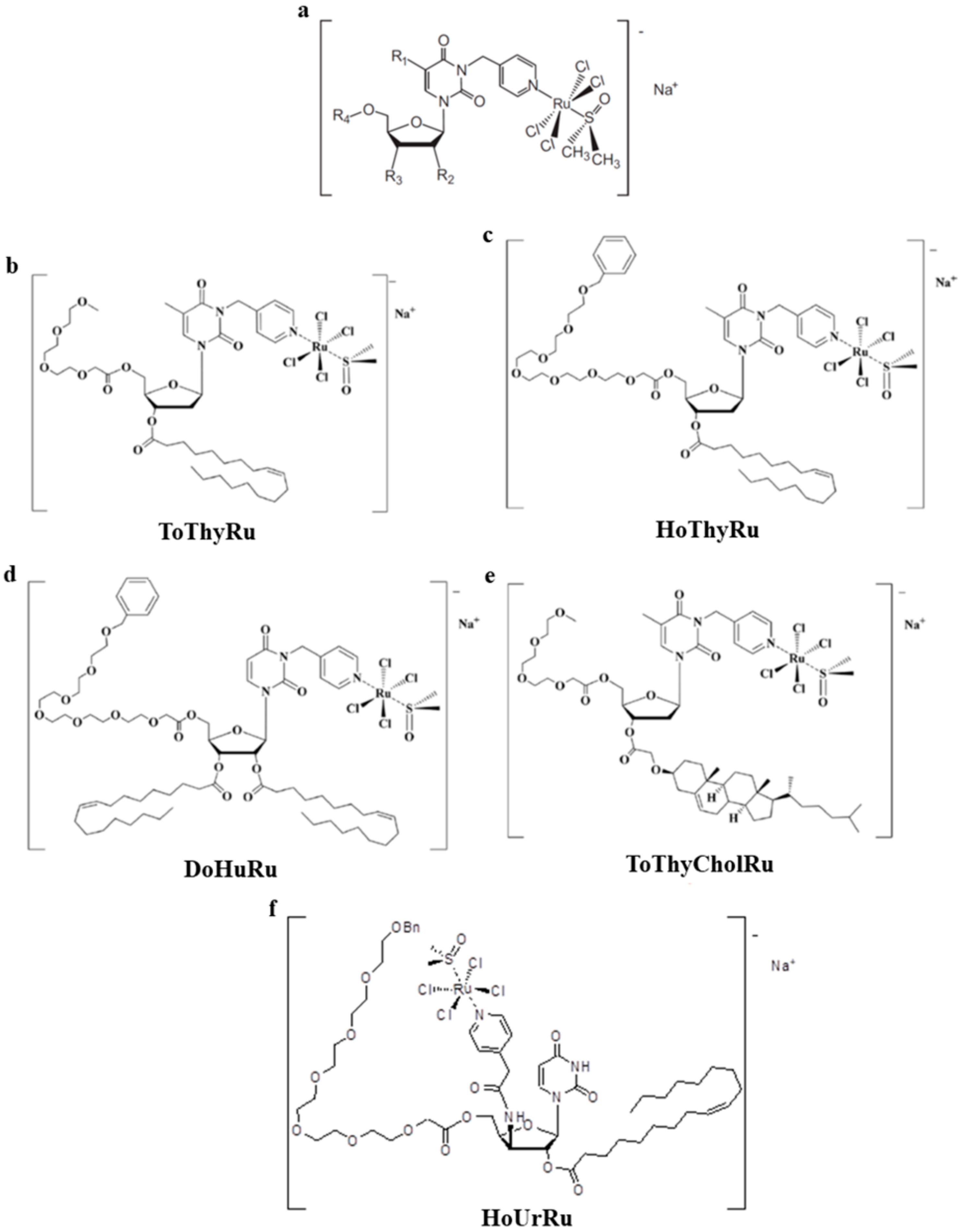
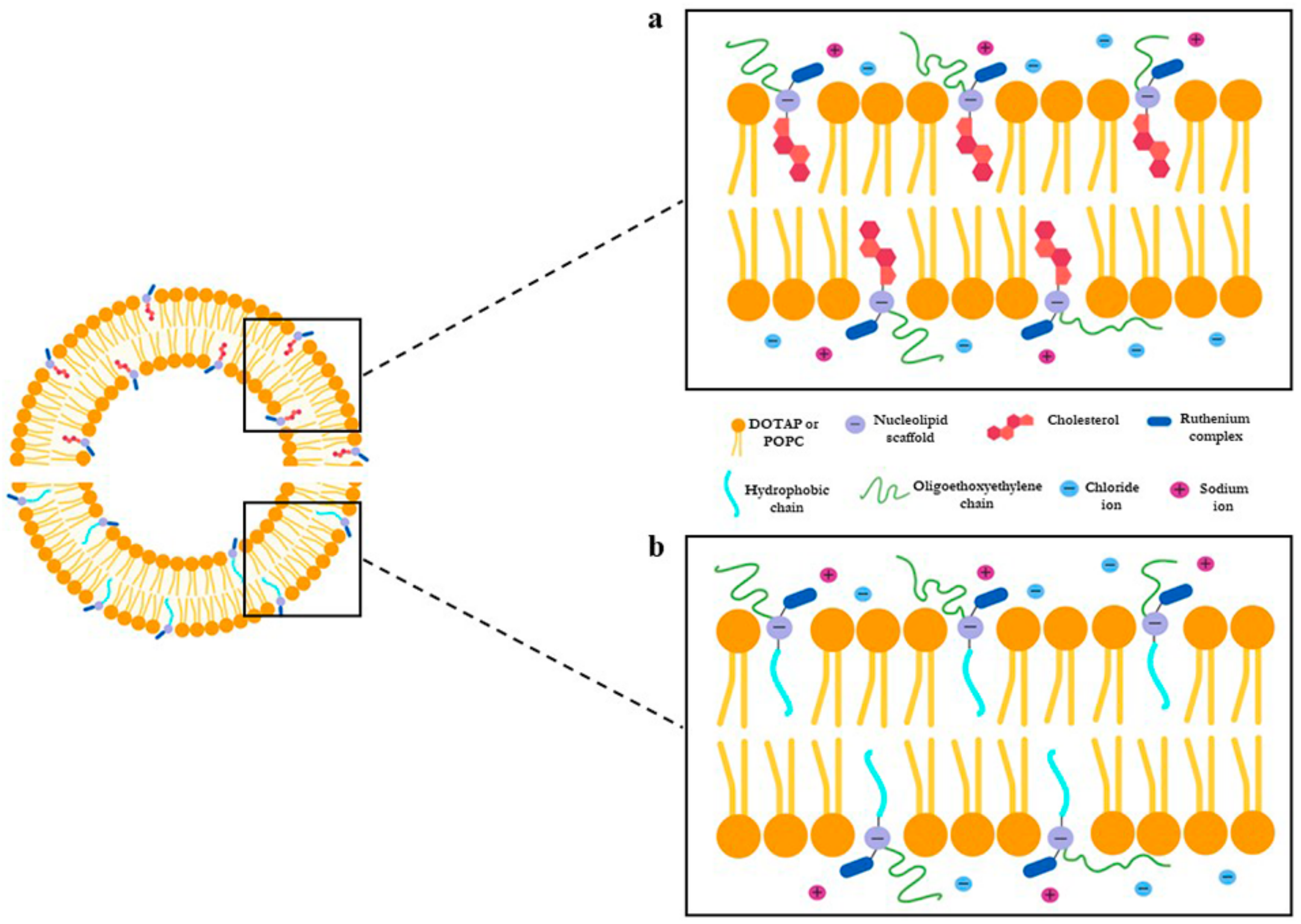{kind=link}
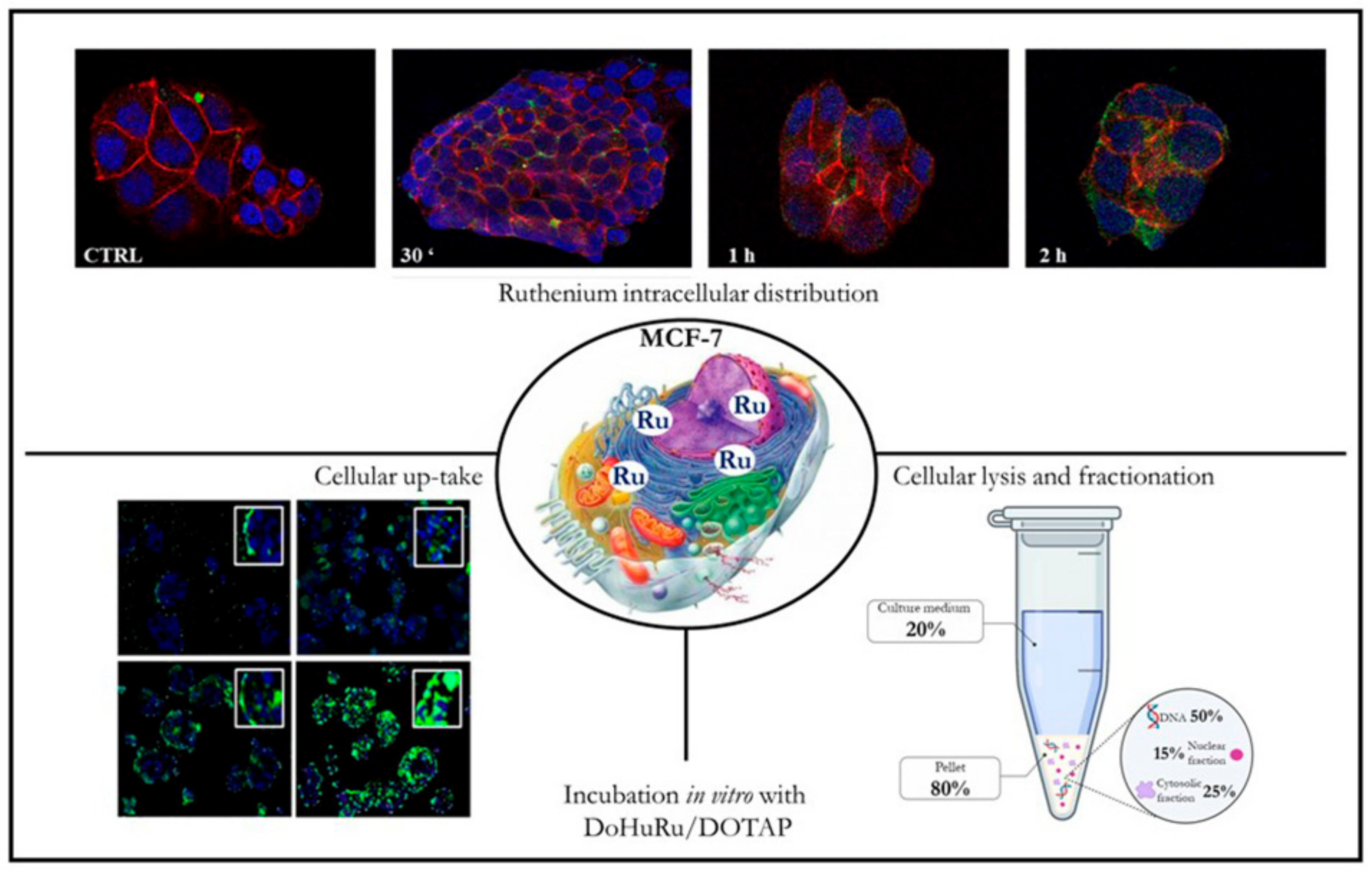{kind=link}
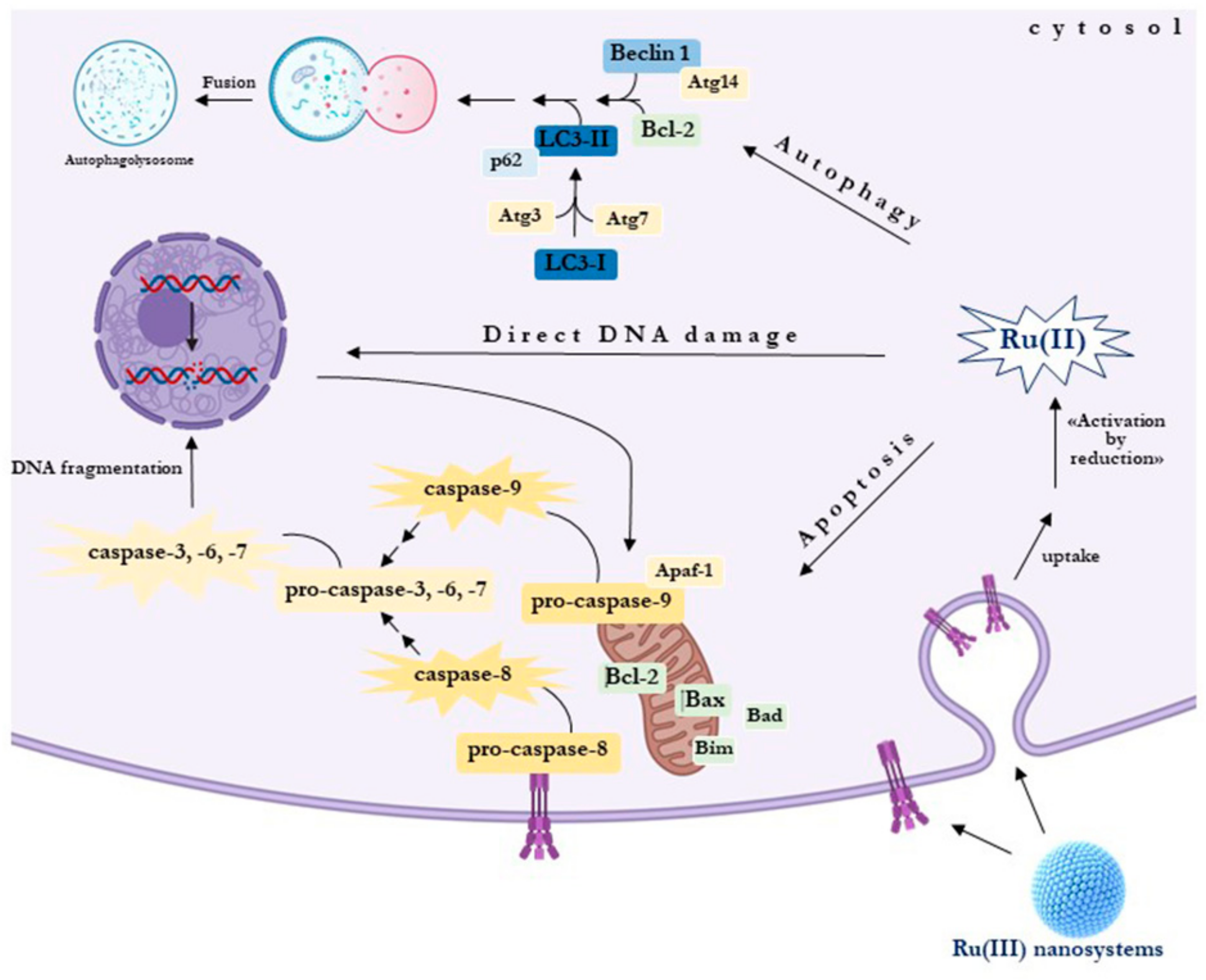{kind=link}
| IC50 (µM) | |||
|---|---|---|---|
| POPC Liposomes | DOTAP Liposomes | cDDP | |
| Breast cancer cells | |||
| ER-positive | |||
| MCF-7 | 18.9 ± 0.1 [DoHuRu] | 10.1 ± 0.1 [ToThyRu] | 17 ± 5 |
| CG-5 | 19.4 ± 0.2 [ToThyRu] | 3.3 ± 0.2 [DoHuRu] | n.a. |
| TNBC | |||
| MDA-MB-231 | 15 ± 1 [DoHuRu] | 10.8 ± 0.2 [ToThyRu] | 19 ± 4 |
| MDA-MB-436 | 37 ± 1 [DoHuRu] | 15 ± 0.2 [ToThyRu] | n.a. |
| MDA-MB-468 | 15.7 ± 0.1 [ToThyRu] | 14.2 ± 0.1 [DoHuRu] | 24 ± 1 |
| Other cancer cells | |||
| WiDr | 20 ± 8 [HoUrRu] | 12 ± 5 [HoUrRu] | n.a. |
| HeLa | 25 ± 3 [ToThyCholRu] | 34 ± 4 [ToThyCholRu] | 10.1 ± 3 |
| LN-229 | >75 [ToThyRu] | 7.7 ± 1 [ToThyRu] | n.a. |
| U87-MG | 19.8 ± 0.1 [DoHuRu] | 11.7 ± 0.1 [ToThyRu] | 11.7 ± 0.5 |
| C6 | 24 ± 5 [DoHuRu] | 34 ± 9 [DoHuRu] | 6.8 ± 0.3 |
| Ruthenium Nanosystems | Cell Line | IC50 Values (µM) | Main Results |
|---|---|---|---|
| DoHuRu/POPC | MCF-7 | 18.9 ± 0.1 | DNA fragmentation, activation of |
| pro-caspase-9, ↑Bax, ↓Bcl-2 | |||
| DoHuRu/DOTAP | MCF-7 | 10.3 ± 0.2 | DNA fragmentation, activation of |
| pro-caspase-9, ↑Bax, ↓Bcl-2, | |||
| ↑LC3-I, ↑LC3-II, ↑Beclin 1 | |||
| DoHuRu/POPC | MDA-MB-231 | 15.0 ± 1 | DNA fragmentation, activation of |
| pro-caspase-9, activation of pro- | |||
| caspase-3, ↑Bax, ↓Bcl-2 | |||
| DoHuRu/DOTAP | MDA-MB-231 | 12.1 ± 0.3 | DNA fragmentation, activation of |
| pro-caspase-9, activation of pro- | |||
| caspase-8, activation of pro-caspase-3, | |||
| ↑Bax, ↓Bcl-2, ↑LC3-I, ↑LC3-II, ↑Beclin 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferraro, M.G.; Piccolo, M.; Misso, G.; Maione, F.; Montesarchio, D.; Caraglia, M.; Paduano, L.; Santamaria, R.; Irace, C. Breast Cancer Chemotherapeutic Options: A General Overview on the Preclinical Validation of a Multi-Target Ruthenium(III) Complex Lodged in Nucleolipid Nanosystems. Cells 2020, 9, 1412. https://doi.org/10.3390/cells9061412
Ferraro MG, Piccolo M, Misso G, Maione F, Montesarchio D, Caraglia M, Paduano L, Santamaria R, Irace C. Breast Cancer Chemotherapeutic Options: A General Overview on the Preclinical Validation of a Multi-Target Ruthenium(III) Complex Lodged in Nucleolipid Nanosystems. Cells. 2020; 9(6):1412. https://doi.org/10.3390/cells9061412
Chicago/Turabian StyleFerraro, Maria Grazia, Marialuisa Piccolo, Gabriella Misso, Francesco Maione, Daniela Montesarchio, Michele Caraglia, Luigi Paduano, Rita Santamaria, and Carlo Irace. 2020. "Breast Cancer Chemotherapeutic Options: A General Overview on the Preclinical Validation of a Multi-Target Ruthenium(III) Complex Lodged in Nucleolipid Nanosystems" Cells 9, no. 6: 1412. https://doi.org/10.3390/cells9061412
APA StyleFerraro, M. G., Piccolo, M., Misso, G., Maione, F., Montesarchio, D., Caraglia, M., Paduano, L., Santamaria, R., & Irace, C. (2020). Breast Cancer Chemotherapeutic Options: A General Overview on the Preclinical Validation of a Multi-Target Ruthenium(III) Complex Lodged in Nucleolipid Nanosystems. Cells, 9(6), 1412. https://doi.org/10.3390/cells9061412