DHA Affects Microtubule Dynamics Through Reduction of Phospho-TCTP Levels and Enhances the Antiproliferative Effect of T-DM1 in Trastuzumab-Resistant HER2-Positive Breast Cancer Cell Lines

 , , , , ,
, , , , ,
Abstract
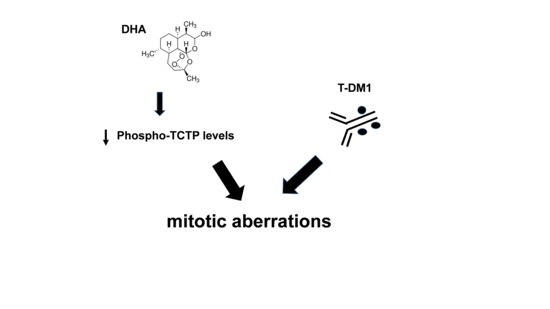
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture and Treatments
2.3. Antibodies
2.4. Cell Viability Assay
2.5. ROS Production Assay
2.6. Western Blot Analysis
2.7. Colony Formation Assays
2.8. Flow Cytometry
2.9. Immunofluorescence Staining
2.10. Quantification of TCTP Distribution
2.11. Quantification of Ki-67 Positive Cells
2.12. Quantification of Microtubule Density
2.13. Growth Curve
2.14. Vector Construction
2.15. Mutagenesis
2.16. Recombinant Retroviral Vectors
2.17. Cell Transfection
2.18. Evaluation of Cell Sensitivity to Combined Treatment
2.19. Immunodeficient Mice Study
2.20. Statistical Analysis
3. Results
3.1. DHA Affects Mitosis of HER2+ BC Cell Lines with Aberrant PI3K/AKT Signalling
3.2. DHA Induces a Decrease in AKT Phosphorylation Levels and DNA Damage Through the Increase of ROS in HER2+ BC Cell Lines
3.3. Phosphorylation of TCTP is Required for Correct Mitotic Progression in Human Mammary Cells
3.4. DHA Enhances T-DM1 Efficacy in Breast Cancer Cells Resistant to Trastuzumab Therapy
3.5. The Effects of Two-Drug Combination on HER2-Mediated Cell Signalling
3.6. DHA in Combination with T-DM1 Led to Mitotic Catastrophe
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [PubMed]
- Larionov, A. Current Therapies for Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer Patients. Front. Oncol. 2018, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Vernieri, C.; Milano, M.; Brambilla, M.; Mennitto, A.; Maggi, C.; Cona, M.S.; Prisciandaro, M.; Fabbroni, C.; Celio, L.; Mariani, G.; et al. Resistance mechanisms to anti-HER2 therapies in HER2-positive breast cancer: Current knowledge, new research directions and therapeutic perspectives. Crit. Rev. Oncol. 2019, 139, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yu, K.; Zhang, K.; Liu, L.; Li, Y. Efficacy and safety of trastuzumab emtansine (T-DM1) in the treatment of HER2-positive metastatic breast cancer (MBC): A meta-analysis of randomized controlled trial. Oncotarget 2017, 8, 102458–102467. [Google Scholar] [CrossRef]
- Diéras, V.; Miles, D.; Verma, S.; Pegram, M.; Welslau, M.; Baselga, J.; E Krop, I.; Blackwell, K.; Hoersch, S.; Xu, J.; et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): A descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 732–742. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2018, 380, 617–628. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Procter, M.; De Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef]
- García-Alonso, S.; Ocana, A.; Pandiella, A. Resistance to Antibody–Drug Conjugates. Cancer Res. 2018, 78, 2159–2165. [Google Scholar] [CrossRef]
- Li, G.; Guo, J.; Shen, B.-Q.; Yadav, D.B.; Sliwkowski, M.X.; Crocker, L.M.; Lacap, J.A.; Phillips, G.D.L. Mechanisms of Acquired Resistance to Trastuzumab Emtansine in Breast Cancer Cells. Mol. Cancer Ther. 2018, 17, 1441–1453. [Google Scholar] [CrossRef]
- Lucibello, M.; Adanti, S.; Antelmi, E.; Dezi, D.; Ciafre, S.; Carcangiu, M.L.; Zonfrillo, M.; Nicotera, G.; Sica, L.; De Braud, F.; et al. Phospho-TCTP as a therapeutic target of dihydroartemisinin for aggressive breast cancer cells. Oncotarget 2015, 6, 5275–5291. [Google Scholar] [CrossRef]
- Bommer, U. The Translational Controlled Tumour Protein TCTP: Biological Functions and Regulation. Results Probl. Cell Differ. 2017, 64, 69–126. [Google Scholar] [CrossRef]
- Chen, S.H.; Wu, P.-S.; Chou, C.-H.; Yan, Y.-T.; Liu, H.; Weng, S.-Y.; Yang-Yen, H.-F. A Knockout Mouse Approach Reveals that TCTP Functions as an Essential Factor for Cell Proliferation and Survival in a Tissue- or Cell Type–specific Manner. Mol. Biol. Cell 2007, 18, 2525–2532. [Google Scholar] [CrossRef] [PubMed]
- Lucibello, M.; Gambacurta, A.; Zonfrillo, M.; Pierimarchi, P.; Serafino, A.; Rasi, G.; Rubartelli, A.; Garaci, E. TCTP is a critical survival factor that protects cancer cells from oxidative stress-induced cell-death. Exp. Cell Res. 2011, 317, 2479–2489. [Google Scholar] [CrossRef] [PubMed]
- Amson, R.; Karp, J.E.; Telerman, A. Lessons from tumor reversion for cancer treatment. Curr. Opin. Oncol. 2013, 25, 59–65. [Google Scholar] [CrossRef]
- Tuynder, M.; Susini, L.; Prieur, S.; Besse, S.; Fiucci, G.; Amson, R.; Telerman, A. Biological models and genes of tumor reversion: Cellular reprogramming through tpt1/TCTP and SIAH-1. Proc. Natl. Acad. Sci. USA 2002, 99, 14976–14981. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Chern, J.J.; Cai, Y.; Liu, M.; Choi, K.-W. Drosophila TCTP is essential for growth and proliferation through regulation of dRheb GTPase. Nature 2007, 445, 785–788. [Google Scholar] [CrossRef]
- Tuynder, M.; Fiucci, G.; Prieur, S.; Lespagnol, A.; Géant, A.; Beaucourt, S.; Duflaut, D.; Besse, S.; Susini, L.; Cavarelli, J.; et al. Translationally controlled tumor protein is a target of tumor reversion. Proc. Natl. Acad. Sci. USA 2004, 101, 15364–15369. [Google Scholar] [CrossRef]
- Amson, R.; Pece, S.; Marine, J.-C.; Di Fiore, P.P.; Telerman, A. TPT1/ TCTP-regulated pathways in phenotypic reprogramming. Trends Cell Biol. 2013, 23, 37–46. [Google Scholar] [CrossRef]
- Amson, R.; Auclair, C.; Andre, F.; Karp, J.; Telerman, A. Targeting TCTP with Sertraline and Thioridazine in Cancer Treatment. Results Probl. Cell Differ. 2017, 64, 283–290. [Google Scholar] [CrossRef]
- Amson, R.; Pece, S.; Lespagnol, A.; Vyas, R.; Mazzarol, G.; Tosoni, D.; Colaluca, I.; Viale, G.; Rodrigues-Ferreira, S.; Wynendaele, J.; et al. Reciprocal repression between P53 and TCTP. Nat. Med. 2011, 18, 91–99. [Google Scholar] [CrossRef]
- Bae, S.-Y.; Kim, H.J.; Lee, K.-J.; Lee, K. Translationally Controlled Tumor Protein induces epithelial to mesenchymal transition and promotes cell migration, invasion and metastasis. Sci. Rep. 2015, 5, 8061. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.K.; Srivastava, P.; Sharma, A.; Prasad, R.; Bhuyan, S.K.; Malage, R.; Kumar, P.; Yadava, P. Translationally controlled tumor protein (TCTP) is required for TGF-β1 induced epithelial to mesenchymal transition and influences cytoskeletal reorganization. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2018, 1865, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Chen, Y.-B.; Xu, S.-L.; Zhao, T.; Liu, J.-Y.; Li, Y.-R.; Wang, J.; Zhang, J.; Guo, G.-Z. TCTP overexpression is associated with the development and progression of glioma. Tumor Biol. 2013, 34, 3357–3361. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Deng, Y.; Hua, M.; Xi, Q.; Liu, R.; Yang, S.; Liu, J.; Zhong, J.; Tang, M.; Lu, S.; et al. Expression and clinical role of TCTP in epithelial ovarian cancer. J. Mol. Histol. 2015, 46, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Baylot, V.; Karaki, S.; Rocchi, P. TCTP Has a Crucial Role in the Different Stages of Prostate Cancer Malignant Progression. Results Probl. Cell Differ. 2017, 64, 255–261. [Google Scholar] [CrossRef]
- Giusiano, S.; Garcia, S.; Andrieu, C.; Dusetti, N.; Bastide, C.; Gleave, M.; Iovanna, J.; Rocchi, P.; Taranger-Charpin, C. TP53INP1 overexpression in prostate cancer correlates with poor prognostic factors and is predictive of biological cancer relapse. Prostate 2011, 72, 117–128. [Google Scholar] [CrossRef]
- Combes, G.; Alharbi, I.; Braga, L.G.; Elowe, S. Playing polo during mitosis: PLK1 takes the lead. Oncogene 2017, 36, 4819–4827. [Google Scholar] [CrossRef]
- Li, S.; Ge, F. Current Understanding of the TCTP Interactome. Plant Promot. Transcr. Factors 2017, 64, 127–136. [Google Scholar] [CrossRef]
- Liu, H.; Peng, H.-W.; Cheng, Y.-S.; Yuan, H.S.; Yang-Yen, H.-F. Stabilization and Enhancement of the Antiapoptotic Activity of Mcl-1 by TCTP. Mol. Cell. Biol. 2005, 25, 3117–3126. [Google Scholar] [CrossRef]
- Yarm, F.R. Plk Phosphorylation Regulates the Microtubule-Stabilizing Protein TCTP. Mol. Cell. Biol. 2002, 22, 6209–6221. [Google Scholar] [CrossRef]
- Cucchi, U.; Gianellini, L.M.; De Ponti, A.; Sola, F.; Alzani, R.; Patton, V.; Pezzoni, A.; Troiani, S.; Saccardo, M.B.; Rizzi, S.; et al. Phosphorylation of TCTP as a marker for polo-like kinase-1 activity in vivo. Anticancer. Res. 2010, 30, 4973–4985. [Google Scholar]
- Lemmens, B.; Hégarat, N.; Akopyan, K.; Sala-Gaston, J.; Bartek, J.; Hochegger, H.; Lindqvist, A. DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation. Mol. Cell 2018, 71, 117–128.e3. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.-J.; You, S.Y.; Park, Y.S.; Chang, J.W.; Kim, J.-S.; Oh, J.S. TCTP regulates spindle microtubule dynamics by stabilizing polar microtubules during mouse oocyte meiosis. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2016, 1863, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Gachet, Y.; Tournier, S.; Lee, M.; Lazaris-Karatzas, A.; Poulton, T.; A Bommer, U. The growth-related, translationally controlled protein P23 has properties of a tubulin binding protein and associates transiently with microtubules during the cell cycle. J. Cell Sci. 1999, 112, 1257–1271. [Google Scholar] [PubMed]
- Bhisutthibhan, J.; Meshnick, S.R. Immunoprecipitation of [3H]Dihydroartemisinin Translationally Controlled Tumor Protein (TCTP) Adducts from Plasmodium falciparum-Infected Erythrocytes by Using Anti-TCTP Antibodies. Antimicrob. Agents Chemother. 2001, 45, 2397–2399. [Google Scholar] [CrossRef]
- Efferth, T. Mechanistic perspectives for 1,2,4-trioxanes in anti-cancer therapy. Drug Resist. Updat. 2005, 8, 85–97. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, Q.; Xu, Z.; Liang, H.; Li, H.; Ye, Y.; Xiang, S.; Zhang, Y.; Jiang, L.; Hu, Y.; et al. Dihydroartemisinin inhibits TCTP-dependent metastasis in gallbladder cancer. J. Exp. Clin. Cancer Res. 2017, 36, 68. [Google Scholar] [CrossRef]
- Tu, Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220. [Google Scholar] [CrossRef]
- Jansen, F.H.; Adoubi, I.; C, K.C.J.; De Cnodder, T.; Jansen, N.; Tschulakow, A.; Efferth, T. First study of oral Artenimol-R in advanced cervical cancer: Clinical benefit, tolerability and tumor markers. Anticancer. Res. 2011, 31, 4417–4422. [Google Scholar]
- Ericsson, T.; Blank, A.; Von Hagens, C.; Ashton, M.; Äbelö, A. Population pharmacokinetics of artesunate and dihydroartemisinin during long-term oral administration of artesunate to patients with metastatic breast cancer. Eur. J. Clin. Pharmacol. 2014, 70, 1453–1463. [Google Scholar] [CrossRef]
- Von Hagens, C.; Walter-Sack, I.; Goeckenjan, M.; Osburg, J.; Storch-Hagenlocher, B.; Sertel, S.; Elsässer, M.; Remppis, B.A.; Edler, L.; Munzinger, J.; et al. Prospective open uncontrolled phase I study to define a well-tolerated dose of oral artesunate as add-on therapy in patients with metastatic breast cancer (ARTIC M33/2). Breast Cancer Res. Treat. 2017, 164, 359–369. [Google Scholar] [CrossRef] [PubMed]
- A Morris, C.; Duparc, S.; Borghini-Fuhrer, I.; Jung, D.; Shin, C.-S.; Fleckenstein, L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar. J. 2011, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Remich, S.; Miller, R.S.; Ogutu, B.; Otieno, W.; Melendez, V.; Teja-Isavadharm, P.; Weina, P.J.; Hickman, M.; Smith, B.; et al. Pharmacokinetic evaluation of intravenous artesunate in adults with uncomplicated falciparum malaria in Kenya: A phase II study. Malar. J. 2014, 13, 281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Efferth, T.; Li, P.C.; Konkimalla, B.; Kaina, B. From traditional Chinese medicine to rational cancer therapy. Trends Mol. Med. 2007, 13, 353–361. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Di Rocco, G.; Gentile, A.; Antonini, A.; Truffa, S.; Piaggio, G.; Capogrossi, M.C.; Toietta, G. Analysis of Biodistribution and Engraftment into the Liver of Genetically Modified Mesenchymal Stromal Cells Derived from Adipose Tissue. Cell Transplant. 2012, 21, 1997–2008. [Google Scholar] [CrossRef]
- Holen, I.; Speirs, V.; Morrissey, B.; Blyth, K. In vivo models in breast cancer research: Progress, challenges and future directions. Dis. Model. Mech. 2017, 10, 359–371. [Google Scholar] [CrossRef]
- Goel, S.; Krop, I.E. Deciphering the Role of Phosphatidylinositol 3-Kinase Mutations in Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer. J. Clin. Oncol. 2015, 33, 1407–1409. [Google Scholar] [CrossRef]
- Berns, K.; Horlings, H.; Halfwerk, J.; Hennessy, B.; Linn, S.; Hauptmann, M.; Mills, G.; Van De Vijver, M.; Bernards, R. 57 A functional genetic approach identifies the PI3K pathway as a major determinant of Trastuzumab resistance in breast cancer. Eur. J. Cancer Suppl. 2009, 7, 17. [Google Scholar] [CrossRef]
- O’Brien, N.A.; Browne, B.C.; Chow, L.; Wang, Y.; Ginther, C.; Arboleda, J.; Duffy, M.J.; Crown, J.; O’Donovan, N.; Slamon, D.J. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol. Cancer Ther. 2010, 9, 1489–1502. [Google Scholar] [CrossRef]
- Tan, B.; Fleckenstein, L.; Yu, K.S.; Jang, I.J. Population Pharmacokinetics of Artesunate and Dihydroartemisinin in Healthy Volunteers. Am. J. Trop. Med. Hyg. 2008, 79, 135. [Google Scholar]
- Valsasina, B.; Beria, I.; Alli, C.; Alzani, R.; Avanzi, N.; Ballinari, D.; Cappella, P.; Caruso, M.; Casolaro, A.; Ciavolella, A.; et al. NMS-P937, an Orally Available, Specific Small-Molecule Polo-like Kinase 1 Inhibitor with Antitumor Activity in Solid and Hematologic Malignancies. Mol. Cancer Ther. 2012, 11, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gerhard, G.S. Heme Mediates Cytotoxicity from Artemisinin and Serves as a General Anti-Proliferation Target. PLoS ONE 2009, 4, e7472. [Google Scholar] [CrossRef] [PubMed]
- Jumbe, N.L.; Xin, Y.; Leipold, D.D.; Crocker, L.; Dugger, D.; Mai, E.; Sliwkowski, M.X.; Fielder, P.J.; Tibbitts, J. Modeling the efficacy of trastuzumab-DM1, an antibody drug conjugate, in mice. J. Pharmacokinet. Pharmacodyn. 2010, 37, 221–242. [Google Scholar] [CrossRef]
- Schwarz, L.; Hutchinson, K.E.; Rexer, B.N.; Estrada, M.V.; Gonzalez-Ericsson, P.I.; Sanders, M.E.; Dugger, T.C.; Formisano, L.; Guerrero-Zotano, A.; Red-Brewer, M.; et al. An ERBB1-3 Neutralizing Antibody Mixture With High Activity Against Drug-Resistant HER2+ Breast Cancers With ERBB Ligand Overexpression. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Phillips, G.D.L.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-Independent HER2/HER3/PI3K Complex Is Disrupted by Trastuzumab and Is Effectively Inhibited by the PI3K Inhibitor GDC-0941. Cancer Cell 2011, 20, 818. [Google Scholar] [CrossRef]
- Phillips, G.D.L.; Fields, C.T.; Li, G.; Dowbenko, D.; Schaefer, G.; Miller, K.; Andre, F.; Burris, H.A.; Albain, K.S.; Harbeck, N.; et al. Dual Targeting of HER2-Positive Cancer with Trastuzumab Emtansine and Pertuzumab: Critical Role for Neuregulin Blockade in Antitumor Response to Combination Therapy. Clin. Cancer Res. 2013, 20, 456–468. [Google Scholar] [CrossRef]
- Zadra, G.; Batista, J.L.; Loda, M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol. Cancer Res. 2015, 13, 1059–1072. [Google Scholar] [CrossRef]
- Shen, Y.; Sherman, J.W.; Chen, X.; Wang, R. Phosphorylation of CDC25C by AMP-activated protein kinase mediates a metabolic checkpoint during cell-cycle G2/M-phase transition. J. Biol. Chem. 2018, 293, 5185–5199. [Google Scholar] [CrossRef]
- Lindström, L.S.; Karlsson, E.; Wilking, U.; Johansson, U.; Hartman, J.; Lidbrink, E.; Hatschek, T.; Skoog, L.; Bergh, J. Clinically Used Breast Cancer Markers Such As Estrogen Receptor, Progesterone Receptor, and Human Epidermal Growth Factor Receptor 2 Are Unstable Throughout Tumor Progression. J. Clin. Oncol. 2012, 30, 2601–2608. [Google Scholar] [CrossRef]
- Yoshida, A.; Hayashi, N.; Suzuki, K.; Takimoto, M.; Nakamura, S.; Yamauchi, H. Change in HER2 status after neoadjuvant chemotherapy and the prognostic impact in patients with primary breast cancer. J. Surg. Oncol. 2017, 116, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Mc Gee, M.M. Targeting the Mitotic Catastrophe Signaling Pathway in Cancer. Mediat. Inflamm. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Rowinsky, E.K.; Eisenhauer, E.A.; Chaudhry, V.; Arbuck, S.G.; Donehower, R.C. Clinical toxicities encountered with paclitaxel (Taxol). Semin. Oncol. 1993, 20, 1–15. [Google Scholar]
- Shi, J.; Mitchison, T.J. Cell death response to anti-mitotic drug treatment in cell culture, mouse tumor model and the clinic. Endocrine-Related Cancer 2017, 24, T83–T96. [Google Scholar] [CrossRef]
- Bhola, N.E.; Jansen, V.M.; Bafna, S.; Giltnane, J.M.; Balko, J.M.; Estrada, M.V.; Meszoely, I.; Mayer, I.; Abramson, V.; Ye, F.; et al. Kinome-wide Functional Screen Identifies Role of PLK1 in Hormone-Independent, ER-Positive Breast Cancer. Cancer Res. 2015, 75, 405–414. [Google Scholar] [CrossRef]
- Degenhardt, Y.; Lampkin, T. Targeting Polo-like Kinase in Cancer Therapy. Clin. Cancer Res. 2010, 16, 384–389. [Google Scholar] [CrossRef]
- Watanabe, G.; Ishida, T.; Furuta, A.; Takahashi, S.; Watanabe, M.; Nakata, H.; Kato, S.; Ishioka, C.; Ohuchi, N. Combined Immunohistochemistry of PLK1, p21, and p53 for Predicting TP53 Status. Am. J. Surg. Pathol. 2015, 39, 1026–1034. [Google Scholar] [CrossRef]
- De Cárcer, G.; Venkateswaran, S.V.; Salgueiro, L.; El Bakkali, A.; Somogyi, K.; Rowald, K.; Montañés, P.; Sanclemente, M.; Escobar, B.; De Martino, A.; et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat. Commun. 2018, 9, 3012. [Google Scholar] [CrossRef]
- Gutteridge, R.E.A.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Saatci, Ö.; Borgoni, S.; Akbulut, O.; Durmus, S.; Raza, U.; Eyüpoğlu, E.; Alkan, C.; Akyol, A.; Kutuk, O.; Wiemann, S.; et al. Targeting PLK1 overcomes T-DM1 resistance via CDK1-dependent phosphorylation and inactivation of Bcl-2/xL in HER2-positive breast cancer. Oncogene 2018, 37, 2251–2269. [Google Scholar] [CrossRef] [PubMed]
- Jaglarz, M.K.; Bazile, F.; Laskowska, K.; Polański, Z.; Chesnel, F.; Borsuk, E.; Kloc, M.; Kubiak, J.Z. Association of TCTP with Centrosome and Microtubules. Biochem. Res. Int. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ramani, P.; Nash, R.; Sowa-Avugrah, E.; Rogers, C. High levels of polo-like kinase 1 and phosphorylated translationally controlled tumor protein indicate poor prognosis in neuroblastomas. J. Neuro-Oncol. 2015, 125, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.-L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Park, Y.H.; Ahn, J.S.; Im, Y.-H.; Nam, S.J.; Cho, S.Y.; Cho, E.Y. PIK3CA Mutations and Neoadjuvant Therapy Outcome in Patients with Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer: A Sequential Analysis. J. Breast Cancer 2018, 21, 382–390. [Google Scholar] [CrossRef]
- Kotoula, V.; Tsakiri, K.; Koliou, G.-A.; Lazaridis, G.; Papadopoulou, K.; Giannoulatou, E.; Tikas, I.; Christodoulou, C.; Chatzopoulos, K.; Bobos, M.; et al. Relapsed and De Novo Metastatic HER2-positive Breast Cancer Treated With Trastuzumab: Tumor Genotypes and Clinical Measures Associated With Patient Outcome. Clin. Breast Cancer 2019, 19, 113–125.e4. [Google Scholar] [CrossRef]
- Stern, H.M.; Gardner, H.; Burzykowski, T.; Elatre, W.; O’Brien, C.; Lackner, M.; Pestano, G.A.; Santiago, A.; Villalobos, I.; Eiermann, W.; et al. PTEN Loss Is Associated with Worse Outcome in HER2-Amplified Breast Cancer Patients but Is Not Associated with Trastuzumab Resistance. Clin. Cancer Res. 2015, 21, 2065–2074. [Google Scholar] [CrossRef]
- Tang, N.; Marshall, W.F. Centrosome positioning in vertebrate development. J. Cell Sci. 2012, 125, 4951–4961. [Google Scholar] [CrossRef]
- Gnanasekar, M.; Ramaswamy, K. Translationally controlled tumor protein of Brugia malayi functions as an antioxidant protein. Parasitol. Res. 2007, 101, 1533–1540. [Google Scholar] [CrossRef]
- Berdelle, N.; Nikolova, T.; Quirós, S.; Efferth, T.; Kaina, B. Artesunate Induces Oxidative DNA Damage, Sustained DNA Double-Strand Breaks, and the ATM/ATR Damage Response in Cancer Cells. Mol. Cancer Ther. 2011, 10, 2224–2233. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Dunstan, H.; Sauerbrey, A.; Miyachi, H.; Chitambar, C. The anti-malarial artesunate is also active against cancer. Int. J. Oncol. 2001, 18, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Kabeche, L.; Compton, D.A.; Powell, S.N.; Bastians, H. Mitotic DNA Damage Response: At the Crossroads of Structural and Numerical Cancer Chromosome Instabilities. Trends Cancer 2017, 3, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | EC50 (µM) |
|---|---|
| MCF10A-pBabe | 31.65 ± 8.81 |
| MCF10A-AATCTP | 65.95 ± 11.08 |
| MCF10A-WTTCTP | 133.00 ± 30.45 |
| HCC1569 | 8.50 ± 1.70 *** |
| HCC1954 | 7.61 ± 1.86 *** |
| HCC1954 cells | HCC1569 cells | ||||||||||
| DHA (µM) | Fractional inhibition | T-DM1 (µg/mL) | Fractional inhibition | DHA (µM) | Fractional inhibition | T-DM1 (µg/mL) | Fractional inhibition | ||||
| (D1) | (D2) | (D1) | (D2) | ||||||||
| 1 | 0.10 ± 0.08 | 0.01 | 0.04 ± 0.03 | 1.25 | 0.11 ± 0.06 | 1 | 0.03± 0.04 | ||||
| 2.5 | 0.15 ± 0.09 | 0.1 | 0.22 ± 0.07 | 2.5 | 0.26 ± 0.07 | 2.5 | 0.09 ± 0.07 | ||||
| 5 | 0.29 ± 0.14 | 0.25 | 0.38 ± 0.15 | 5 | 0.41 ± 0.06 | 5 | 0.13 ± 0.08 | ||||
| 10 | 0.61 ± 0.16 | 0.5 | 0.68 ± 0.13 | 10 | 0.54 ± 0.05 | 10 | 0.23 ± 0.02 | ||||
| 20 | 0.85 ± 0.08 | 1 | 0.78 ± 0.07 | 20 | 0.65 ± 0.03 | 20 | 0.21 ± 0.12 | ||||
| 50 | 0.57 ± 0.11 | ||||||||||
| 200 | 0.59 ± 0.04 | ||||||||||
| Parameter | Parameter | Parameter | Parameter | ||||||||
| EC50 (µM) | 7.61 ± 1.86 | EC50 (µg/mL) | 0.33 ± 0.18 | EC50 (µM) | 8.5 ± 1.7 | EC50 (µg/mL) | 101.70 ± 44.8 | ||||
| m | 1.88 ± 0.47 | m | 0.88 ± 0.26 | m | 0.90 ± 0.13 | m | 0.78 ± 0.24 | ||||
| r | 0.98 ± 0.01 | r | 0.96 ± 0.02 | r | 0.98 ± 0.02 | r | 0.97 ± 0.01 | ||||
| (D1) + (D2) Ratio 10:1 | (D1) + (D2) Ratio 1:1 | ||||||||||
| Fractional inhibition | CI | DRI (D1) | DRI (D2) | Fractional inhibition | CI | DRI (D1) | DRI (D2) | ||||
| 2.5 | 0.25 | 0.74 ± 0.08 | 0.53 ± 0.11 | 6.1 | 3.5 | 2.5 | 2.5 | 0.30 ± 0.10 | 0.35 ± 0.24 | 1.47 | 9.32 |
| 5 | 0.5 | 0.81 ± 0.06 | 0.77 ± 0.31 | 4.1 | 2.65 | 5 | 5 | 0.51 ± 0.02 | 0.57 ± 0.18 | 1.84 | 15.55 |
| 10 | 1 | 0.90 ± 0.04 | 0.99 ± 0.53 | 10 | 10 | 0.59 ± 0.03 | 0.59 ± 0.37 | 1.28 | 12.07 | ||
| 20 | 2 | 0.94 ± 0.02 | 1.28 ± 0.80 | 20 | 20 | 0.70 ± 0.01 | 0.90 ± 0.37 | 1.06 | 11.63 | ||
| (D1) + (D2) No constant ratio | (D1) + (D2) No constant ratio | ||||||||||
| Fractional inhibition | CI | DRI (D1) | DRI (D2) | Fractional inhibition | CI | DRI (D1) | DRI (D2) | ||||
| 1.25 | 0.1 | 0.21 ± 0.10 | 2.15 ± 0.35 | 1.25 | 2.5 | 0.26 ± 0.03 | 0.71 ± 0.03 | 2.39 | 7.10 | ||
| 1.25 | 0.25 | 0.44 ± 0.14 | 1.14 ± 0.30 | 1.25 | 5 | 0.28 ± 0.01 | 0.84 ± 0.11 | 2.66 | 4.08 | ||
| 2.5 | 0.1 | 0.26 ± 0.01 | 1.80 ± 0.70 | 2.5 | 2.5 | 0.43 ± 0.09 | 0.56 ± 0.13 | 2.64 | 20.00 | ||
| 2.5 | 0.25 | 0.64 ± 0.14 | 0.48 ± 0.17 | 4.35 | 2.16 | 2.5 | 5 | 0.44 ± 0.07 | 0.58 ± 0.02 | 2.75 | 10.61 |
| 2.5 | 0.5 | 0.74 ± 0.11 | 0.64 ± 0.14 | 6.18 | 1.75 | 2.5 | 10 | 0.45 ± 0.08 | 0.59 ± 0.13 | 2.87 | 5.60 |
| 5 | 0.1 | 0.40 ± 0.06 | 1.51 ± 0.45 | 5 | 2.5 | 0.50 ± 0.03 | 0.63 ± 0.11 | 1.76 | 29.45 | ||
| 5 | 0.25 | 0.75 ± 0.17 | 1.60 ± 0.45 | 5 | 5 | 0.50 ± 0.06 | 0.67 ± 0.13 | 1.76 | 14.72 | ||
| 5 | 0.5 | 0.79 ± 0.09 | 0.96 ± 0.31 | 5 | 10 | 0.54 ± 0.07 | 0.61 ± 0.13 | 2.08 | 9.15 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amico, S.; Krasnowska, E.K.; Manni, I.; Toietta, G.; Baldari, S.; Piaggio, G.; Ranalli, M.; Gambacurta, A.; Vernieri, C.; Di Giacinto, F.; et al. DHA Affects Microtubule Dynamics Through Reduction of Phospho-TCTP Levels and Enhances the Antiproliferative Effect of T-DM1 in Trastuzumab-Resistant HER2-Positive Breast Cancer Cell Lines. Cells 2020, 9, 1260. https://doi.org/10.3390/cells9051260
D’Amico S, Krasnowska EK, Manni I, Toietta G, Baldari S, Piaggio G, Ranalli M, Gambacurta A, Vernieri C, Di Giacinto F, et al. DHA Affects Microtubule Dynamics Through Reduction of Phospho-TCTP Levels and Enhances the Antiproliferative Effect of T-DM1 in Trastuzumab-Resistant HER2-Positive Breast Cancer Cell Lines. Cells. 2020; 9(5):1260. https://doi.org/10.3390/cells9051260
Chicago/Turabian StyleD’Amico, Silvia, Ewa Krystyna Krasnowska, Isabella Manni, Gabriele Toietta, Silvia Baldari, Giulia Piaggio, Marco Ranalli, Alessandra Gambacurta, Claudio Vernieri, Flavio Di Giacinto, and et al. 2020. "DHA Affects Microtubule Dynamics Through Reduction of Phospho-TCTP Levels and Enhances the Antiproliferative Effect of T-DM1 in Trastuzumab-Resistant HER2-Positive Breast Cancer Cell Lines" Cells 9, no. 5: 1260. https://doi.org/10.3390/cells9051260
APA StyleD’Amico, S., Krasnowska, E. K., Manni, I., Toietta, G., Baldari, S., Piaggio, G., Ranalli, M., Gambacurta, A., Vernieri, C., Di Giacinto, F., Bernassola, F., de Braud, F., & Lucibello, M. (2020). DHA Affects Microtubule Dynamics Through Reduction of Phospho-TCTP Levels and Enhances the Antiproliferative Effect of T-DM1 in Trastuzumab-Resistant HER2-Positive Breast Cancer Cell Lines. Cells, 9(5), 1260. https://doi.org/10.3390/cells9051260