Central Modulation of Selective Sphingosine-1-Phosphate Receptor 1 Ameliorates Experimental Multiple Sclerosis

 , , ,
, , ,  , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Drug Formulation
2.2. Mice
2.3. EAE Model
2.4. Minipump and Surgery
2.5. Ex Vivo Experiments
2.6. In Vitro Experiments
2.7. Electrophysiology
2.8. Real Time PCR (qPCR)
2.9. Immunohistochemistry and Confocal Microscopy
2.10. Murine CD3+ Cell Isolation
2.11. T-Cell Absolute Count
2.12. Statistical Analysis
3. Results
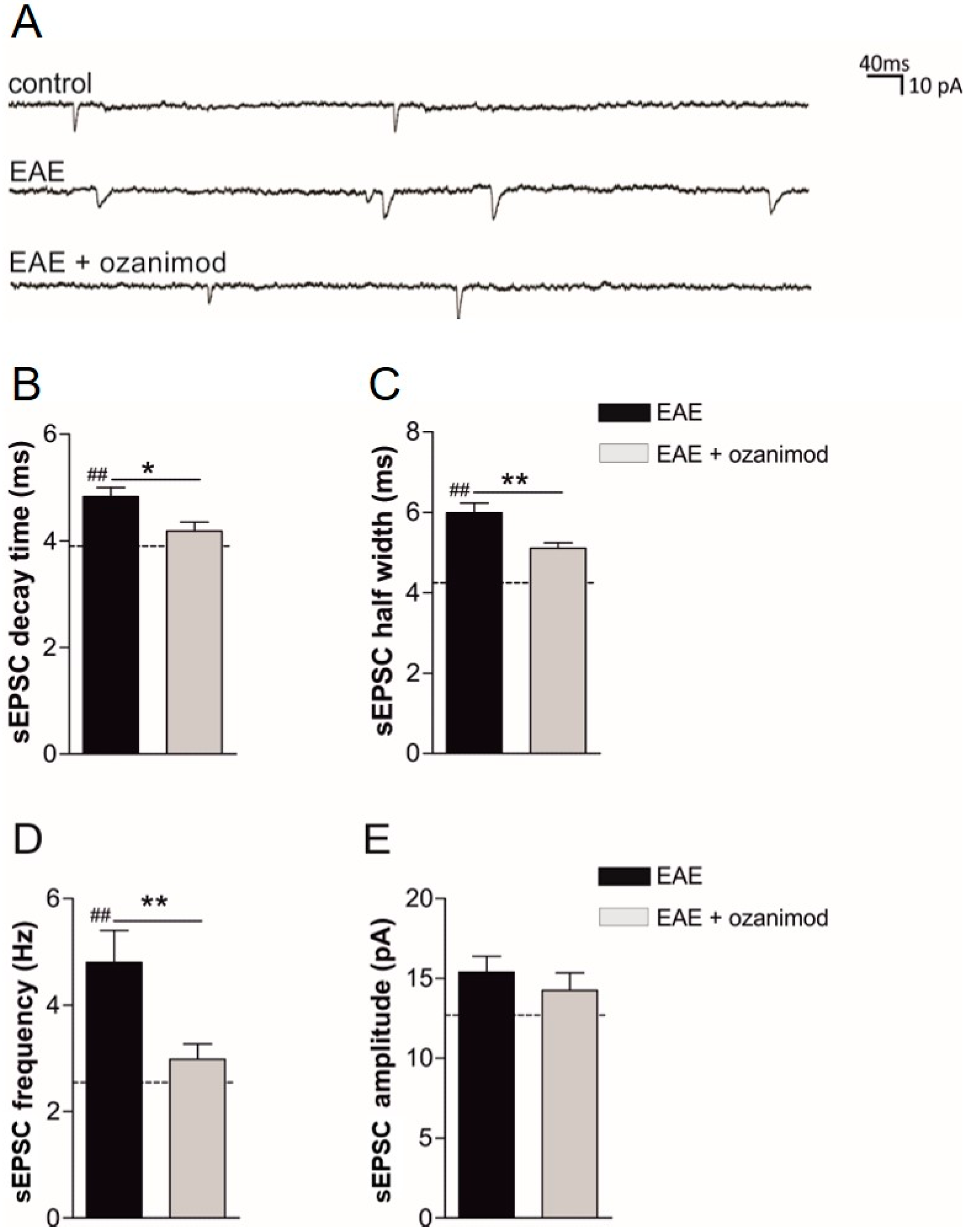
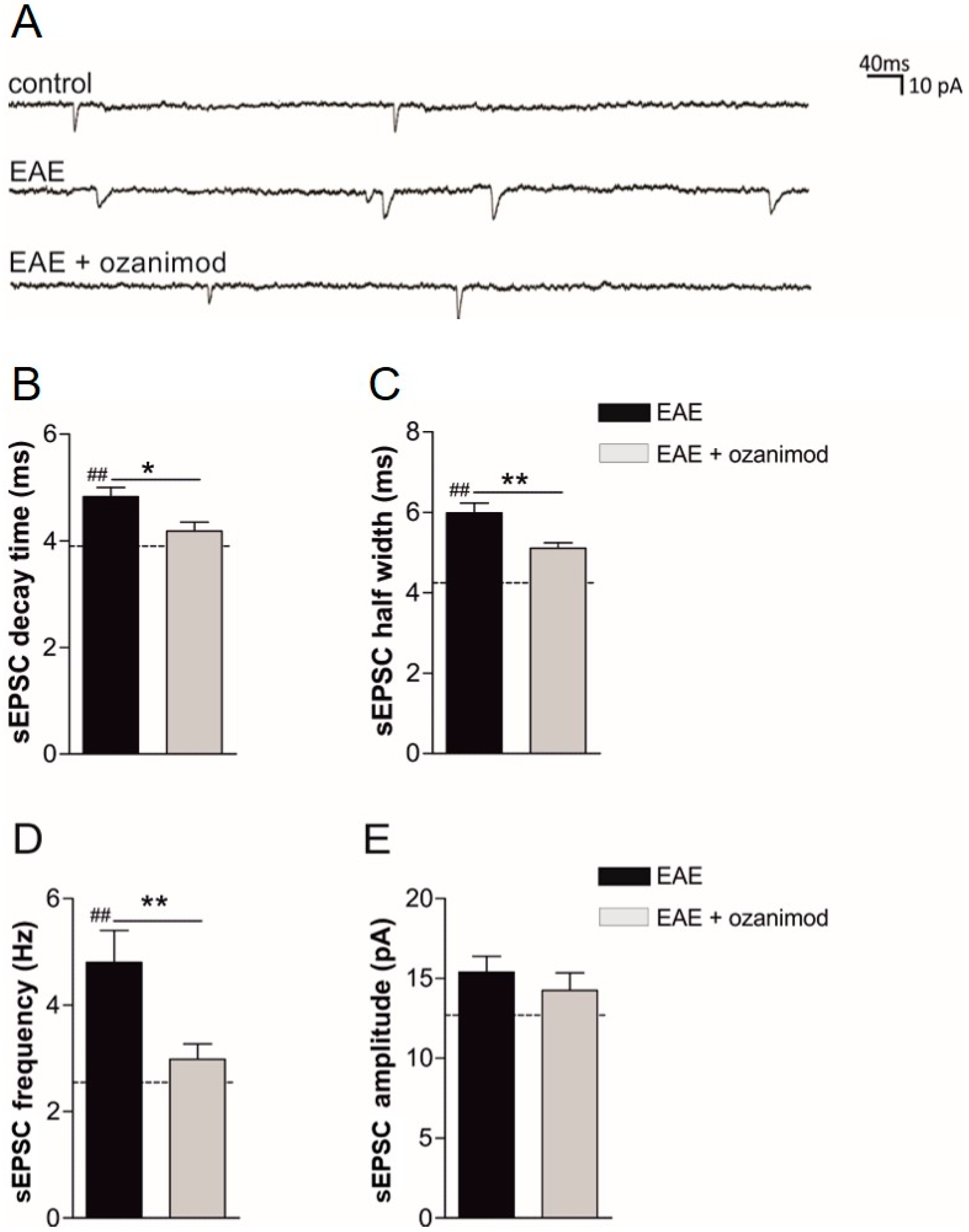
3.1. Ex Vivo Ozanimod Treatment Restores Normal Glutamatergic Transmission in EAE Striatum
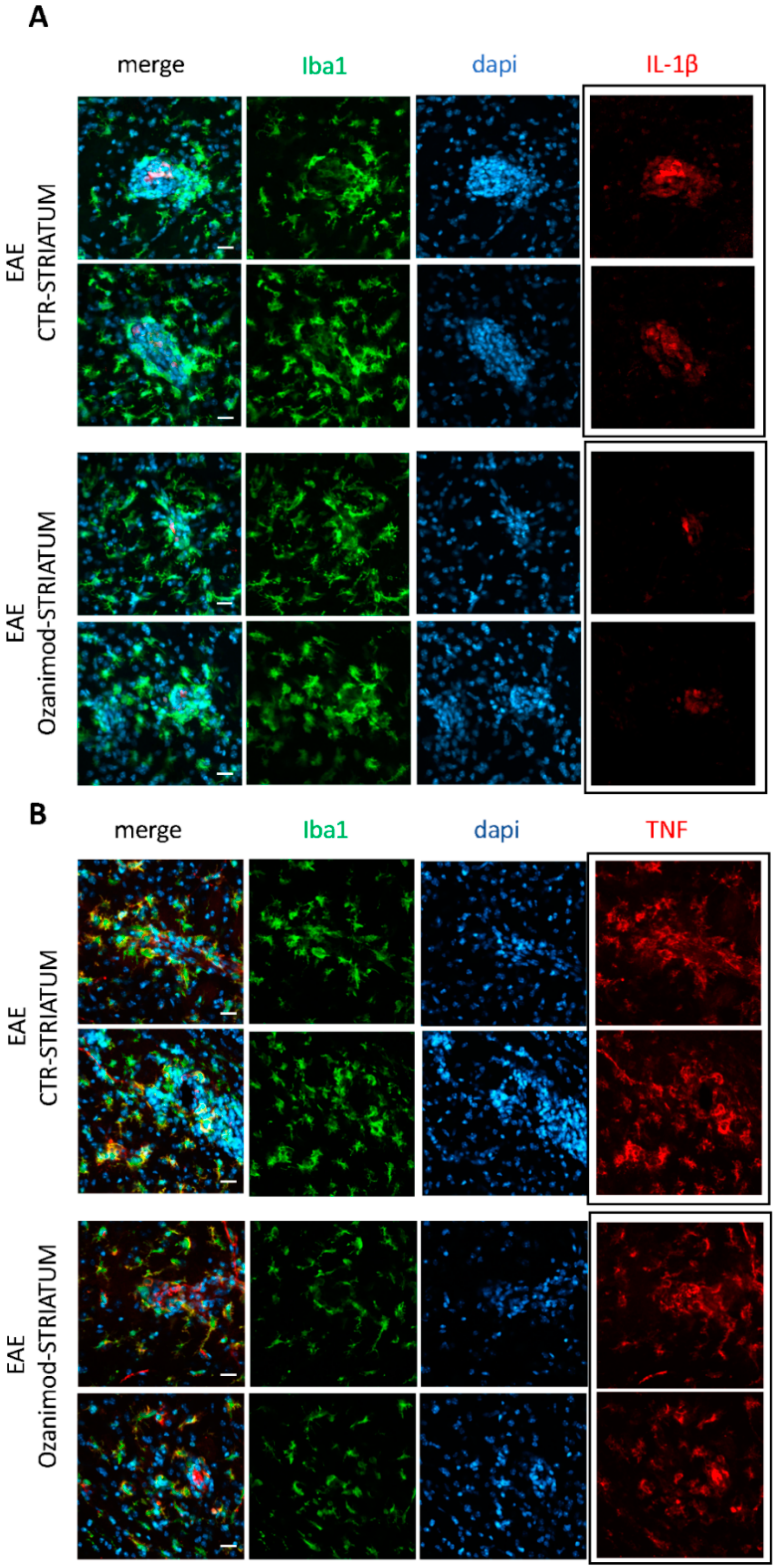
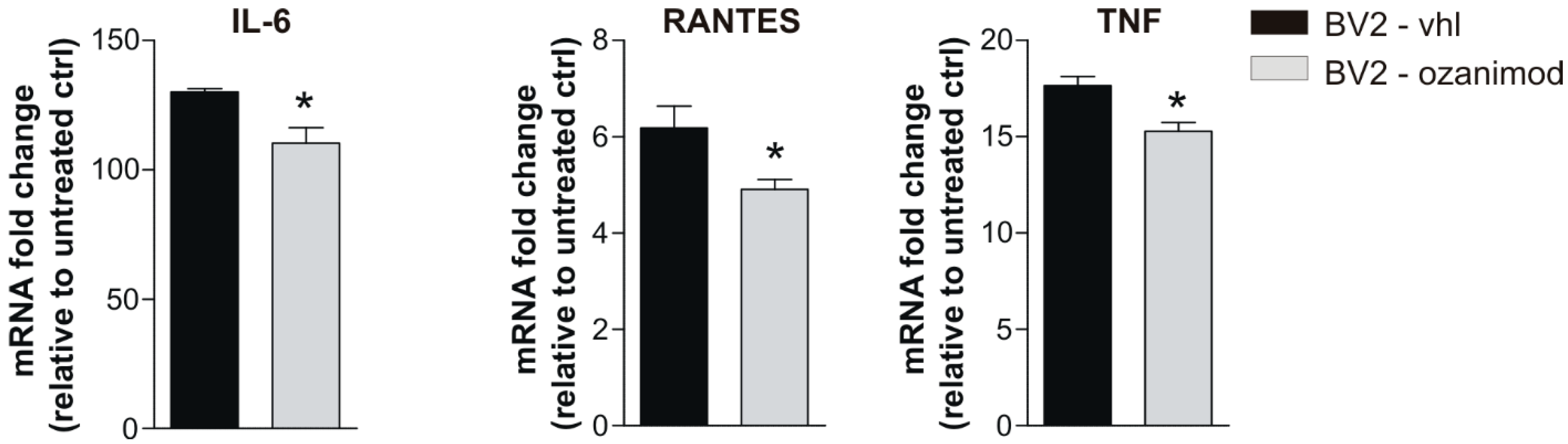
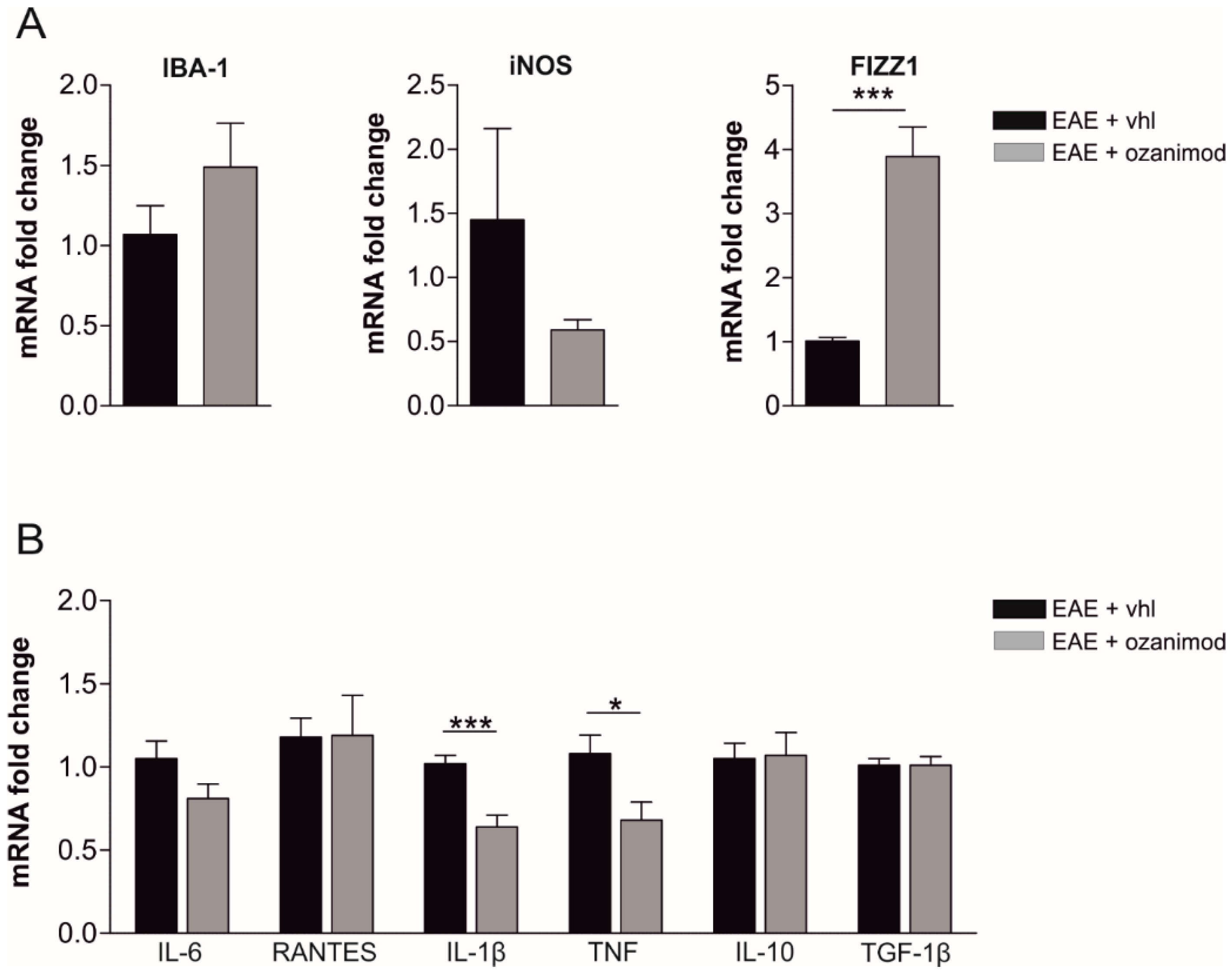
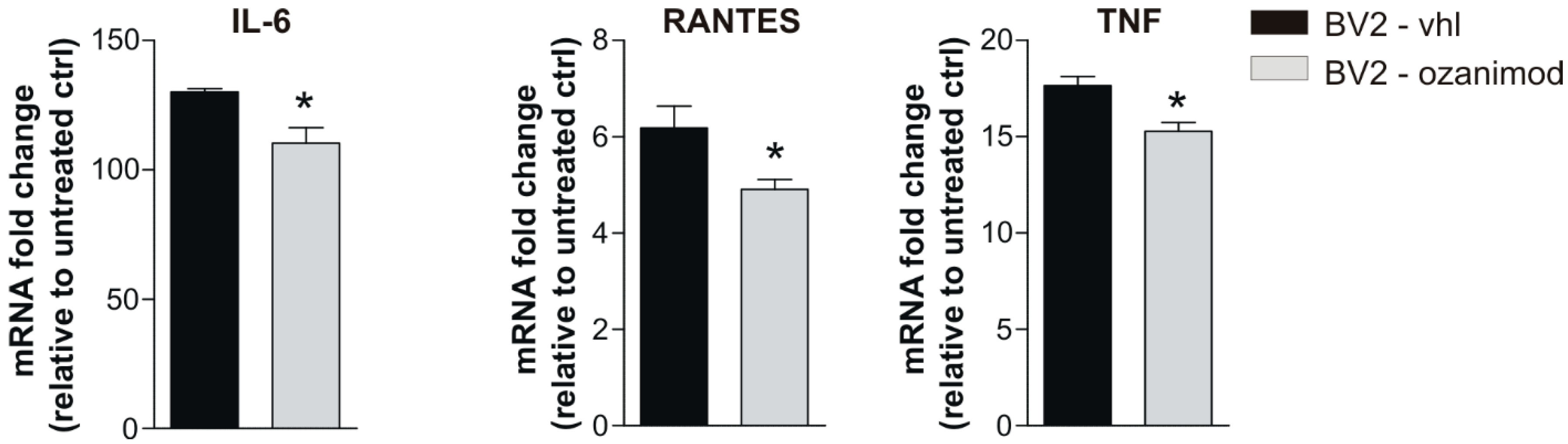
3.2. Ozanimod Treatment Exerts an Anti-Inflammatory Action on EAE Striatum and on Activated Microglial Cell Line
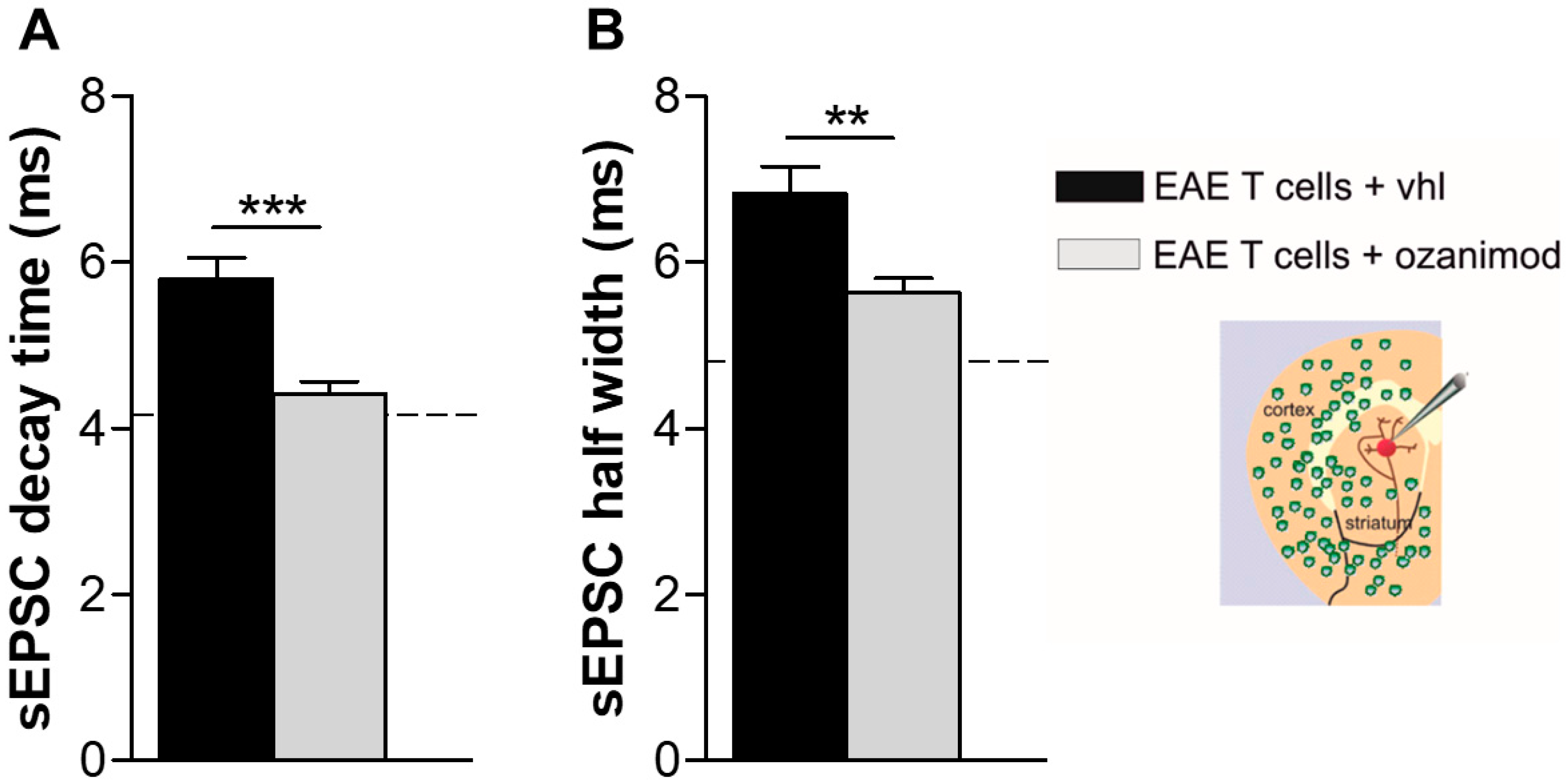
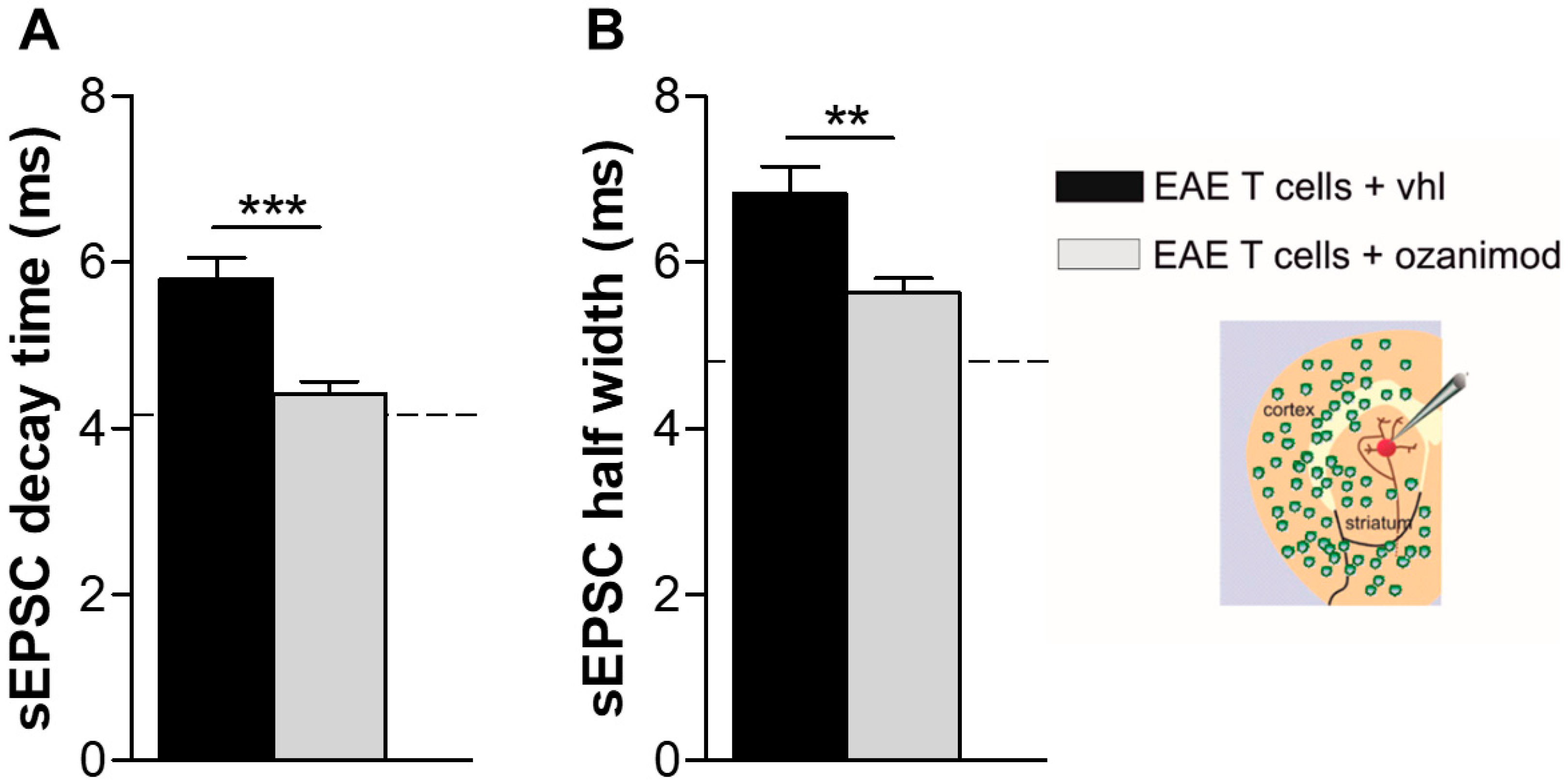
3.3. Ozanimod Pre-Treatment of EAE T Lymphocytes Rescues Striatal Glutamatergic Alterations In Ex Vivo EAE Model
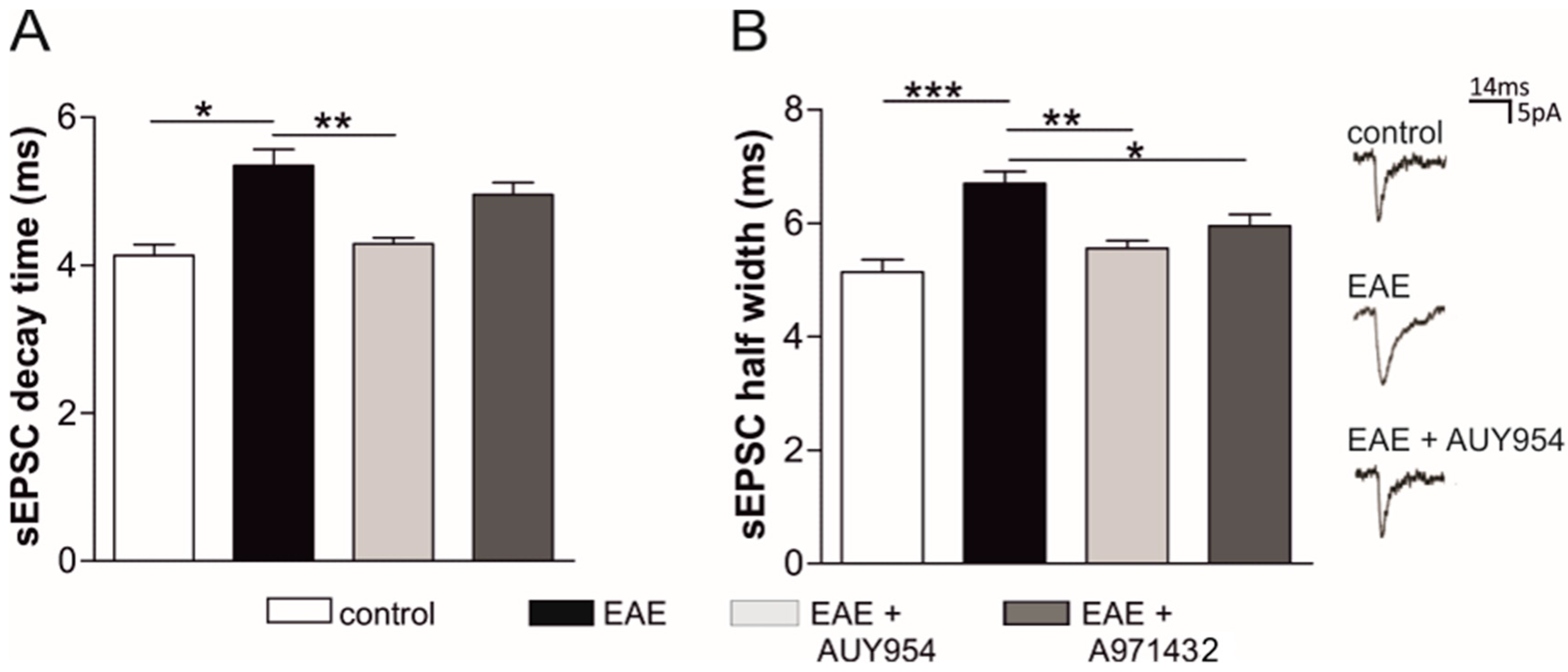
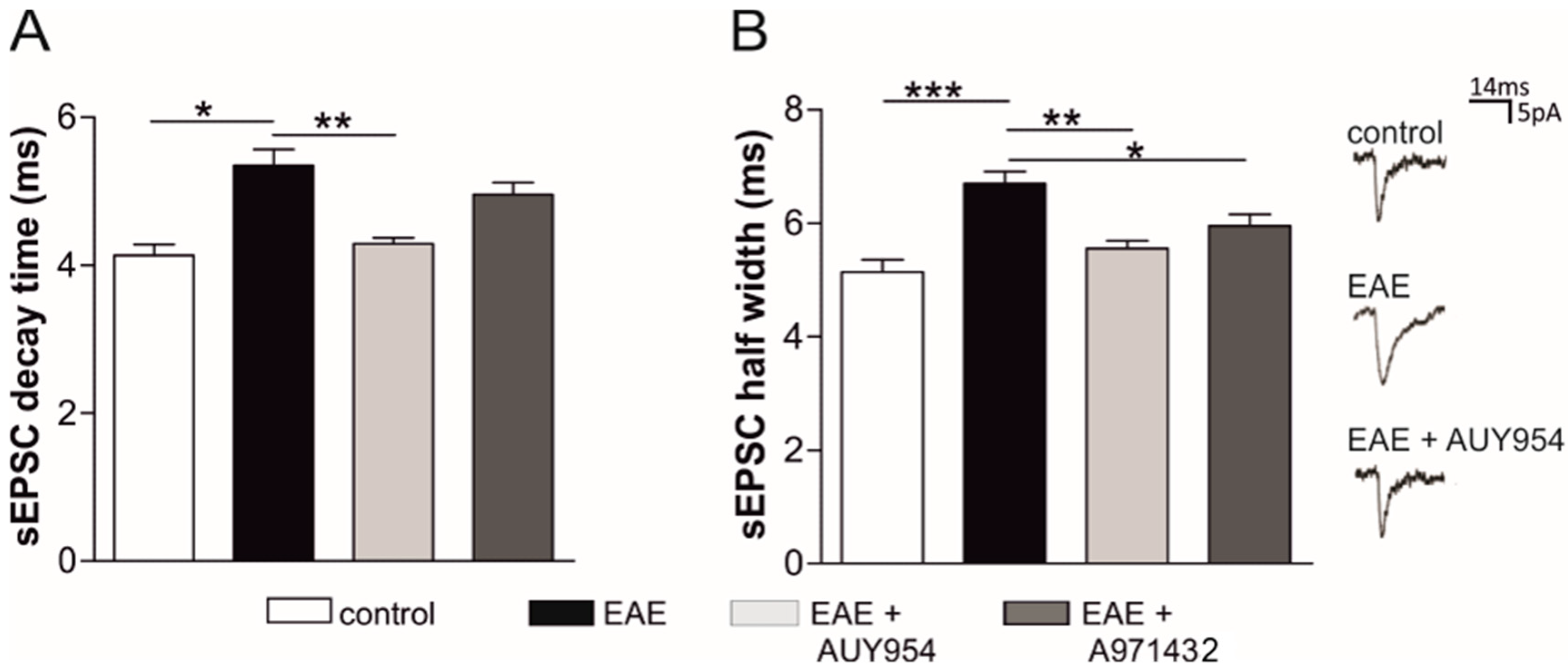
3.4. Selective Agonists of Central S1P1 and S1P5 Differently Modulate EAE Striatal Glutamatergic Alterations
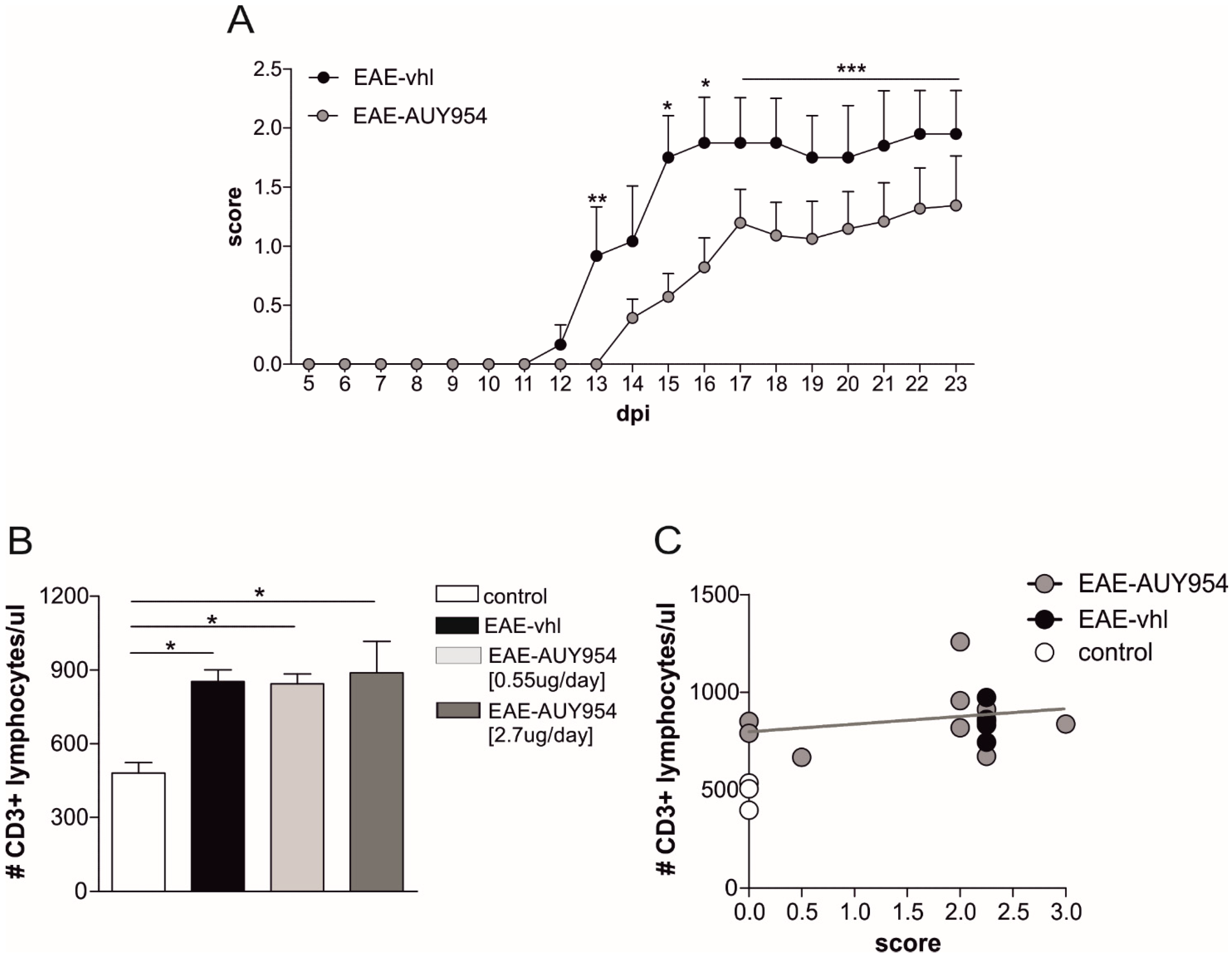
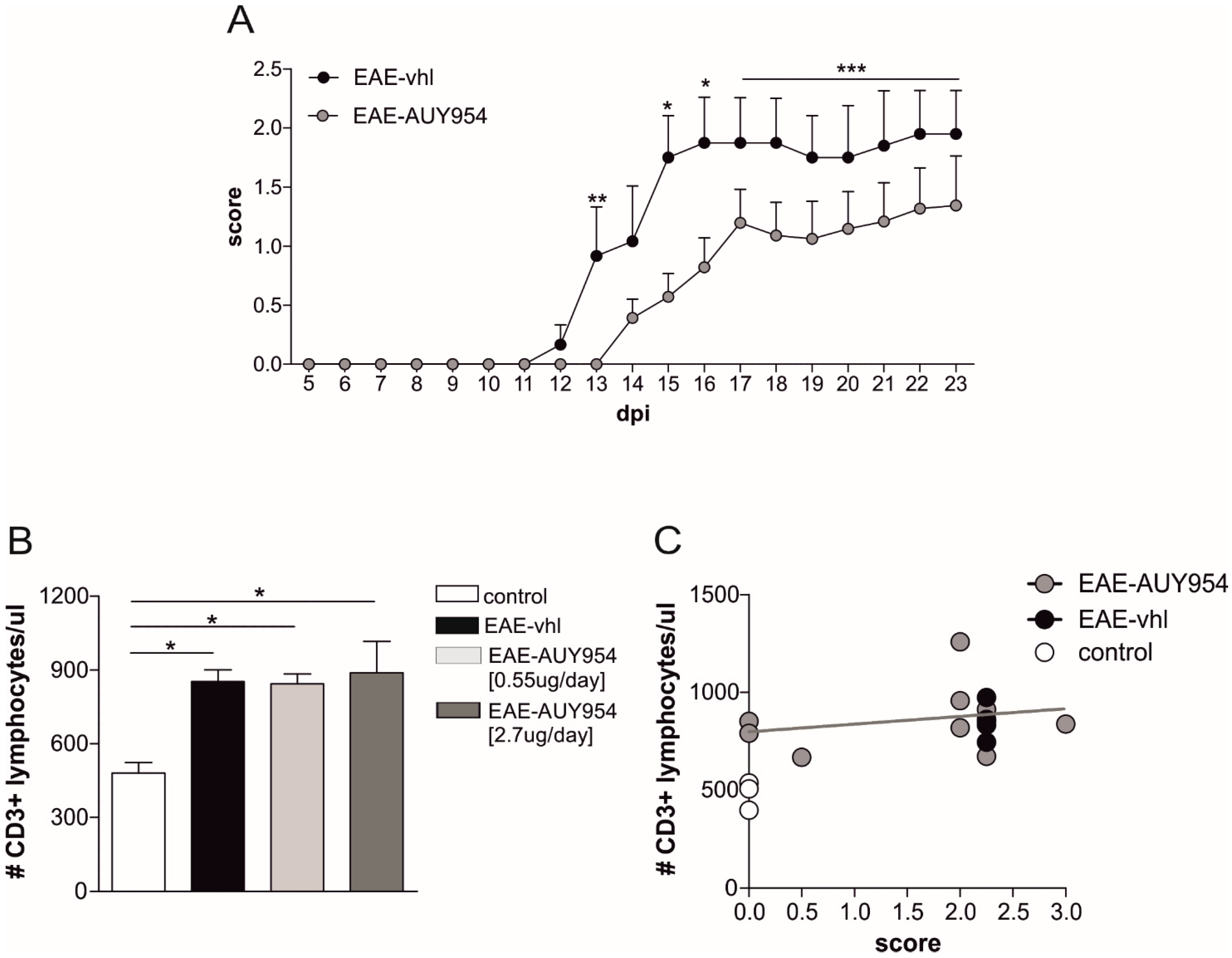
3.5. In Vivo Treatment with S1P1 Selective Agonist Ameliorates EAE Disease
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [Green Version]
- Centonze, D.; Muzio, L.; Rossi, S.; Furlan, R.; Bernardi, G.; Martino, G. The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ. 2010, 17, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Rossi, S.; Furlan, R.; De Chiara, V.; Motta, C.; Studer, V.; Mori, F.; Musella, A.; Bergami, A.; Muzio, L.; Bernardi, G.; et al. Interleukin-1β causes synaptic hyperexcitability in multiple sclerosis. Ann. Neurol. 2012, 71, 76–83. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef]
- Mandolesi, G.; Gentile, A.; Musella, A.; Fresegna, D.; De Vito, F.; Bullitta, S.; Sepman, H.; Marfia, G.A.; Centonze, D. Synaptopathy connects inflammation and neurodegeneration in multiple sclerosis. Nat. Rev. Neurol. 2015, 11, 711. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Mandolesi, G.; Grasselli, G.; Musella, A.; Gentile, A.; Musumeci, G.; Sepman, H.; Haji, N.; Fresegna, D.; Bernardi, G.; Centonze, D. GABAergic signaling and connectivity on Purkinje cells are impaired in experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2012, 46, 414–424. [Google Scholar] [CrossRef]
- Mandolesi, G.; Musella, A.; Gentile, A.; Grasselli, G.; Haji, N.; Sepman, H.; Fresegna, D.; Bullitta, S.; De Vito, F.; Musumeci, G.; et al. Interleukin-1β alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. J. Neurosci. 2013, 33, 12105–12121. [Google Scholar] [CrossRef]
- Rossi, S.; Muzio, L.; De Chiara, V.; Grasselli, G.; Musella, A.; Musumeci, G.; Mandolesi, G.; De Ceglia, R.; Maida, S.; Biffi, E.; et al. Impaired striatal GABA transmission in experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2011, 25, 947–956. [Google Scholar] [CrossRef]
- Azevedo, C.J.; Kornak, J.; Chu, P.; Sampat, M.; Okuda, D.T.; Cree, B.A.; Nelson, S.J.; Hauser, S.L.; Pelletier, D. In vivo evidence of glutamate toxicity in multiple sclerosis. Ann. Neurol. 2014, 76, 269–278. [Google Scholar] [CrossRef]
- Gentile, A.; De Vito, F.; Fresegna, D.; Rizzo, F.R.; Bullitta, S.; Guadalupi, L.; Vanni, V.; Buttari, F.; Stampanoni Bassi, M.; Leuti, A.; et al. Peripheral T cells from multiple sclerosis patients trigger synaptotoxic alterations in central neurons. Neuropathol. Appl. Neurobiol. 2019, 46, 160–170. [Google Scholar] [CrossRef]
- Rossi, S.; Lo Giudice, T.; De Chiara, V.; Musella, A.; Studer, V.; Motta, C.; Bernardi, G.; Martino, G.; Furlan, R.; Martorana, A.; et al. Oral fingolimod rescues the functional deficits of synapses in experimental autoimmune encephalomyelitis. Br. J. Pharmacol. 2012, 165, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Gentile, A.; Musella, A.; Bullitta, S.; Fresegna, D.; De Vito, F.; Fantozzi, R.; Piras, E.; Gargano, F.; Borsellino, G.; Battistini, L.; et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J. Neuroinflamm. 2016, 13, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi, B.; Rossi, S.; Morando, S.; Cordano, C.; Bragoni, A.; Motta, C.; Usai, C.; Wipke, B.T.; Scannevin, R.H.; Mancardi, G.L.; et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015, 130, 279–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.A.; Schmid, C.; Zurbruegg, S.; Jivkov, M.; Doelemeyer, A.; Theil, D.; Dubost, V.; Beckmann, N. Fingolimod inhibits brain atrophy and promotes brain-derived neurotrophic factor in an animal model of multiple sclerosis. J. Neuroimmunol. 2018, 318, 103–113. [Google Scholar] [CrossRef]
- Derfuss, T.; Mehling, M.; Papadopoulou, A.; Bar-Or, A.; Cohen, J.A.; Kappos, L. Advances in oral immunomodulating therapies in relapsing multiple sclerosis. Lancet Neurol. 2020, 19, 336–347. [Google Scholar] [CrossRef]
- Colombo, E.; Di Dario, M.; Capitolo, E.; Chaabane, L.; Newcombe, J.; Martino, G.; Farina, C. Fingolimod may support neuroprotection via blockade of astrocyte nitric oxide. Ann. Neurol. 2014, 76, 325–337. [Google Scholar] [CrossRef]
- O’Sullivan, S.; Dev, K.K. Sphingosine-1-phosphate receptor therapies: Advances in clinical trials for CNS-related diseases. Neuropharmacology 2017, 113, 597–607. [Google Scholar] [CrossRef]
- Subei, A.M.; Cohen, J.A. Sphingosine 1-phosphate receptor modulators in multiple sclerosis. CNS Drugs 2015, 29, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- De Stefano, N.; Silva, D.G.; Barnett, M.H. Effect of Fingolimod on Brain Volume Loss in Patients with Multiple Sclerosis. CNS Drugs 2017, 31, 289–305. [Google Scholar] [CrossRef] [Green Version]
- Noda, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J. Neuroimmunol. 2013, 256, 13–18. [Google Scholar] [CrossRef]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.-W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Di Menna, L.; Molinaro, G.; Di Nuzzo, L.; Riozzi, B.; Zappulla, C.; Pozzilli, C.; Turrini, R.; Caraci, F.; Copani, A.; Battaglia, G.; et al. Fingolimod protects cultured cortical neurons against excitotoxic death. Pharmacol. Res. 2013, 67, 1–9. [Google Scholar] [CrossRef]
- Cipriani, R.; Chara, J.C.; Rodriguez-Antiguedad, A.; Matute, C. FTY720 attenuates excitotoxicity and neuroinflammation. J. Neuroinflamm. 2015, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Luchtman, D.; Gollan, R.; Ellwardt, E.; Birkenstock, J.; Robohm, K.; Siffrin, V.; Zipp, F. In vivo and in vitro effects of multiple sclerosis immunomodulatory therapeutics on glutamatergic excitotoxicity. J. Neurochem. 2016, 136, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Centonze, D.; Muzio, L.; Rossi, S.; Cavasinni, F.; De Chiara, V.; Bergami, A.; Musella, A.; D’Amelio, M.; Cavallucci, V.; Martorana, A.; et al. Inflammation Triggers Synaptic Alteration and Degeneration in Experimental Autoimmune Encephalomyelitis. J. Neurosci. 2009, 29, 3442–3452. [Google Scholar] [CrossRef] [Green Version]
- Scott, F.L.; Clemons, B.; Brooks, J.; Brahmachary, E.; Powell, R.; Dedman, H.; Desale, H.G.; Timony, G.A.; Martinborough, E.; Rosen, H.; et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br. J. Pharmacol. 2016, 173, 1778–1792. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.A.; Comi, G.; Selmaj, K.W.; Bar-Or, A.; Arnold, D.L.; Steinman, L.; Hartung, H.P.; Montalban, X.; Kubala Havrdová, E.; Cree, B.A.C.; et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): A multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 2019, 18, 1021–1033. [Google Scholar] [CrossRef]
- Hobson, A.D.; Harris, C.M.; van der Kam, E.L.; Turner, S.C.; Abibi, A.; Aguirre, A.L.; Bousquet, P.; Kebede, T.; Konopacki, D.B.; Gintant, G.; et al. Discovery of A-971432, An Orally Bioavailable Selective Sphingosine-1-Phosphate Receptor 5 (S1P5) Agonist for the Potential Treatment of Neurodegenerative Disorders. J. Med. Chem. 2015, 58, 9154–9170. [Google Scholar] [CrossRef]
- Pan, S.; Mi, Y.; Pally, C.; Beerli, C.; Chen, A.; Guerini, D.; Hinterding, K.; Nuesslein-Hildesheim, B.; Tuntland, T.; Lefebvre, S.; et al. A monoselective sphingosine-1-phosphate receptor-1 agonist prevents allograft rejection in a stringent rat heart transplantation model. Chem. Biol. 2006, 13, 1227–1234. [Google Scholar] [CrossRef] [Green Version]
- Mandolesi, G.; De Vito, F.; Musella, A.; Gentile, A.; Bullitta, S.; Fresegna, D.; Sepman, H.; Di Sanza, C.; Haji, N.; Mori, F.; et al. miR-142-3p Is a Key Regulator of IL-1beta-Dependent Synaptopathy in Neuroinflammation. J. Neurosci. 2017, 37, 546–561. [Google Scholar] [CrossRef]
- Gentile, A.; Fresegna, D.; Federici, M.; Musella, A.; Rizzo, F.R.; Sepman, H.; Bullitta, S.; De Vito, F.; Haji, N.; Rossi, S.; et al. Dopaminergic dysfunction is associated with IL-1β-dependent mood alterations in experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2015, 74, 347–358. [Google Scholar] [CrossRef]
- Chu, F.; Shi, M.; Zheng, C.; Shen, D.; Zhu, J.; Zheng, X.; Cui, L. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2018, 318, 1–7. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H.G. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef]
- Macrez, R.; Stys, P.K.; Vivien, D.; Lipton, S.A.; Docagne, F. Mechanisms of glutamate toxicity in multiple sclerosis: Biomarker and therapeutic opportunities. Lancet Neurol. 2016, 15, 1089–1102. [Google Scholar] [CrossRef]
- Pitt, D.; Nagelmeier, I.E.; Wilson, H.C.; Raine, C.S. Glutamate uptake by oligodendrocytes: Implications for excitotoxicity in multiple sclerosis. Neurology 2003, 61, 1113–1120. [Google Scholar] [CrossRef]
- Chaudhry, B.Z.; Cohen, J.A.; Conway, D.S. Sphingosine 1-Phosphate Receptor Modulators for the Treatment of Multiple Sclerosis. Neurotherapeutics 2017, 14, 859–873. [Google Scholar] [CrossRef]
- Landi, D.; Vollaro, S.; Pellegrino, G.; Mulas, D.; Ghazaryan, A.; Falato, E.; Pasqualetti, P.; Rossini, P.M.; Filippi, M.M. Oral fingolimod reduces glutamate-mediated intracortical excitability in relapsing-remitting multiple sclerosis. Clin. Neurophysiol. 2015, 126, 165–169. [Google Scholar] [CrossRef]
- Behrangi, N.; Fischbach, F.; Kipp, M. Mechanism of Siponimod: Anti-Inflammatory and Neuroprotective Mode of Action. Cells 2019, 8, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deogracias, R.; Yazdani, M.; Dekkers, M.P.J.; Guy, J.; Ionescu, M.C.S.; Vogt, K.E.; Barde, Y.-A. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 14230–14235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pardo, A.; Castaldo, S.; Amico, E.; Pepe, G.; Marracino, F.; Capocci, L.; Giovannelli, A.; Madonna, M.; van Bergeijk, J.; Buttari, F.; et al. Stimulation of S1PR5 with A-971432, a selective agonist, preserves blood-brain barrier integrity and exerts therapeutic effect in an animal model of Huntington’s disease. Hum. Mol. Genet. 2018, 27, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Han, M.; Wei, X.; Guo, Y.; Shi, H.; Zhang, X.; Perez, R.G.; Lou, H. FTY720 Attenuates 6-OHDA-Associated Dopaminergic Degeneration in Cellular and Mouse Parkinsonian Models. Neurochem. Res. 2017, 42, 686–696. [Google Scholar] [CrossRef]
- Comi, G.; Kappos, L.; Selmaj, K.W.; Bar-Or, A.; Arnold, D.L.; Steinman, L.; Hartung, H.P.; Montalban, X.; Kubala Havrdová, E.; Cree, B.A.C.; et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): A multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 2019, 18, 1009–1020. [Google Scholar] [CrossRef]
- Brana, C.; Frossard, M.J.; Pescini Gobert, R.; Martinier, N.; Boschert, U.; Seabrook, T.J. Immunohistochemical detection of sphingosine-1-phosphate receptor 1 and 5 in human multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 2014, 40, 564–578. [Google Scholar] [CrossRef]
- Mullershausen, F.; Craveiro, L.M.; Shin, Y.; Cortes-Cros, M.; Bassilana, F.; Osinde, M.; Wishart, W.L.; Guerini, D.; Thallmair, M.; Schwab, M.E.; et al. Phosphorylated FTY720 promotes astrocyte migration through sphingosine-1-phosphate receptors. J. Neurochem. 2007, 102, 1151–1161. [Google Scholar] [CrossRef]
- O’Sullivan, C.; Schubart, A.; Mir, A.K.; Dev, K.K. The dual S1PR1/S1PR5 drug BAF312 (Siponimod) attenuates demyelination in organotypic slice cultures. J. Neuroinflamm. 2016, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Tham, C.-S.; Lin, F.-F.; Rao, T.S.; Yu, N.; Webb, M. Microglial activation state and lysophospholipid acid receptor expression. Int. J. Dev. Neurosci. 2003, 21, 431–443. [Google Scholar] [CrossRef]
- Oo, M.L.; Thangada, S.; Wu, M.-T.; Liu, C.H.; Macdonald, T.L.; Lynch, K.R.; Lin, C.-Y.; Hla, T. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J. Biol. Chem. 2007, 282, 9082–9089. [Google Scholar] [CrossRef] [Green Version]
- Gaire, B.P.; Lee, C.-H.; Sapkota, A.; Lee, S.Y.; Chun, J.; Cho, H.J.; Nam, T.-G.; Choi, J.W. Identification of Sphingosine 1-Phosphate Receptor Subtype 1 (S1P1) as a Pathogenic Factor in Transient Focal Cerebral Ischemia. Mol. Neurobiol. 2018, 55, 2320–2332. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musella, A.; Gentile, A.; Guadalupi, L.; Rizzo, F.R.; De Vito, F.; Fresegna, D.; Bruno, A.; Dolcetti, E.; Vanni, V.; Vitiello, L.; et al. Central Modulation of Selective Sphingosine-1-Phosphate Receptor 1 Ameliorates Experimental Multiple Sclerosis. Cells 2020, 9, 1290. https://doi.org/10.3390/cells9051290
Musella A, Gentile A, Guadalupi L, Rizzo FR, De Vito F, Fresegna D, Bruno A, Dolcetti E, Vanni V, Vitiello L, et al. Central Modulation of Selective Sphingosine-1-Phosphate Receptor 1 Ameliorates Experimental Multiple Sclerosis. Cells. 2020; 9(5):1290. https://doi.org/10.3390/cells9051290
Chicago/Turabian StyleMusella, Alessandra, Antonietta Gentile, Livia Guadalupi, Francesca Romana Rizzo, Francesca De Vito, Diego Fresegna, Antonio Bruno, Ettore Dolcetti, Valentina Vanni, Laura Vitiello, and et al. 2020. "Central Modulation of Selective Sphingosine-1-Phosphate Receptor 1 Ameliorates Experimental Multiple Sclerosis" Cells 9, no. 5: 1290. https://doi.org/10.3390/cells9051290
APA StyleMusella, A., Gentile, A., Guadalupi, L., Rizzo, F. R., De Vito, F., Fresegna, D., Bruno, A., Dolcetti, E., Vanni, V., Vitiello, L., Bullitta, S., Sanna, K., Caioli, S., Balletta, S., Nencini, M., Buttari, F., Stampanoni Bassi, M., Centonze, D., & Mandolesi, G. (2020). Central Modulation of Selective Sphingosine-1-Phosphate Receptor 1 Ameliorates Experimental Multiple Sclerosis. Cells, 9(5), 1290. https://doi.org/10.3390/cells9051290