Transposon-Based CAR T Cells in Acute Leukemias: Where Are We Going?

 ,
,
Abstract
:1. Background on CAR T-Cell Therapy in Acute Leukemias: Gene Transfer Strategies, Clinical Settings and Accessibility
2. Preclinical Evolution of Sleeping Beauty Vector Design
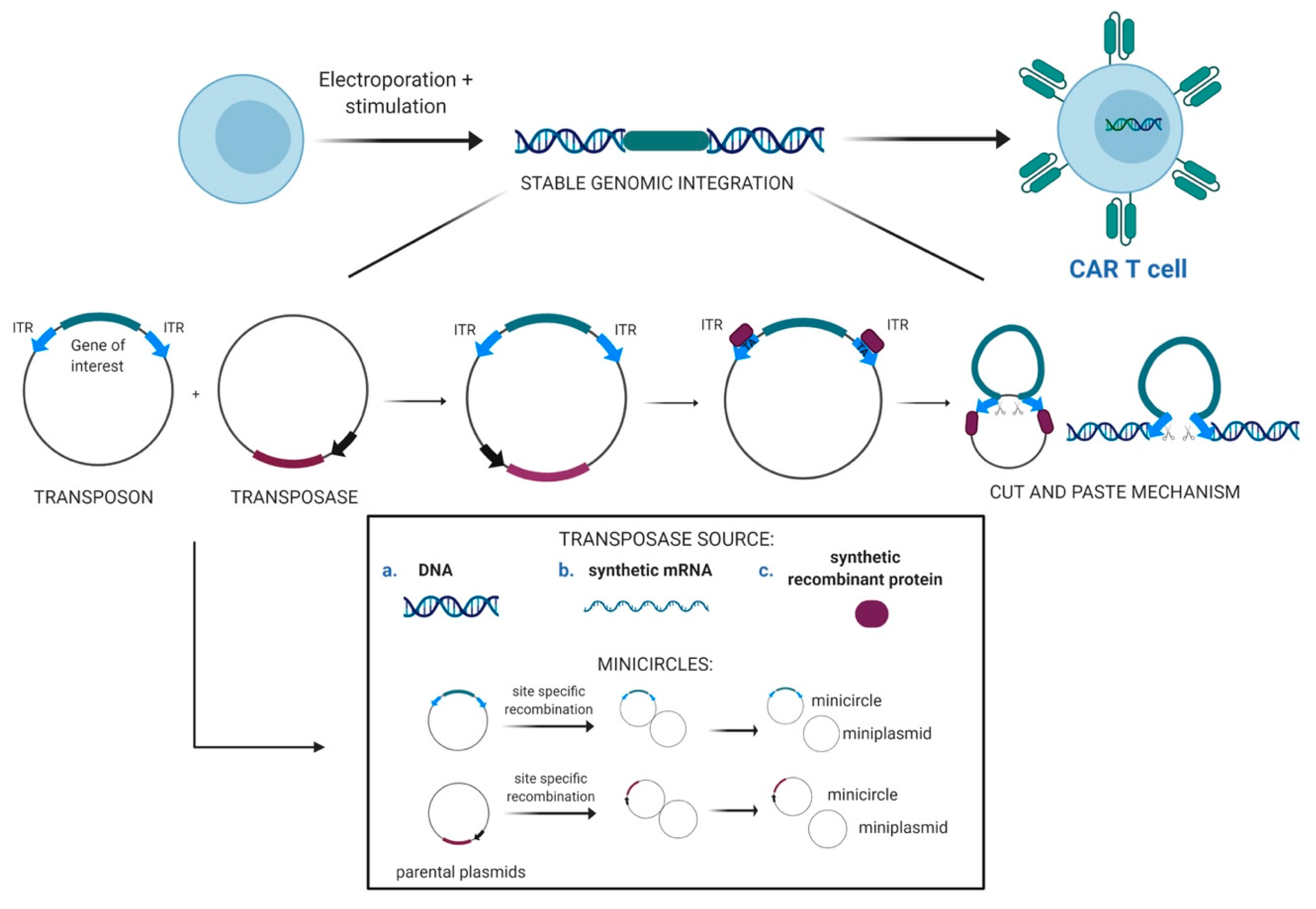
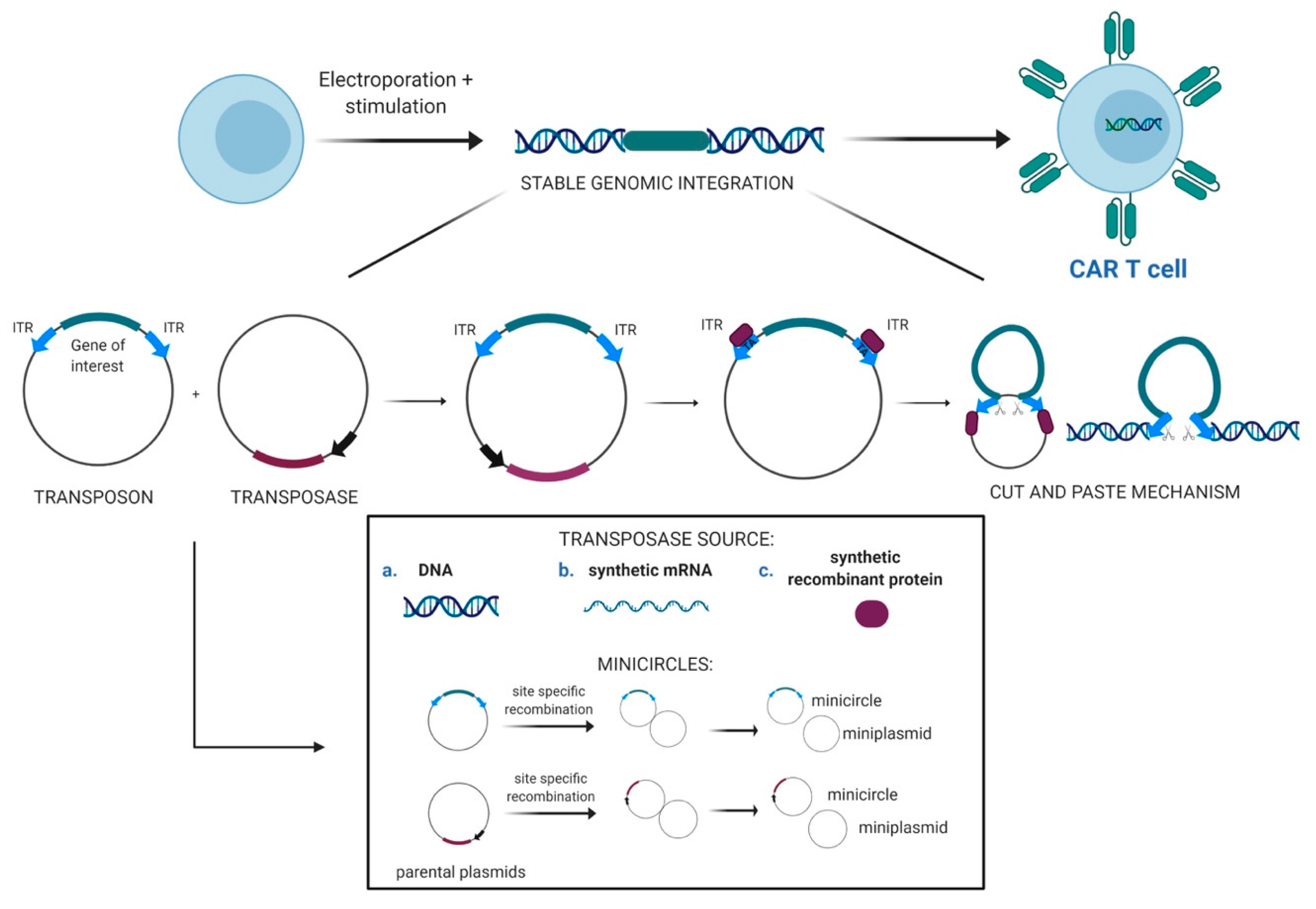
2.1. Transposons as Vectors for Gene Therapy
2.2. Evolution of the SB Transposon System and Delivery Technologies
2.3. CAR T-Cells Engineered With Sleeping Beauty Transposon Vectors
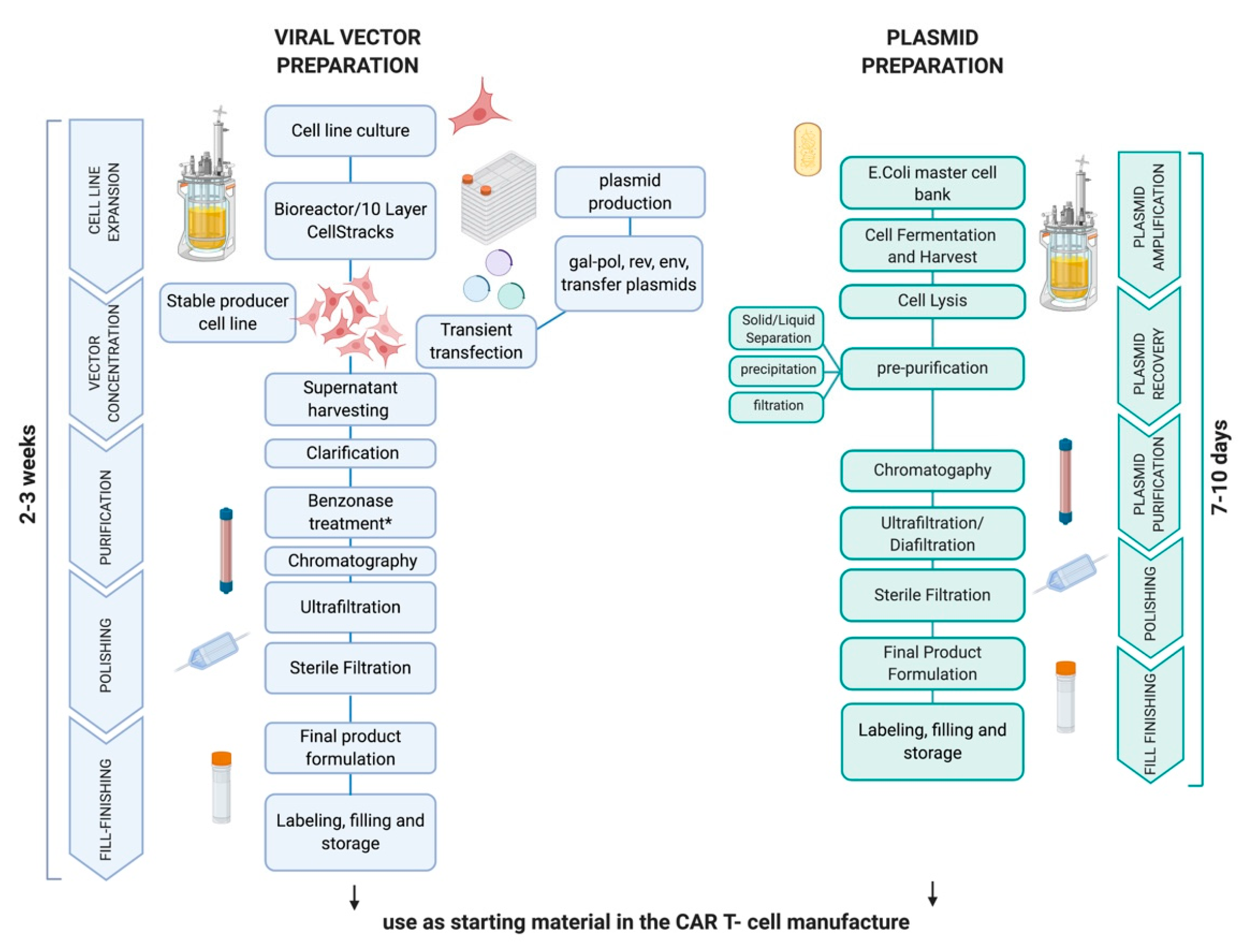
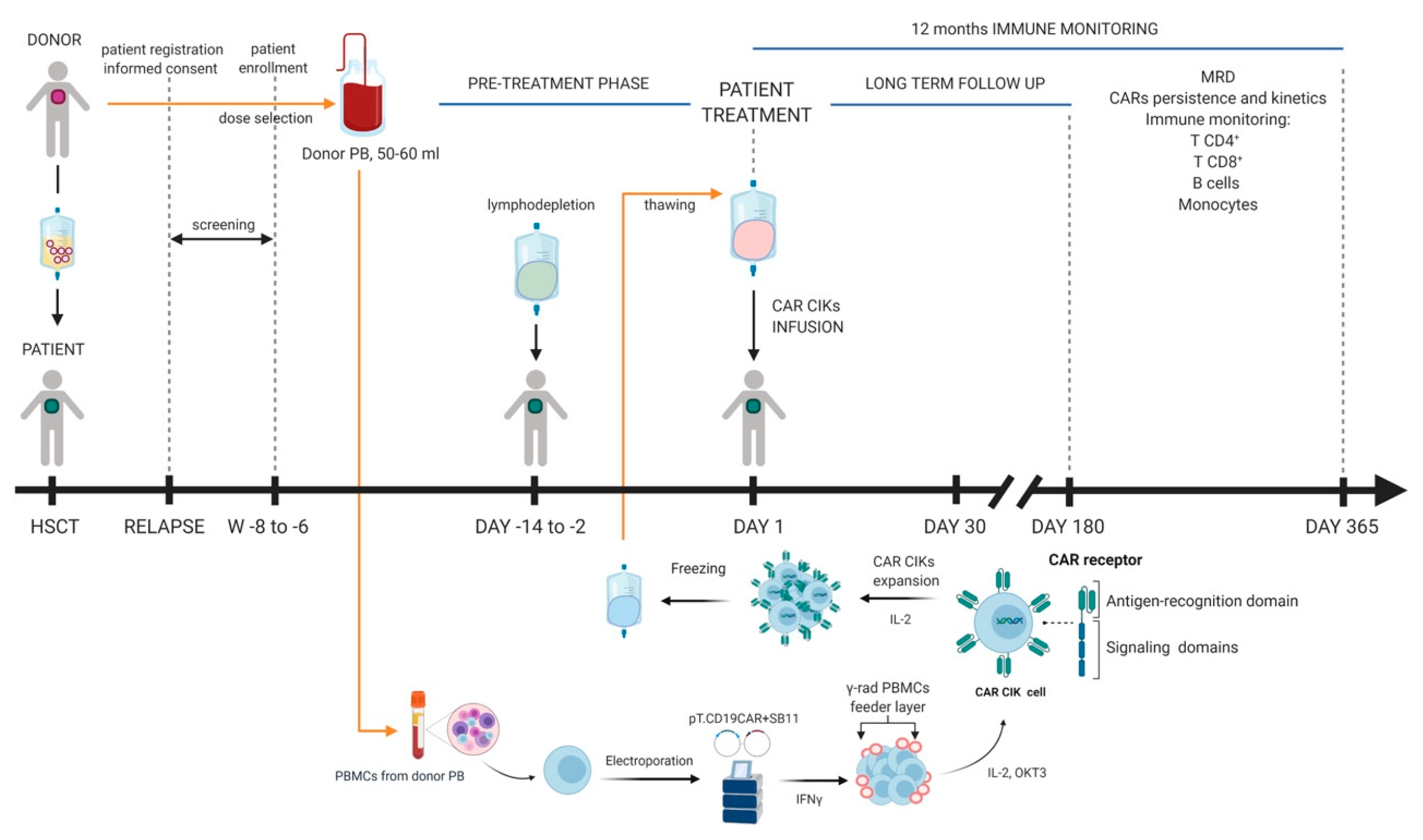
3. Clinical Grade Production of SB-Engineered CAR T-Cells: Cell Processing and Release Testing
4. Early Clinical Experiences
5. Future Perspectives of Non-Viral CAR T Platforms
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dickinson, A.M.; Norden, J.; Li, S.; Hromadnikova, I.; Schmid, C.; Schmetzer, H.; Jochem-Kolb, H. Graft-versus-Leukemia Effect Following Hematopoietic Stem Cell Transplantation for Leukemia. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Blazar, B.R.; Murphy, W.J.; Abedi, M. Advances in graft-versus-host disease biology and therapy. Nat. Rev. Immunol. 2012, 12, 443–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; A Feldman, S.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+: CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [Green Version]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. New Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+and CD4+CD19-specific chimeric antigen receptor–modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J.N.; Somerville, R.P.; Lu, T.; Yang, J.C.; Sherry, R.M.; Feldman, S.A.; McIntyre, L.; Bot, A.; Rossi, J.; Lam, N.; et al. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma after Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol. Ther. 2017, 25, 2245–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudno, J.N.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2017, 15, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor–Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. New Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Frigault, M.J.; Maus, M.V. State of the art in CAR T cell therapy for CD19+ B cell malignancies. J. Clin. Investig. 2020, 130, 1586–1594. [Google Scholar] [CrossRef]
- Ghani, K.; Wang, X.; de Campos-Lima, P.O.; Olszewska, M.; Kamen, A.; Rivière, I.; Caruso, M. Efficient Human Hematopoietic Cell Transduction Using RD114- and GALV-Pseudotyped Retroviral Vectors Produced in Suspension and Serum-Free Media. Hum. Gene Ther. 2009, 20, 966–974. [Google Scholar] [CrossRef] [Green Version]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Bedrosian, T.A.; Verma, I.M.; Trono, D. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral vectors: A look back and ahead on gene transfer technology. New Microbiol. 2013, 36, 1–22. [Google Scholar] [PubMed]
- Throm, R.E.; Ouma, A.A.; Zhou, S.; Chandrasekaran, A.; Lockey, T.; Greene, M.; de Ravin, S.S.; Moayeri, M.; Malech, H.L.; Sorrentino, B.P.; et al. Efficient construction of producer cell lines for a SIN lentiviral vector for SCID-X1 gene therapy by concatemeric array transfection. Blood 2009, 113, 5104–5110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, J.; Schüsler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells—Challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef]
- Wang, X.; Rivière, I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther. Oncol. 2016, 3, 16015. [Google Scholar] [CrossRef] [Green Version]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular Reconstruction of Sleeping Beauty, a Tc1-like Transposon from Fish, and Its Transposition in Human Cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Hackett, P.B.; Largaespada, D.; Cooper, L.J. A Transposon and Transposase System for Human Application. Mol. Ther. 2010, 18, 674–683. [Google Scholar] [CrossRef]
- Boehme, P.; Doerner, J.; Solanki, M.; Jing, L.; Zhang, W.; Ehrhardt, A. The sleeping beauty transposon vector system for treatment of rare genetic diseases: An unrealized hope? Curr. Gene Ther. 2015, 15, 255–265. [Google Scholar] [CrossRef]
- Ivics, Z.; Izsvák, Z. The expanding universe of transposon technologies for gene and cell engineering. Mob. DNA 2010, 1, 25. [Google Scholar] [CrossRef] [Green Version]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef]
- Woodard, L.E.; Wilson, M.H. PiggyBac-ing models and new therapeutic strategies. Trends Biotechnol. 2015, 33, 525–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izsvák, Z.; Khare, D.; Behlke, J.; Heinemann, U.; Plasterk, R.H.; Ivics, Z. Involvement of a Bifunctional, Paired-like DNA-binding Domain and a Transpositional Enhancer inSleeping BeautyTransposition. J. Boil. Chem. 2002, 277, 34581–34588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Pryputniewicz-Dobrinska, D.; Nagy, E.E.; Kaufman, C.D.; Singh, M.; Yant, S.; Wang, J.; Dalda, A.; Kay, M.A.; Ivics, Z.; et al. Regulated complex assembly safeguards the fidelity of Sleeping Beauty transposition. Nucleic Acids Res. 2016, 45, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.C.-Y.; Meir, Y.-J.J.; Coates, C.J.; Handler, A.M.; Pelczar, P.; Moisyadi, S.; Kaminski, J.M. piggyBac is a flexible and highly active transposon as compared to Sleeping Beauty, Tol2, and Mos1 in mammalian cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15008–15013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zayed, H.; Izsvák, Z.; Walisko, O.; Ivics, Z.; Izsv, Z. Aacute Development of Hyperactive Sleeping Beauty Transposon Vectors by Mutational Analysis. Mol. Ther. 2004, 9, 292–304. [Google Scholar] [CrossRef]
- Turchiano, G.; Latella, M.C.; Döring, A.G.; Cattoglio, C.; Mavilio, F.; Izsvák, Z.; Ivics, Z.; Recchia, A. Correction: Genomic Analysis of Sleeping Beauty Transposon Integration in Human Somatic Cells. PLoS ONE 2020, 15, e0228703. [Google Scholar] [CrossRef] [Green Version]
- Izsvák, Z. Efficient stable gene transfer into human cells by the Sleeping Beauty transposon vectors. Methods 2009, 49, 287–297. [Google Scholar] [CrossRef]
- Wilber, A.; Wangensteen, K.J.; Chen, Y.; Zhuo, L.; Frandsen, J.L.; Bell, J.B.; Chen, Z.J.; Ekker, S.C.; McIvor, R.S.; Wang, X. Messenger RNA as a Source of Transposase for Sleeping Beauty Transposon–mediated Correction of Hereditary Tyrosinemia Type I. Mol. Ther. 2007, 15, 1280–1287. [Google Scholar] [CrossRef]
- Querques, I.; Mades, A.; Zuliani, C.; Miskey, C.; Alb, M.; Grueso, E.; Machwirth, M.; Rausch, T.; Einsele, H.; Ivics, Z.; et al. A highly soluble Sleeping Beauty transposase improves control of gene insertion. Nat. Biotechnol. 2019, 37, 1502–1512. [Google Scholar] [CrossRef]
- Cui, Z.; Geurts, A.M.; Liu, G.; Kaufman, C.D.; Hackett, P.B. Structure–Function Analysis of the Inverted Terminal Repeats of the Sleeping Beauty Transposon. J. Mol. Boil. 2002, 318, 1221–1235. [Google Scholar] [CrossRef]
- Geurts, A.M.; Yang, Y.; Clark, K.; Liu, G.; Cui, Z.; Dupuy, A.J.; Bell, J.B.; Largaespada, D.A.; Hackett, P.B. Gene transfer into genomes of human cells by the sleeping beauty transposon system. Mol. Ther. 2003, 8, 108–117. [Google Scholar] [CrossRef]
- Kowarz, E.; Loescher, D.; Marschalek, R.; Löscher, D. Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol. J. 2015, 10, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Mátés, L.; Chuah, M.K.L.; Belay, E.; Jerchow, B.; Manoj, N.; Acosta-Sanchez, A.; Grzela, D.; Schmitt, A.; Becker, K.; Matrai, J.; et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009, 41, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.C.; Popplewell, L.; di Giusto, D.; Kalos, M.; Raubitschek, A.; Forman, S.J. A First-In-Human Clinical Trial of Adoptive Therapy Using CD19-Specific Chimeric Antigen Receptor Re-Directed T-Cells for Recurrent/Refractory Follicular Lymphoma. Blood 2007, 110, 288. [Google Scholar] [CrossRef]
- Huang, X.; Guo, H.; Kang, J.; Choi, S.; Zhou, T.; Tammana, S.; Lees, C.J.; Li, Z.; Milone, M.; Levine, B.L.; et al. Sleeping Beauty Transposon-mediated Engineering of Human Primary T Cells for Therapy of CD19+ Lymphoid Malignancies. Mol. Ther. 2008, 16, 580–589. [Google Scholar] [CrossRef]
- Singh, H.; Manuri, P.R.; Olivares, S.; Dara, N.; Dawson, M.J.; Huls, H.; Hackett, P.B.; Kohn, D.B.; Shpall, E.J.; Champlin, R.E.; et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008, 68, 2961–2971. [Google Scholar] [CrossRef] [Green Version]
- Deniger, D.C.; Yu, J.; Huls, M.H.; Figliola, M.J.; Mi, T.; Maiti, S.N.; Widhopf, G.F.; Hurton, L.V.; Thokala, R.; Singh, H.; et al. Sleeping Beauty Transposition of Chimeric Antigen Receptors Targeting Receptor Tyrosine Kinase-Like Orphan Receptor-1 (ROR1) into Diverse Memory T-Cell Populations. PLoS ONE 2015, 10, e0128151. [Google Scholar] [CrossRef]
- Magnani, C.F.; Turazzi, N.; Benedicenti, F.; Calabria, A.; Tenderini, E.; Tettamanti, S.; Attianese, G.M.G.; Cooper, L.J.; Aiuti, A.; Montini, E.; et al. Immunotherapy of acute leukemia by chimeric antigen receptor-modified lymphocytes using an improved Sleeping Beauty transposon platform. Oncotarget 2016, 7, 51581–51597. [Google Scholar] [CrossRef]
- Rambaldi, A.; Biagi, E.; Bonini, C.; Biondi, A.; Introna, M.; Bonini, C. Cell-based strategies to manage leukemia relapse: Efficacy and feasibility of immunotherapy approaches. Leukemia 2014, 29, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Introna, M.; Borleri, G.; Conti, E.; Franceschetti, M.; Barbui, A.M.; Broady, R.; Dander, E.; Gaipa, G.; D’Amico, G.; Biagi, E.; et al. Repeated infusions of donor-derived cytokine-induced killer cells in patients relapsing after allogeneic stem cell transplantation: A phase I study. Haematology 2007, 92, 952–959. [Google Scholar] [CrossRef]
- Schmeel, F.; Schmeel, L.C.; Gast, S.-M.; Schmidt-Wolf, I.G. Adoptive Immunotherapy Strategies with Cytokine-Induced Killer (CIK) Cells in the Treatment of Hematological Malignancies. Int. J. Mol. Sci. 2014, 15, 14632–14648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanayake, S.; Bilmon, I.; Bishop, D.; Dubosq, M.-C.; Blyth, E.; Clancy, L.; Gottlieb, D.; Micklethwaite, K. Low-cost generation of Good Manufacturing Practice–grade CD19-specific chimeric antigen receptor–expressing T cells using piggyBac gene transfer and patient-derived materials. Cytotherapy 2015, 17, 1251–1267. [Google Scholar] [CrossRef] [PubMed]
- Klös, S.; Oberschmidt, O.; Morgan, M.A.; Dahlke, J.; Arseniev, L.; Huppert, V.; Granzin, M.; Gardlowski, T.; Matthies, N.; Soltenborn, S.; et al. Optimization of Human NK Cell Manufacturing: Fully Automated Separation, ImprovedEx VivoExpansion Using IL-21 with Autologous Feeder Cells, and Generation of Anti-CD123-CAR-Expressing Effector Cells. Hum. Gene Ther. 2017, 28, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Holstein, M.; Mesa-Nuñez, C.; Miskey, C.; Almarza, E.; Poletti, V.; Schmeer, M.; Grueso, E.; Flores, J.C.O.; Kobelt, D.; Walther, W.; et al. Efficient Non-viral Gene Delivery into Human Hematopoietic Stem Cells by Minicircle Sleeping Beauty Transposon Vectors. Mol. Ther. 2018, 26, 1137–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monjezi, R.; Miskey, C.; Gogishvili, T.; Schleef, M.; Schmeer, M.; Einsele, H.; Ivics, Z.; Hudecek, M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2016, 31, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Shankar, R.; Schmeer, M.; Schleef, M. Minicircles: Next-generation gene vectors. Cell Gene Ther. Insights 2017, 3, 285–300. [Google Scholar] [CrossRef]
- Chicaybam, L.; Abdo, L.; Viegas, M.; Marques, L.V.C.; de Sousa, P.; Batista-Silva, L.R.; Alves-Monteiro, V.; Bonecker, S.; Monte-Mór, B.; Bonamino, M.H. Transposon-mediated generation of CAR-T cells shows efficient anti B-cell leukemia response after ex vivo expansion. Gene Ther. 2020, 27, 85–95. [Google Scholar] [CrossRef]
- Chan, T.; Gallagher, J.; Cheng, N.-L.; Carvajal-Borda, F.; Plummer, J.; Govekung, A.; Barrett, J.A.; Khare, P.D.; Cooper, L.J.N.; Shah, R.R. CD19-Specific Chimeric Antigen Receptor-Modified T Cells with Safety Switch Produced Under Point-of-Care Using the Sleeping Beauty System for the Very Rapid Manufacture and Treatment of B-Cell Malignancies. Blood 2017, 130, 1324. [Google Scholar] [CrossRef]
- Abdo, L.D.M.; Barros, L.R.C.; Viegas, M.S.; Marques, L.V.C.; Ferreira, P.D.S.; Chicaybam, L.; Bonamino, M.H. Development of CAR-T cell therapy for B-ALL using a point-of-care approach. OncoImmunology 2020, 9, 1752592. [Google Scholar] [CrossRef] [Green Version]
- Roddie, C.; O’Reilly, M.; Pinto, J.D.A.; Vispute, K.; Lowdell, M. Manufacturing chimeric antigen receptor T cells: Issues and challenges. Cytotherapy 2019, 21, 327–340. [Google Scholar] [CrossRef]
- Lamers, C.H.J.; van Elzakker, P.; A Luider, B.; van Steenbergen, S.C.L.; Sleijfer, S.; Debets, R.; Gratama, J.W. Retroviral vectors for clinical immunogene therapy are stable for up to 9 years. Cancer Gene Ther. 2008, 15, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Przybylowski, M.; Hakakha, A.; Stefanski, J.; Hodges, J.; Sadelain, M.; Rivière, I. Production scale-up and validation of packaging cell clearance of clinical-grade retroviral vector stocks produced in Cell Factories. Gene Ther. 2005, 13, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Olszewska, M.; Qu, J.; Wasielewska, T.; Bartido, S.; Hermetet, G.; Sadelain, M.; Rivière, I. Large-scale Clinical-grade Retroviral Vector Production in a Fixed-Bed Bioreactor. J. Immunother. 2015, 38, 127–135. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2016, 4, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Narayanavari, S.; Izsvák, Z. Sleeping Beauty transposon vectors for therapeutic applications: Advances and challenges. Cell Gene Ther. Insights 2017, 3, 131–158. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Figliola, M.J.; Dawson, M.J.; Huls, H.; Olivares, S.; Switzer, K.; Mi, T.; Maiti, S.; Kebriaei, P.; Lee, D.-J.; et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res. 2011, 71, 3516–3527. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Huls, H.; Kebriaei, P.; Cooper, L.J.N. A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol. Rev. 2014, 257, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Huls, M.H.; Figliola, M.J.; Dawson, M.J.; Olivares, S.; Kebriaei, P.; Shpall, E.J.; Champlin, R.E.; Singh, H.; Cooper, L.J.N. Clinical application of Sleeping Beauty and artificial antigen presenting cells to genetically modify T cells from peripheral and umbilical cord blood. J. Vis. Exp. 2013, e50070. [Google Scholar] [CrossRef]
- Singh, H.; Figliola, M.J.; Dawson, M.J.; Olivares, S.; Zhang, L.; Yang, G.; Maiti, S.; Manuri, P.; Senyukov, V.; Jena, B.; et al. Manufacture of Clinical-Grade CD19-Specific T Cells Stably Expressing Chimeric Antigen Receptor Using Sleeping Beauty System and Artificial Antigen Presenting Cells. PLoS ONE 2013, 8, e64138. [Google Scholar] [CrossRef]
- Gaipa, G.; Introna, M.; Golay, J.; Nolli, M.L.; Vallanti, G.; Parati, E.; Giordano, R.; Romagnoli, L.; Melazzini, M.; Biondi, A.; et al. Development of advanced therapies in Italy: Management models and sustainability in six Italian cell factories. Cytotherapy 2016, 18, 481–486. [Google Scholar] [CrossRef]
- Magnani, C.F.; Gaipa, G.; Belotti, D.; Matera, G.; Tettamanti, S.; Cabiati, B.; Buracchi, C.; Fazio, G.; Zaninelli, S.; Rigamonti, S.; et al. Donor-Derived CD19 CAR Cytokine Induced Killer (CIK) Cells Engineered with Sleeping Beauty Transposon for Relapsed B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood 2019, 134, 200. [Google Scholar] [CrossRef]
- Levine, B.L. Performance-enhancing drugs: Design and production of redirected chimeric antigen receptor (CAR) T cells. Cancer Gene Ther. 2015, 22, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Somerville, R.P.; Dudley, M.E. Bioreactors get personal. OncoImmunology 2012, 1, 1435–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somerville, R.P.; de Villier, L.; Parkhurst, M.R.; Rosenberg, S.A.; Dudley, M.E. Clinical scale rapid expansion of lymphocytes for adoptive cell transfer therapy in the WAVE® bioreactor. J. Transl. Med. 2012, 10, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Sabatino, M.; Somerville, R.; Wilson, J.R.; Dudley, M.E.; Ascierto, P.A.; Rosenberg, S.A. Simplified method of the growth of human tumor infiltrating lymphocytes in gas-permeable flasks to numbers needed for patient treatment. J. Immunother. 2012, 35, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajgain, P.; Mucharla, R.; Wilson, J.; Welch, D.; Anurathapan, U.; Liang, B.; Lü, X.; Ripple, K.; Centanni, J.M.; Hall, C.; et al. Optimizing the production of suspension cells using the G-Rex “M” series. Mol. Ther. Methods Clin. Dev. 2014, 1, 14015. [Google Scholar] [CrossRef]
- Abramowski-Mock, U.; Nickolay, L.; Philip, B.; Cheung, G.W.-K.; Zhan, H.; Johnston, I.C.; Kaiser, A.D.; Peggs, K.; Pule, M.; Thrasher, A.J.; et al. Automated manufacturing of chimeric antigen receptor T cells for adoptive immunotherapy using CliniMACS Prodigy. Cytotherapy 2016, 18, 1002–1011. [Google Scholar] [CrossRef]
- Zhu, F.; Shah, N.; Xu, H.; Schneider, D.; Orentas, R.; Dropulić, B.; Hari, P.; Keever-Taylor, C.A. Closed-system manufacturing of CD19 and dual-targeted CD20/19 chimeric antigen receptor T cells using the CliniMACS Prodigy device at an academic medical center. Cytotherapy 2018, 20, 394–406. [Google Scholar] [CrossRef]
- Fernández, L.; Fernández, A.; Mirones, I.; Escudero, A.; Cardoso, L.; Vela, M.; Lanzarot, D.; de Paz, R.; Leivas, A.; Gallardo, M.; et al. GMP-Compliant Manufacturing of NKG2D CAR Memory T Cells Using CliniMACS Prodigy. Front. Immunol. 2019, 10, 2361. [Google Scholar] [CrossRef] [Green Version]
- Hodge, R.; Narayanavari, S.; Izsvák, Z.; Ivics, Z. Wide Awake and Ready to Move: 20 Years of Non-Viral Therapeutic Genome Engineering with the Sleeping Beauty Transposon System. Hum. Gene Ther. 2017, 28, 842–855. [Google Scholar] [CrossRef] [Green Version]
- Srour, S.; Singh, H.; Mccarty, J.; de Groot, E.; Huls, H.; Rondon, G.; Qazilbash, M.H.; Ciurea, S.O.; Bardelli, G.; Buck, J.; et al. Long-term outcomes of Sleeping Beauty–generated CD19-specific CAR T-cell therapy for relapsed-refractory B-cell lymphomas. Blood 2020, 135, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Kebriaei, P.; Huls, H.; Neel, S.L.; Olivares, S.; Orozco, A.F.; Su, S.; Maiti, S.N.; Smith, A.; de Groot, E.; Kantarjian, H.M.; et al. Shortening the Time to Manufacture CAR+ T Cells with Sleeping Beauty System Supports T-Cell Engraftment and Anti-Tumor Effects in Patients with Refractory CD19+ Tumors. Blood 2017, 130, 2060. [Google Scholar] [CrossRef]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.-A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, 7788–7797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyjolfsdottir, H.; Eriksdotter, M.; Linderoth, B.; Lind, G.; Juliusson, B.; Kusk, P.; Almkvist, O.; Andreasen, N.; Blennow, K.; Ferreira, D.; et al. Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: Application of a second-generation encapsulated cell biodelivery device. Alzheimer’s Res. Ther. 2016, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Magnani, C.F.; Mezzanotte, C.; Cappuzzello, C.; Bardini, M.; Tettamanti, S.; Fazio, G.; Cooper, L.J.; Dastoli, G.; Cazzaniga, G.; Biondi, A.; et al. Preclinical Efficacy and Safety of CD19CAR Cytokine-Induced Killer Cells Transfected with Sleeping Beauty Transposon for the Treatment of Acute Lymphoblastic Leukemia. Hum. Gene Ther. 2018, 29, 602–613. [Google Scholar] [CrossRef]
- Introna, M.; Lussana, F.; Algarotti, A.; Gotti, E.; Valgardsdottir, R.; Micò, C.; Grassi, A.; Pavoni, C.; Ferrari, M.L.; Delaini, F.; et al. Phase II Study of Sequential Infusion of Donor Lymphocyte Infusion and Cytokine-Induced Killer Cells for Patients Relapsed after Allogeneic Hematopoietic Stem Cell Transplantation. Boil. Blood Marrow Transplant. 2017, 23, 2070–2078. [Google Scholar] [CrossRef] [Green Version]
- Magnani, C.F.; Biondi, A.; Biagi, E. Donor-derived CD19-targeted T cells in allogeneic transplants. Curr. Opin. Hematol. 2015, 22, 497–502. [Google Scholar] [CrossRef]
- Gogishvili, T.; Danhof, S.; Prommersberger, S.; Rydzek, J.; Schreder, M.; Brede, C.; Einsele, H.; Hudecek, M. SLAMF7-CAR T cells eliminate myeloma and confer selective fratricide of SLAMF7+ normal lymphocytes. Blood 2017, 130, 2838–2847. [Google Scholar] [CrossRef] [Green Version]
- SLAMF7-CAR T Cell Treatment of Multiple Myeloma Patients. Available online: https://www.caramba-cart.eu/ (accessed on 30 April 2020).
- Rotiroti, M.C.; Buracchi, C.; Arcangeli, S.; Galimberti, S.; Valsecchi, M.G.; Perriello, V.M.; Rasko, T.; Alberti, G.; Magnani, C.F.; Cappuzzello, C.; et al. Targeting CD33 in chemoresistant AML patient-derived xenografts by CAR-CIK cells engineered with an optimized Sleeping Beauty transposon version. Mol. Ther. 2020, 11. [Google Scholar] [CrossRef]
- Bishop, D.; Xu, N.; Tse, B.; O’Brien, T.A.; Gottlieb, D.; Dolnikov, A.; Micklethwaite, K. PiggyBac-Engineered T Cells Expressing CD19-Specific CARs that Lack IgG1 Fc Spacers Have Potent Activity against B-ALL Xenografts. Mol. Ther. 2018, 26, 1883–1895. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.; Clancy, L.; Burgess, J.; Mathew, G.; Atkins, E.; Advic, S.; Maddock, K.; Street, J.; Moezzi, L.; Simms, R.; et al. Matched sibling donor-derived piggybac CAR19 T cells induce remission of relapsed/refractory CD19+ malignancy following haematopoietic stem cell transplant. Cytotherapy 2019, 21, 9. [Google Scholar] [CrossRef]
- Costello, C.L.; Gregory, T.K.; Ali, S.A.; Berdeja, J.G.; Patel, K.K.; Shah, N.D.; Ostertag, E.; Martin, C.; Ghoddusi, M.; Shedlock, D.J.; et al. Phase 2 Study of the Response and Safety of P-Bcma-101 CAR-T Cells in Patients with Relapsed/Refractory (r/r) Multiple Myeloma (MM) (PRIME). Blood 2019, 134, 3184. [Google Scholar] [CrossRef]
- Papathanasiou, M.M.; Stamatis, C.; Lakelin, M.; Farid, S.; Titchener-Hooker, N.; Shah, N. Autologous CAR T-cell therapies supply chain: Challenges and opportunities? Cancer Gene Ther. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Dong, X.; Xu, Q.; Zhu, L.; Wang, F.; Hou, Y.; Chao, C.-C. Therapeutic potential of CRISPR/Cas9 gene editing in engineered T-cell therapy. Cancer Med. 2019, 8, 4254–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charrot, S.; Hallam, S. CAR-T Cells. HemaSphere 2019, 3, e188. [Google Scholar] [CrossRef]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2018, 120, 26–37. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
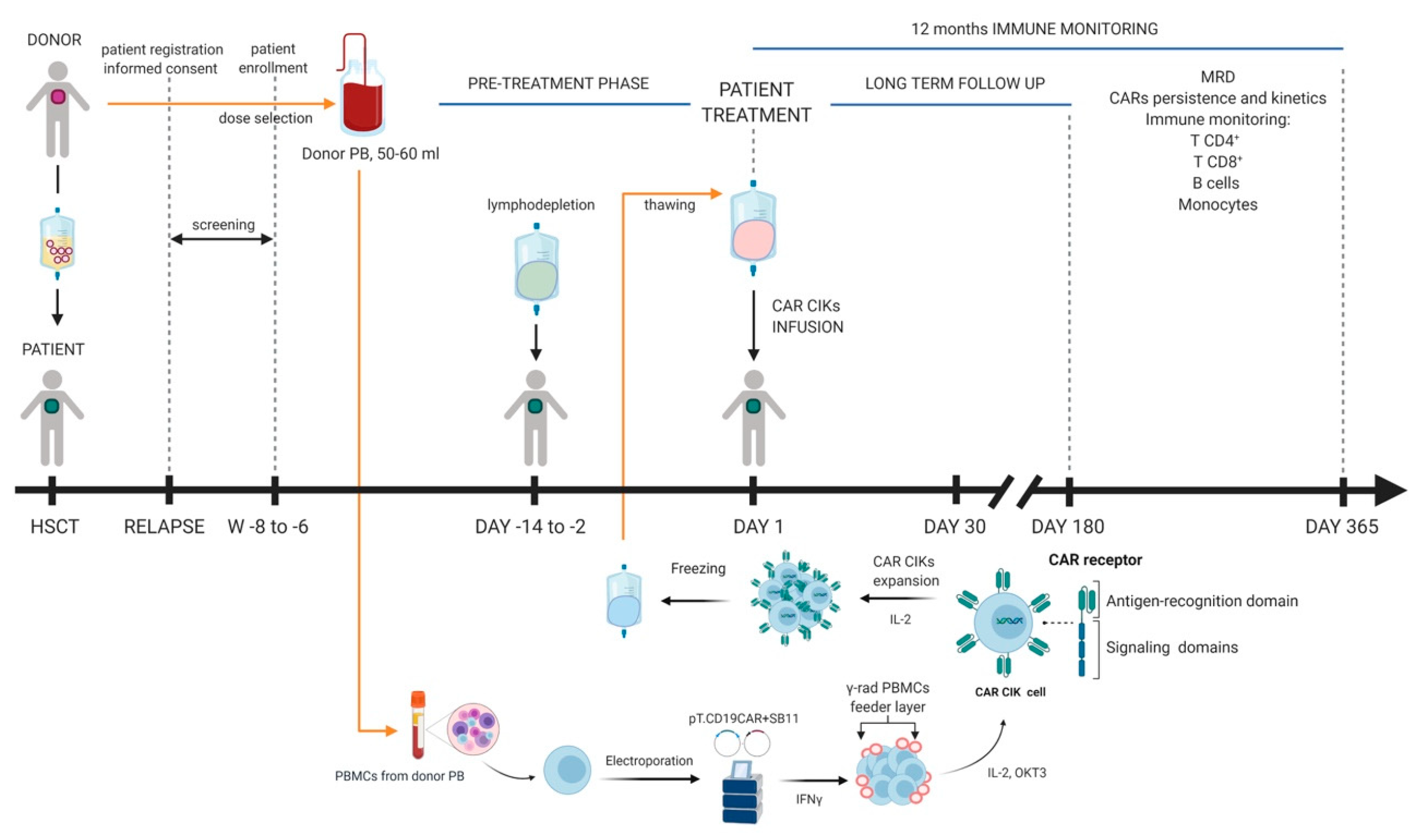{kind=link}
| Significance | Test | Methods |
|---|---|---|
| Quality | Cell Viability | Trypan blue dye exclusion; Flow cytometry |
| Purity/identity | %CD3+ cells | Flow cytometry |
| Identity | %CD3+/CAR+ cells | Flow cytometry |
| Potency | Cytotoxicity/Cytokine production toward target cell lines | Flow cytometry/ detection of cytokines |
| Safety | Mycoplasma | Culture assay/PCR assay |
| Safety | Bacterial sterility (aerobic, anaerobic and fungal testing) | BacT/ALERT 3D |
| Safety/purity | Endotoxin | Different methods |
| Purity | Contamination of beads, cytokines, serum, etc | Different methods |
| Safety | Vector Copy number/cell | PCR |
| Safety | Transposase detection (only for transposon-based CAR T-cells) | PCR |
| Safety | Replication competent retroviruses/lentiviruses (RCRs/RCLs) (only for viral-based CAR T-cells) | PCR |
| Disease | Clinical Trial ID/Alias | Location | Population Studied; Phase | Transgene | Vector | Status | Reference |
|---|---|---|---|---|---|---|---|
| B-cell lymphoma | NCT00968760 | MDACC 8 (Texas, USA) | Adult; Phase 1 | Autologous anti-CD19.CD28.z CAR T-cells | SB | Active, not recruiting | [30] |
| CD19+ lymphoma, B-ALL 1 | NCT01497184 | MDACC | Children and adult; Phase 1 | Allogeneic anti-CD19.CD28.z CAR T-cells | SB | Active, not recruiting | [30] |
| CD19+ lymphoma,B-ALL | NCT01492036 | MDACC | Children and adult; Phase 1 | Long-term follow-up | SB | Recruiting | [81] |
| B-CLL 2 | NCT01653717 | MDACC | Adult; Phase 1 | Allogeneic anti-CD19.CD28.z CAR T-cells | SB | Completed | |
| CD19+ lymphoma, B-ALL, B-CLL | NCT02529813 | MDACC | Children and adult; Phase 1 | Autologous anti-CD19.CD28.z CAR T-cells | SB | Active, not recruiting | [82] |
| B-ALL | NCT03389035 | MBBM 9/PGXXIII 10 | Children and adult; Phase 1 and 2 | Allogeneic anti-CD19.CD28.OX40.z CAR T-cells | SB | Recruiting | [71] |
| B-ALL, B-cell lymphoma, B-CLL | NCT03579888 | MDACC | Adult; Phase 1 | CD19.CD8.CD28.CD3.zCAR-mbIL15-HER1t T-cells | SB | Not yet recruiting | [58] |
| GBM 3, NSCLC 4, Breast Cancer, GI 5/GU 6, Ovarian Cancer | NCT04102436 | NCI 11 (Maryland, USA) | Adult; Phase 2 | Autologous neoantigen-specific TCR T-cells | SB | Recruiting | [83] |
| Alzheimer’s Disease | NCT01163825 | KU 12 (Sweden) | Adult; Phase 1 | Encapsulated Cell Biodelivery of Nerve Growth Factor | SB | Unknown | [84] |
| MPS IH 7 | NCT04284254 | MCC 13 (USA) | Adult; Phase 1 and 2 | Autologous IDUA plasmablasts | SB | Not yet recruiting | |
| B-ALL, B-cell lymphoma, B-CLL | The CARTELL Study | WH, WHC, SCH 14 (Australia) | Children and adult; Phase 1 | Allogeneic anti-CD19 CAR T-cells | PB 17 | Recruiting | |
| B-ALL | UMIN000030984 | NUG 15 (Japan) | Children and adult; Phase 1 | Autologous anti-CD19 CAR T-cells | PB | Recruiting | |
| B-ALL, B-cell lymphoma | NCT04289220 | YAHKMU 16 (China) | Adult; Phase 1 | anti-CD19.CD28.41BB.z CAR T-cells | PB | Not yet recruiting |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magnani, C.F.; Tettamanti, S.; Alberti, G.; Pisani, I.; Biondi, A.; Serafini, M.; Gaipa, G. Transposon-Based CAR T Cells in Acute Leukemias: Where Are We Going? Cells 2020, 9, 1337. https://doi.org/10.3390/cells9061337
Magnani CF, Tettamanti S, Alberti G, Pisani I, Biondi A, Serafini M, Gaipa G. Transposon-Based CAR T Cells in Acute Leukemias: Where Are We Going? Cells. 2020; 9(6):1337. https://doi.org/10.3390/cells9061337
Chicago/Turabian StyleMagnani, Chiara F., Sarah Tettamanti, Gaia Alberti, Ilaria Pisani, Andrea Biondi, Marta Serafini, and Giuseppe Gaipa. 2020. "Transposon-Based CAR T Cells in Acute Leukemias: Where Are We Going?" Cells 9, no. 6: 1337. https://doi.org/10.3390/cells9061337
APA StyleMagnani, C. F., Tettamanti, S., Alberti, G., Pisani, I., Biondi, A., Serafini, M., & Gaipa, G. (2020). Transposon-Based CAR T Cells in Acute Leukemias: Where Are We Going? Cells, 9(6), 1337. https://doi.org/10.3390/cells9061337