The Network of Angiotensin Receptors in Breast Cancer


Abstract
1. Introduction
2. AT1R and AT2R Signaling
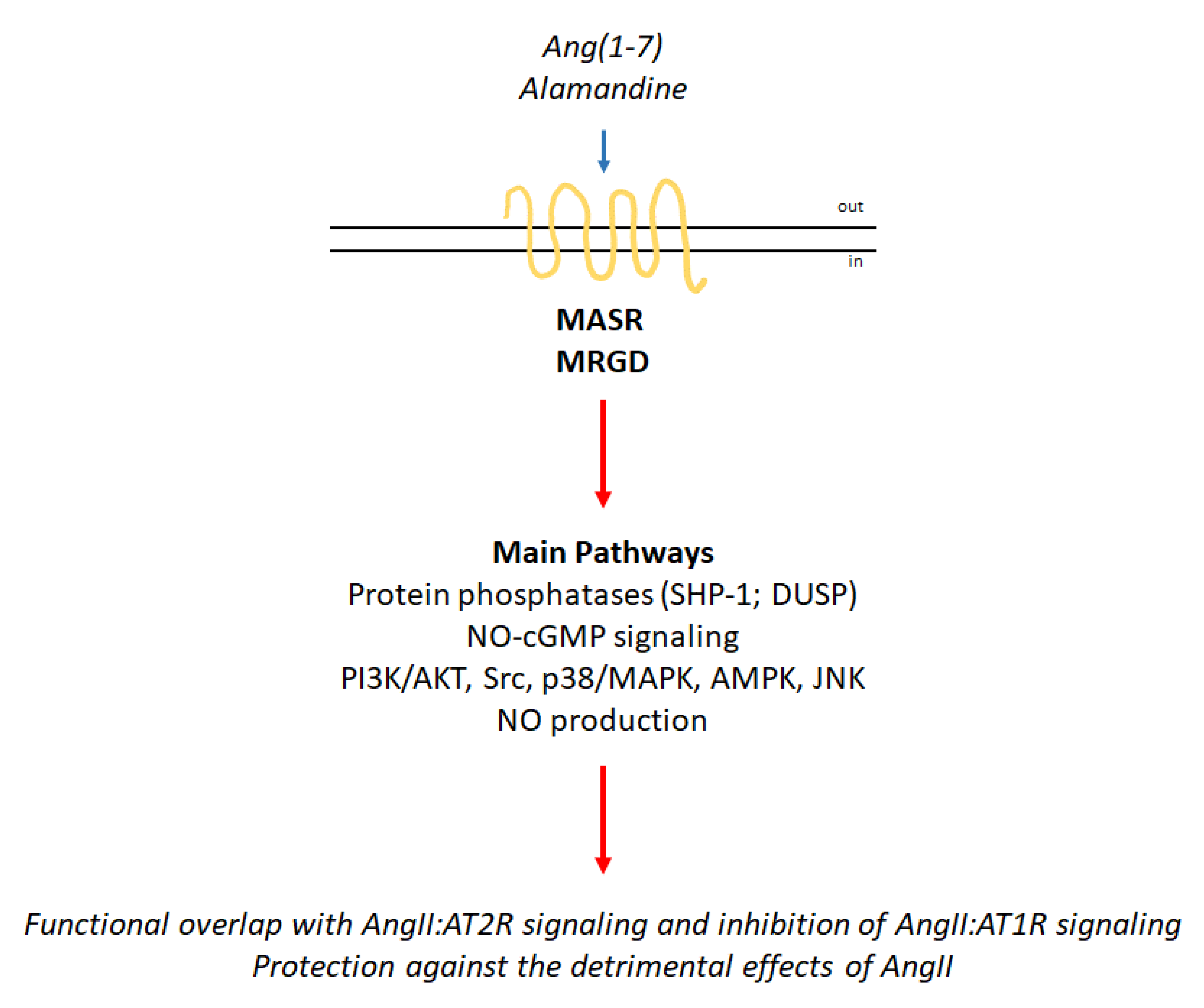
3. Proto-Oncogene Mas Receptor (MASR) Activation and Mas-Related GPCR Member D (MRGD) Signaling
4. The RAS Endocrine System in Cancer
4.1. Breast Cancer
4.1.1. AT1R and Breast Cancer
4.1.2. AT2R and Breast Cancer
4.1.3. MASR and MRGD in Breast Cancer
5. Discussion and Conclusions
Funding
Conflicts of Interest
References
- Rodrigues-Ferreira, S.; Nahmias, C. G-protein coupled receptors of the renin-angiotensin system: New targets against breast cancer? Front Pharm. 2015, 6, 24. [Google Scholar] [CrossRef]
- Crowley, S.D.; Coffman, T.M. Recent advances involving the renin-angiotensin system. Exp. Cell Res. 2012, 318, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Segin, S.; Berlin, M.; Richter, C.; Flockerzi, R.M.V.; Worley, P.; Freichel, M.; Londono, J.E.C. Cardiomyocyte-Specific Deletion of Orai1 Reveals Its Protective Role in Angiotensin-II-Induced Pathological Cardiac Remodeling. Cells 2020, 9, 1092. [Google Scholar] [CrossRef] [PubMed]
- Nouet, S.; Nahmias, C. Signal transduction from the angiotensin II AT2 receptor. Trends Endocrinol Metab. 2000, 11, 1–6. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Nishimura, S.; Uno, M.; Kaneta, Y.; Fukuchi, K.; Nishigohri, H.; Hasegawa, J.; Komori, H.; Takeda, S.; Enomoto, K.; Nara, F.; et al. MRGD, a MAS-related G-protein coupled receptor, promotes tumorigenisis and is highly expressed in lung cancer. PLoS ONE 2012, 7, e38618. [Google Scholar] [CrossRef]
- Jesus, I.C.G.; Scalzo, S.; Alves, F.; Marques, K.; Rocha-Resende, C.; Bader, M.; Santos, R.A.S.; Guatimosim, S. Alamandine acts via MrgD to induce AMPK/NO activation against ANG II hypertrophy in cardiomyocytes. Am. J. Physiol. Cell Physiol. 2018, 314, C702–C711. [Google Scholar] [CrossRef]
- Paz Ocaranza, M.; Riquelme, J.A.; Garcia, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef]
- Rivellese, F.; Prediletto, E. ACE2 at the centre of COVID-19 from paucisymptomatic infections to severe pneumonia. Autoimmun. Rev. 2020, 10, 102536. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, X. scRNA-seq Profiling of Human Testes Reveals the Presence of the ACE2 Receptor, A Target for SARS-CoV-2 Infection in Spermatogonia, Leydig and Sertoli Cells. Cells 2020, 9, 920. [Google Scholar] [CrossRef]
- Tan, D.C.; Roth, I.M.; Wickremesekera, A.C.; Davis, P.F.; Kaye, A.H.; Mantamadiotis, T.; Stylli, S.S.; Tan, S.T. Therapeutic Targeting of Cancer Stem Cells in Human Glioblastoma by Manipulating the Renin-Angiotensin System. Cells 2019, 8, 1364. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Thomas, W.G.; Hannan, R.D. The renin-angiotensin system and cancer: Old dog, new tricks. Nat. Rev. Cancer 2010, 10, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Lumachi, F.; Luisetto, G.; Basso, S.M.; Basso, U.; Brunello, A.; Camozzi, V. Endocrine therapy of breast cancer. Curr. Med. Chem. 2011, 18, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Busonero, C.; Leone, S.; Bartoloni, S.; Acconcia, F. Strategies to degrade estrogen receptor alpha in primary and ESR1 mutant-expressing metastatic breast cancer. Mol. Cell Endocrinol. 2019, 480, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.S.; Oudit, G.Y.; Verano-Braga, T.; Canta, G.; Steckelings, U.M.; Bader, M. The renin-angiotensin system: Going beyond the classical paradigms. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H958–H970. [Google Scholar] [CrossRef]
- Deshayes, F.; Nahmias, C. Angiotensin receptors: A new role in cancer? Trends Endocrinol Metab. 2005, 16, 293–299. [Google Scholar] [CrossRef]
- Young, D.; Waitches, G.; Birchmeier, C.; Fasano, O.; Wigler, M. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell 1986, 45, 711–719. [Google Scholar] [CrossRef]
- Lembo, P.M.; Grazzini, E.; Groblewski, T.; O’Donnell, D.; Roy, M.O.; Zhang, J.; Hoffert, C.; Cao, J.; Schmidt, R.; Pelletier, M.; et al. Proenkephalin A gene products activate a new family of sensory neuron--specific GPCRs. Nat. Neurosci. 2002, 5, 201–209. [Google Scholar] [CrossRef]
- Uchiyama, T.; Okajima, F.; Mogi, C.; Tobo, A.; Tomono, S.; Sato, K. Alamandine reduces leptin expression through the c-Src/p38 MAP kinase pathway in adipose tissue. PLoS ONE 2017, 12, e0178769. [Google Scholar] [CrossRef] [PubMed]
- Qaradakhi, T.; Matsoukas, M.T.; Hayes, A.; Rybalka, E.; Caprnda, M.; Rimarova, K.; Sepsi, M.; Busselberg, D.; Kruzliak, P.; Matsoukas, J.; et al. Alamandine reverses hyperhomocysteinemia-induced vascular dysfunction via PKA-dependent mechanisms. Cardiovasc 2017, 35, e12306. [Google Scholar] [CrossRef]
- Li, P.; Chen, X.R.; Xu, F.; Liu, C.; Li, C.; Liu, H.; Wang, H.; Sun, W.; Sheng, Y.H.; Kong, X.Q. Alamandine attenuates sepsis-associated cardiac dysfunction via inhibiting MAPKs signaling pathways. Life Sci. 2018, 206, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Song, X.D.; Feng, J.P.; Yang, R.X. Alamandine protects rat from myocardial ischemia-reperfusion injury by activating JNK and inhibiting NF-kappaB. Eur. Rev. Med. Pharm. Sci. 2019, 23, 6718–6726. [Google Scholar] [CrossRef]
- Jesus, I.C.G.; Mesquita, T.R.R.; Monteiro, A.L.L.; Parreira, A.B.; Santos, A.K.; Coelho, E.L.X.; Silva, M.M.; Souza, L.A.C.; Campagnole-Santos, M.J.; Santos, R.S. Alamandine enhances cardiomyocyte contractility in hypertensive rats through a nitric oxide-dependent activation of CaMKII. Am. J. Physiol. Cell Physiol. 2020, 318, C740–C750. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fan, J.; Wu, F.; Huang, Q.; Guo, M.; Lv, Z.; Han, J.; Duan, L.; Hu, G.; Chen, L.; et al. The ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Pleiotropic Roles in Cancer. Front Physiol. 2017, 8, 276. [Google Scholar] [CrossRef]
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; de Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. Embo J. 2001, 20, 6050–6059. [Google Scholar] [CrossRef]
- Fiocchetti, M.; Nuzzo, M.T.; Totta, P.; Acconcia, F.; Ascenzi, P.; Marino, M. Neuroglobin, a pro-survival player in estrogen receptor alpha-positive cancer cells. Cell Death Dis. 2014, 5, e1449. [Google Scholar] [CrossRef]
- Fiocchetti, M.; Cipolletti, M.; Leone, S.; Ascenzi, P.; Marino, M. Neuroglobin overexpression induced by the 17beta-Estradiol-Estrogen receptor-alpha Pathway reduces the sensitivity of MCF-7 Breast cancer cell to paclitaxel. Iubmb Life 2016, 68, 645–651. [Google Scholar] [CrossRef]
- Acconcia, F.; Manavathi, B.; Mascarenhas, J.; Talukder, A.H.; Mills, G.; Kumar, R. An inherent role of integrin-linked kinase-estrogen receptor alpha interaction in cell migration. Cancer Res. 2006, 66, 11030–11038. [Google Scholar] [CrossRef]
- Ye, Y.; Xiao, Y.; Wang, W.; Yearsley, K.; Gao, J.X.; Shetuni, B.; Barsky, S.H. ERalpha signaling through slug regulates E-cadherin and EMT. Oncogene 2010, 29, 1451–1462. [Google Scholar] [CrossRef]
- Rugo, H.S.; Rumble, R.B.; Macrae, E.; Barton, D.L.; Connolly, H.K.; Dickler, M.N.; Fallowfield, L.; Fowble, B.; Ingle, J.N.; Jahanzeb, M.; et al. Endocrine Therapy for Hormone Receptor-Positive Metastatic Breast Cancer: American Society of Clinical Oncology Guideline. J. Clin. Oncol. 2016, 34, 3069–3103. [Google Scholar] [CrossRef]
- McDonnell, D.P.; Wardell, S.E.; Norris, J.D. Oral Selective Estrogen Receptor Downregulators (SERDs), a Breakthrough Endocrine Therapy for Breast Cancer. J. Med. Chem. 2015, 58, 4883–4887. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Nass, N.; Kalinski, T. Tamoxifen resistance: From cell culture experiments towards novel biomarkers. Pathol. Res. Pract. 2015, 211, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Rasool, M.; Chaoudhry, H.; Pushparaj, P.N.; Jha, P.; Hafiz, A.; Mahfooz, M.; Sami, G.A.; Kamal, M.A.; Bashir, S.; et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation 2016, 12, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer--An overview and update. Mol. Cell Endocrinol. 2015, 418 Pt 3, 220–234. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef]
- Rondon-Lagos, M.; Villegas, V.E.; Rangel, N.; Sanchez, M.C.; Zaphiropoulos, P.G. Tamoxifen Resistance: Emerging Molecular Targets. Int. J. Mol. Sci. 2016, 17, 1357. [Google Scholar] [CrossRef]
- Tryfonidis, K.; Zardavas, D.; Katzenellenbogen, B.S.; Piccart, M. Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer Treat. Rev. 2016, 50, 68–81. [Google Scholar] [CrossRef]
- Nasrazadani, A.; Thomas, R.A.; Oesterreich, S.; Lee, A.V. Precision Medicine in Hormone Receptor-Positive Breast Cancer. Front. Oncol. 2018, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Li, W.; Wang, X.; Xu, H.; Yang, J.; Wu, Q.; Huang, Y.; Geradts, J.; Jiang, P.; Fei, T.; et al. Estrogen-regulated feedback loop limits the efficacy of estrogen receptor-targeted breast cancer therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 7869–7878. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.; Rao, S.V.; Sutton, J.; Cheeseman, D.; Dunn, S.; Papachristou, E.K.; Prada, J.G.; Couturier, D.L.; Kumar, S.; Kishore, K.; et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat. Genet. 2020, 52, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Knowlden, J.M.; Hutcheson, I.R.; Jones, H.E.; Madden, T.; Gee, J.M.; Harper, M.E.; Barrow, D.; Wakeling, A.E.; Nicholson, R.I. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 2003, 144, 1032–1044. [Google Scholar] [CrossRef]
- Zhao, Y.; Laws, M.J.; Guillen, V.S.; Ziegler, Y.; Min, J.; Sharma, A.; Kim, S.H.; Chu, D.; Park, B.H.; Oesterreich, S.; et al. Structurally Novel Antiestrogens Elicit Differential Responses from Constitutively Active Mutant Estrogen Receptors in Breast Cancer Cells and Tumors. Cancer Res. 2017, 77, 5602–5613. [Google Scholar] [CrossRef]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef]
- Bahreini, A.; Li, Z.; Wang, P.; Levine, K.M.; Tasdemir, N.; Cao, L.; Weir, H.M.; Puhalla, S.L.; Davidson, N.E.; Stern, A.M.; et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res: Bcr 2017, 19, 60. [Google Scholar] [CrossRef]
- Mao, C.; Livezey, M.; Kim, J.E.; Shapiro, D.J. Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor alpha Mutations Upregulate the Unfolded Protein Response and are Killed by BHPI. Sci. Rep. 2016, 6, 34753. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.D.; Darimont, B.; Zhou, W.; Arrazate, A.; Young, A.; Ingalla, E.; Walter, K.; Blake, R.A.; Nonomiya, J.; Guan, Z. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. eLife 2016, 5, e15828. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Srivastava, N.; Amit, S.; Prasad, S.N.; Misra, M.P.; Ateeq, B. Association of AGTR1 (A1166C) and ACE (I/D) Polymorphisms with Breast Cancer Risk in North Indian Population. Transl. Oncol. 2018, 11, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Guo, X.; Niu, L.; Song, D.; Han, B.; Zhang, H. Identification of key molecular targets that correlate with breast cancer through bioinformatic methods. J. Gene Med. 2020, 22, e3141. [Google Scholar] [CrossRef]
- Ekambaram, P.; Lee, J.L.; Hubel, N.E.; Hu, D.; Yerneni, S.; Campbell, P.G.; Pollock, N.; Klei, L.R.; Concel, V.J.; Delekta, P.C.; et al. The CARMA3-Bcl10-MALT1 Signalosome Drives NFkappaB Activation and Promotes Aggressiveness in Angiotensin II Receptor-Positive Breast Cancer. Cancer Res. 2018, 78, 1225–1240. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xia, Z.; Ye, C.; Lu, C.; Zhou, S.; Pan, J.; Liu, C.; Zhang, J.; Liu, T.; Hu, T.; et al. AGTR1 promotes lymph node metastasis in breast cancer by upregulating CXCR4/SDF-1alpha and inducing cell migration and invasion. Aging (Albany Ny) 2019, 11, 3969–3992. [Google Scholar] [CrossRef]
- Coulson, R.; Liew, S.H.; Connelly, A.A.; Yee, N.S.; Deb, S.; Kumar, B.; Vargas, A.C.; O’Toole, S.A.; Parslow, A.C.; Poh, A.; et al. The angiotensin receptor blocker, Losartan, inhibits mammary tumor development and progression to invasive carcinoma. Oncotarget 2017, 8, 18640–18656. [Google Scholar] [CrossRef]
- Oh, E.; Kim, J.Y.; Cho, Y.; An, H.; Lee, N.; Jo, H.; Ban, C.; Seo, J.H. Overexpression of angiotensin II type 1 receptor in breast cancer cells induces epithelial-mesenchymal transition and promotes tumor growth and angiogenesis. Biochim. Biophys. Acta 2016, 1863, 1071–1081. [Google Scholar] [CrossRef]
- Busonero, C.; Leone, S.; Klemm, C.; Acconcia, F. A functional drug re-purposing screening identifies carfilzomib as a drug preventing 17beta-estradiol: ERalpha signaling and cell proliferation in breast cancer cells. Mol. Cell Endocrinol. 2018, 460, 229–237. [Google Scholar] [CrossRef]
- Busonero, C.; Leone, S.; Bianchi, F.; Acconcia, F. In silico screening for ERα downmodulators identifies thioridazine as an anti-proliferative agent in primary, 4OH-tamoxifen-resistant and Y537S ERα-expressing breast cancer cells. Cell. Oncol. 2018, 41, 677–686. [Google Scholar] [CrossRef]
- Leone, S.; Busonero, C.; Acconcia, F. A high throughput method to study the physiology of E2:ERalpha signaling in breast cancer cells. J. Cell Physiol. 2018, 233, 3713–3722. [Google Scholar] [CrossRef] [PubMed]
- Busonero, C.; Leone, S.; Acconcia, F. Emetine induces estrogen receptor alpha degradation and prevents 17beta-estradiol-induced breast cancer cell proliferation. Cell. Oncol. 2017, 40, 299–301. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, B.; Verstraeten, V.M.; De Potter, C.R.; Bullock, G.R. Increased angiotensin II type-2 receptor density in hyperplasia, DCIS and invasive carcinoma of the breast is paralleled with increased iNOS expression. Histochem. Cell Biol. 2002, 117, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Namazi, S.; Sahebi, E.; Rostami-Yalmeh, J.; Jaberipour, M.; Razmkhah, M.; Hosseini, A.; Arabsolghar, R. Effect of angiotensin receptor blockade on prevention and reversion of tamoxifen-resistant phenotype in MCF-7 cells. Tumour. Biol. 2015, 36, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Tovar, H.; Garcia-Herrera, R.; Espinal-Enriquez, J.; Hernandez-Lemus, E. Transcriptional master regulator analysis in breast cancer genetic networks. Comput. Biol. Chem. 2015, 59, 67–77. [Google Scholar] [CrossRef]
- Alhakamy, N.A.; Ishiguro, S.; Uppalapati, D.; Berkland, C.J.; Tamura, M. AT2R Gene Delivered by Condensed Polylysine Complexes Attenuates Lewis Lung Carcinoma after Intravenous Injection or Intratracheal Spray. Mol. Cancer 2016, 15, 209–218. [Google Scholar] [CrossRef]
- Renziehausen, A.; Wang, H.; Rao, B.; Weir, L.; Nigro, C.L.; Lattanzio, L.; Merlano, M.; Vega-Rioja, A.; Del Carmen Fernandez-Carranco, M.; Hajji, N.; et al. The renin angiotensin system (RAS) mediates bifunctional growth regulation in melanoma and is a novel target for therapeutic intervention. Oncogene 2019, 38, 2320–2336. [Google Scholar] [CrossRef]
- Muscella, A.; Greco, S.; Elia, M.G.; Storelli, C.; Marsigliante, S. Angiotensin II stimulation of Na+/K+ATPase activity and cell growth by calcium-independent pathway in MCF-7 breast cancer cells. J. Endocrinol. 2002, 173, 315–323. [Google Scholar] [CrossRef]
- Arrieta, O.; Villarreal-Garza, C.; Vizcaino, G.; Pineda, B.; Hernandez-Pedro, N.; Guevara-Salazar, P.; Wegman-Ostrosky, T.; Villanueva-Rodriguez, G.; Gamboa-Dominguez, A. Association between AT1 and AT2 angiotensin II receptor expression with cell proliferation and angiogenesis in operable breast cancer. Tumour. Biol. 2015, 36, 5627–5634. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol. Asp. Med. 2006, 27, 299–402. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Components of the RAS System | Role in Breast Cancer |
|---|---|
| AT1R | Linked to carcinogenesis, development of the disease and acquisition of the metastatic phenotype. |
| AT2R | Evidence for contrasting connections. |
| MASR | None yet identified. |
| MRGD | Putatively connected with acquisition of resistance to endocrine therapy. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acconcia, F. The Network of Angiotensin Receptors in Breast Cancer. Cells 2020, 9, 1336. https://doi.org/10.3390/cells9061336
Acconcia F. The Network of Angiotensin Receptors in Breast Cancer. Cells. 2020; 9(6):1336. https://doi.org/10.3390/cells9061336
Chicago/Turabian StyleAcconcia, Filippo. 2020. "The Network of Angiotensin Receptors in Breast Cancer" Cells 9, no. 6: 1336. https://doi.org/10.3390/cells9061336
APA StyleAcconcia, F. (2020). The Network of Angiotensin Receptors in Breast Cancer. Cells, 9(6), 1336. https://doi.org/10.3390/cells9061336