Hepatic NAPE-PLD Is a Key Regulator of Liver Lipid Metabolism

 ,
,  , ,
, ,  , , , ,
, , , , 
Abstract

1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Generation of Napepld∆Hep Mice
2.3. Phenotyping in Normal Diet (ND) Condition
2.4. Phenotyping in High-Fat Diet (HFD) Condition
2.5. Oral Glucose Tolerance Test
2.6. Insulin Resistance Index
2.7. LPS Injection Experiment
2.8. Tissue Sampling
2.9. Hepatocyte Isolation
2.10. RNA Preparation and Real-Time qPCR Analysis
2.11. Adipocyte Histological Analysis
2.12. Hepatic Lipid Content Analysis by Oil Red O Staining
2.13. Extraction of Liver Lipids
2.14. Biochemical Analyses
2.15. Lipidomics Analysis
2.16. Ethics Statement
2.17. Statistical Analysis
3. Results
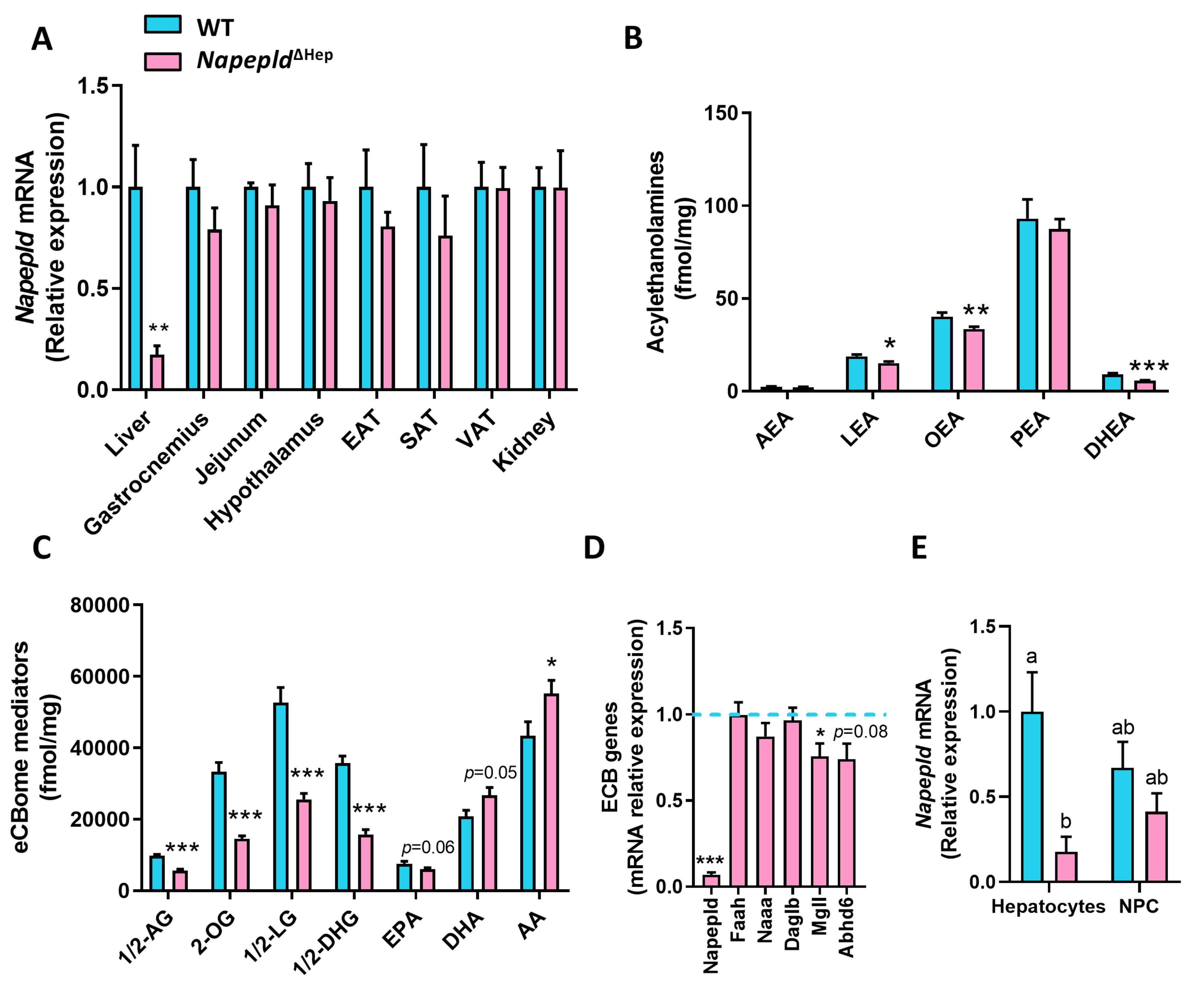
3.1. Hepatocyte-Specific Deletion of Napepld
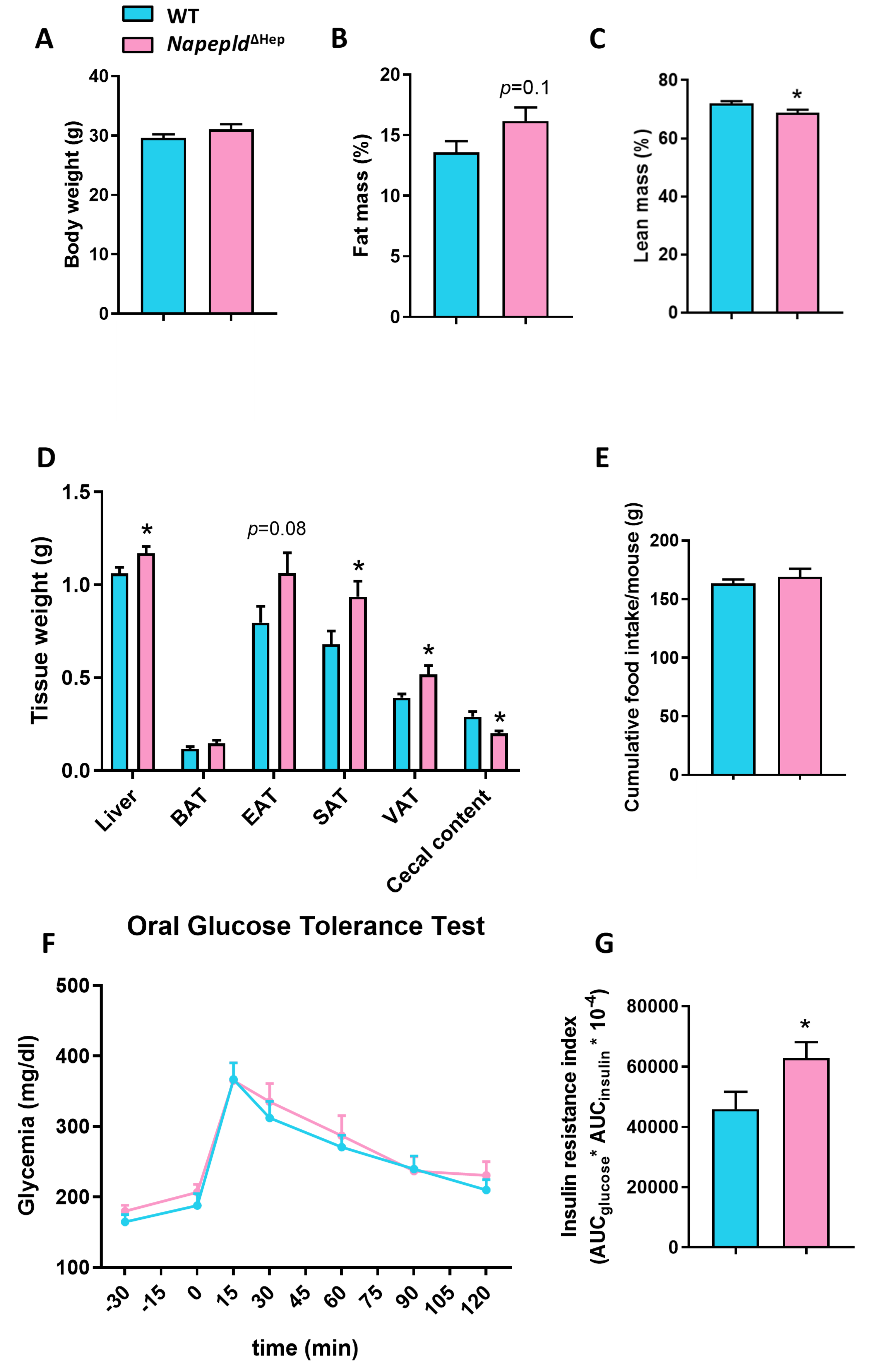
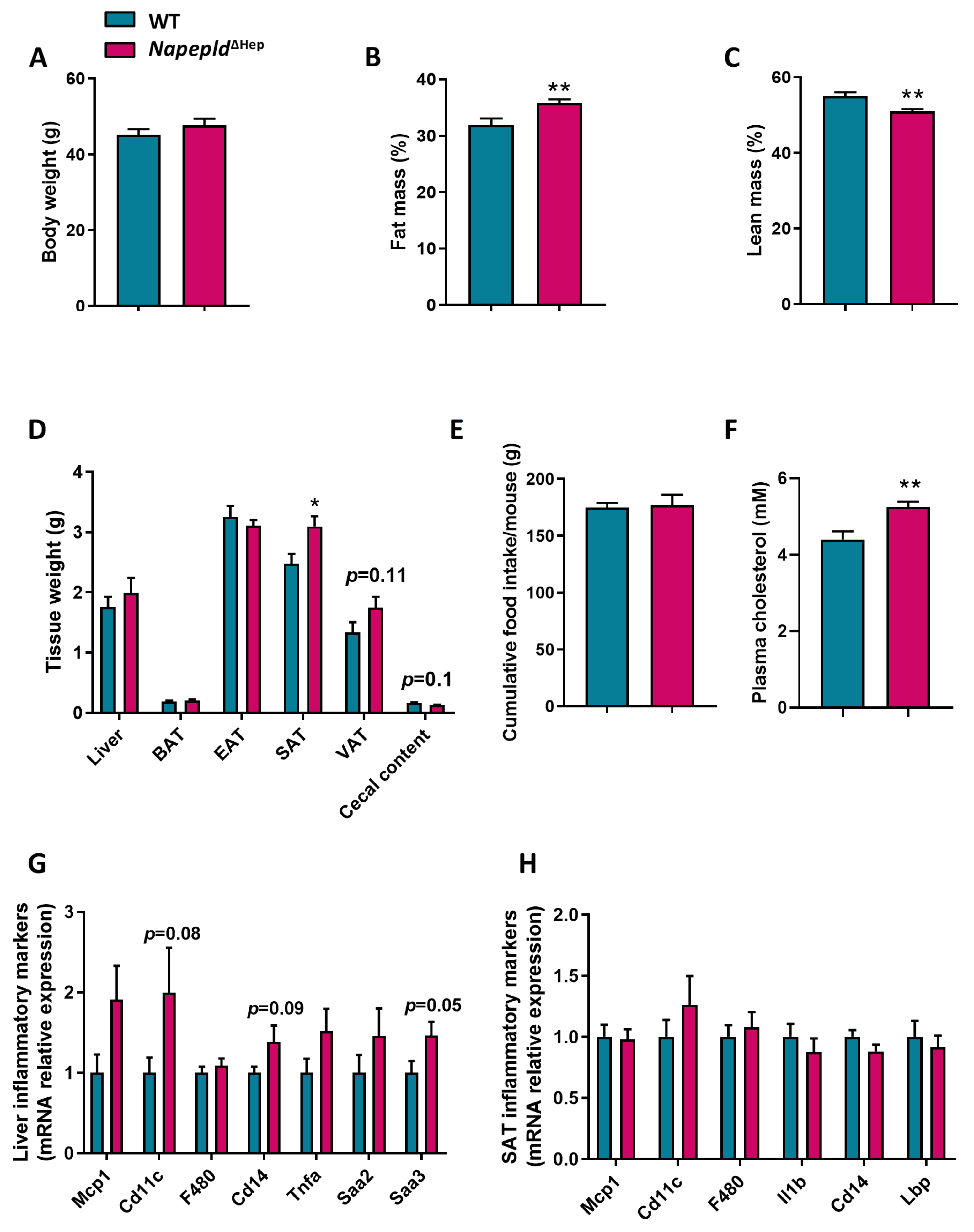
3.2. Napepld∆Hep Mice Develop a High-Fat Diet-Like Phenotype upon Normal Diet
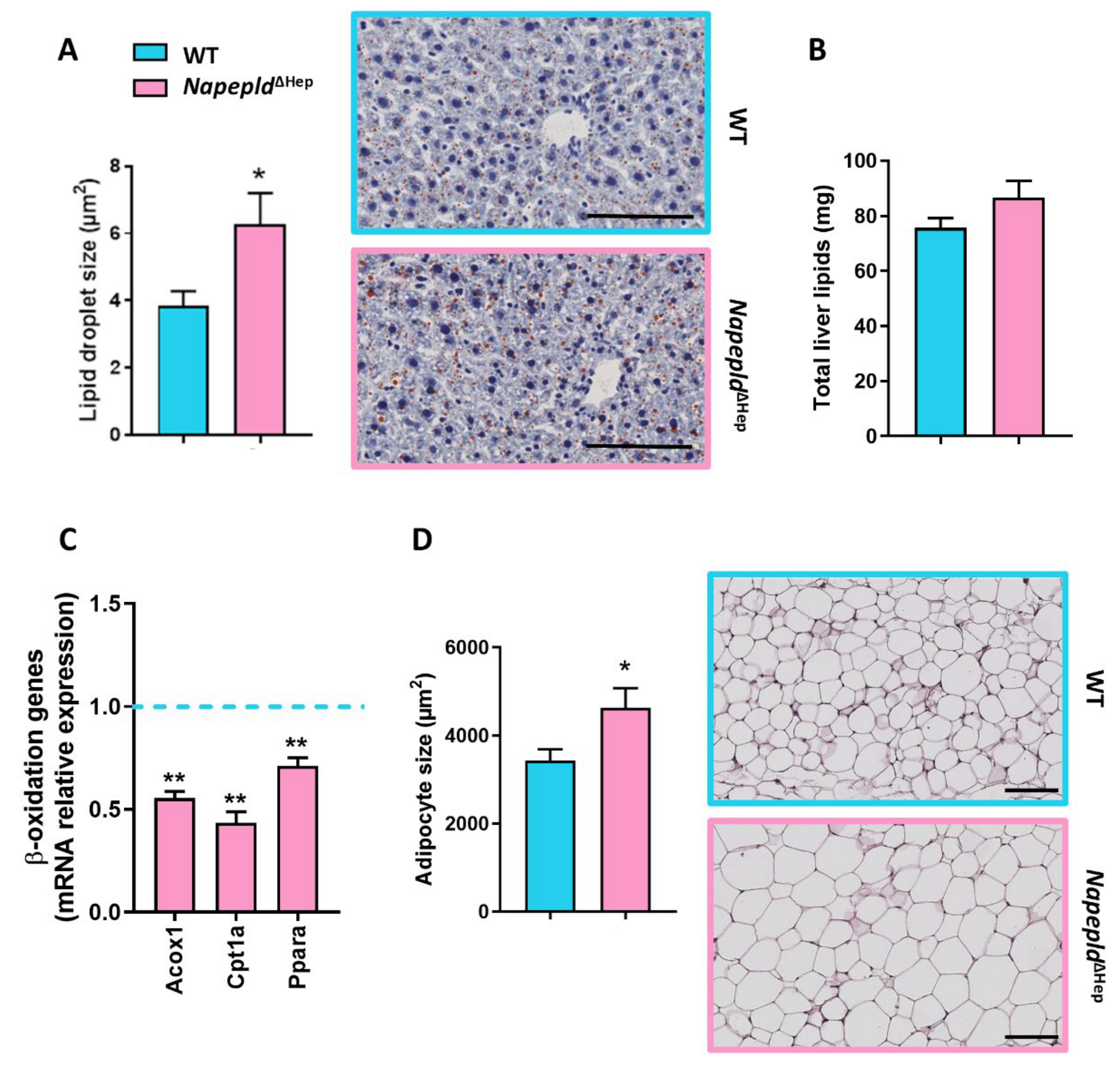
3.3. Napepld∆Hep Mice Are More Sensitive to Liver Lipid Accumulation
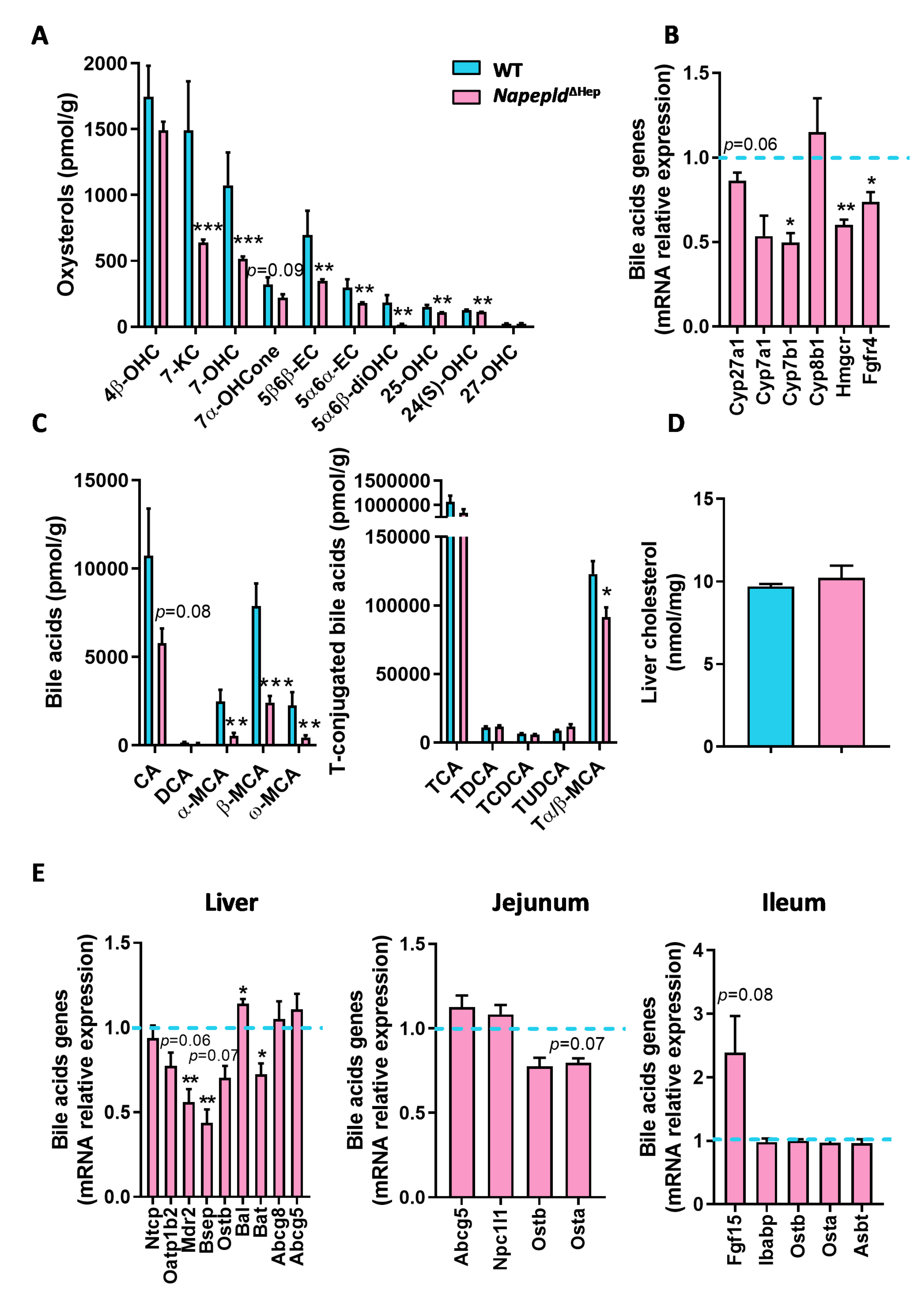
3.4. Hepatocyte Napepld Deletion Modifies Liver Bioactive Lipid Metabolism
3.5. Deletion of Napepld Partially Accentuates the Obese Phenotype Induced by a High-Fat Diet
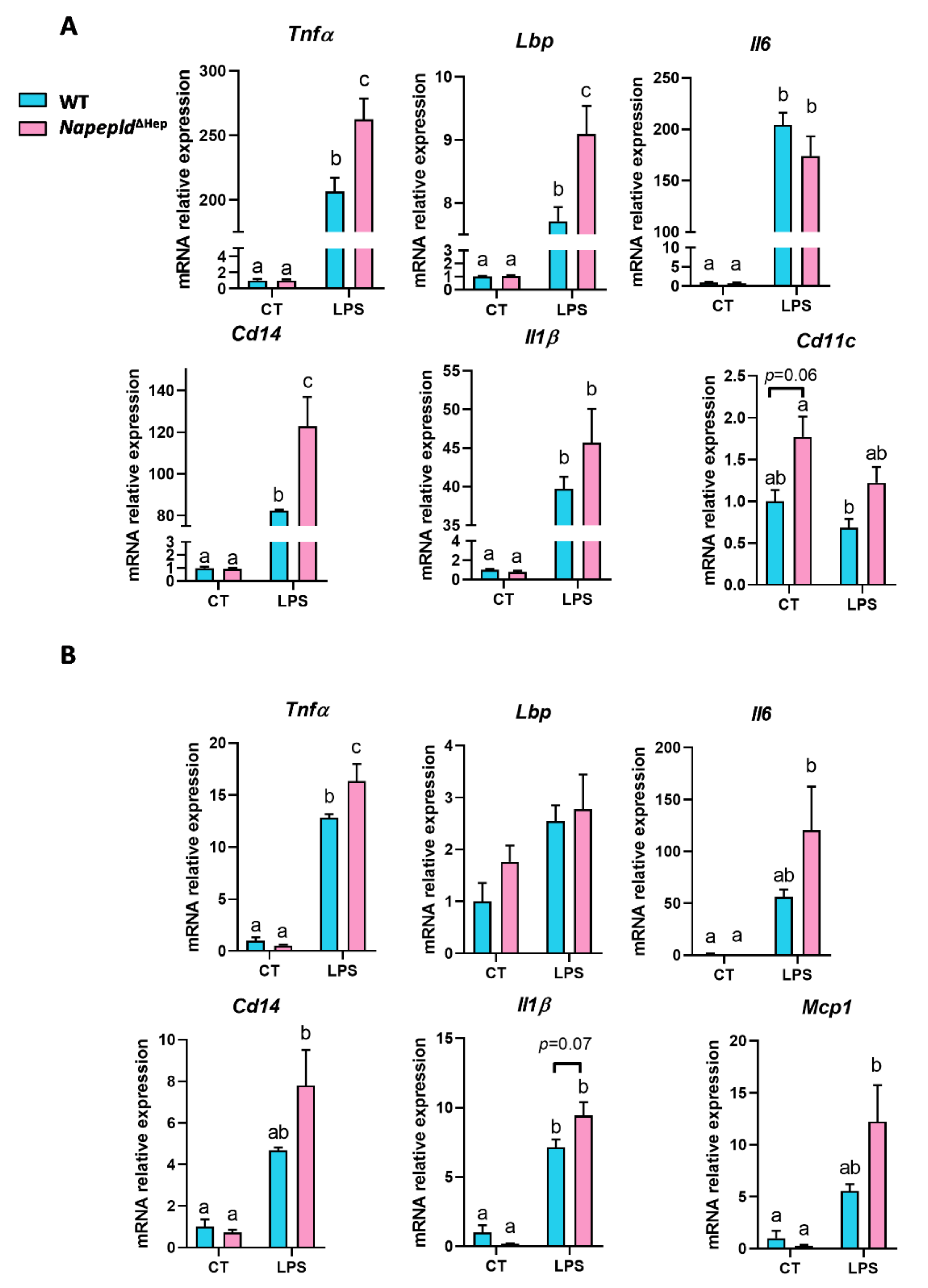
3.6. Napepld∆Hep Mice are More Sensitive to Inflammation
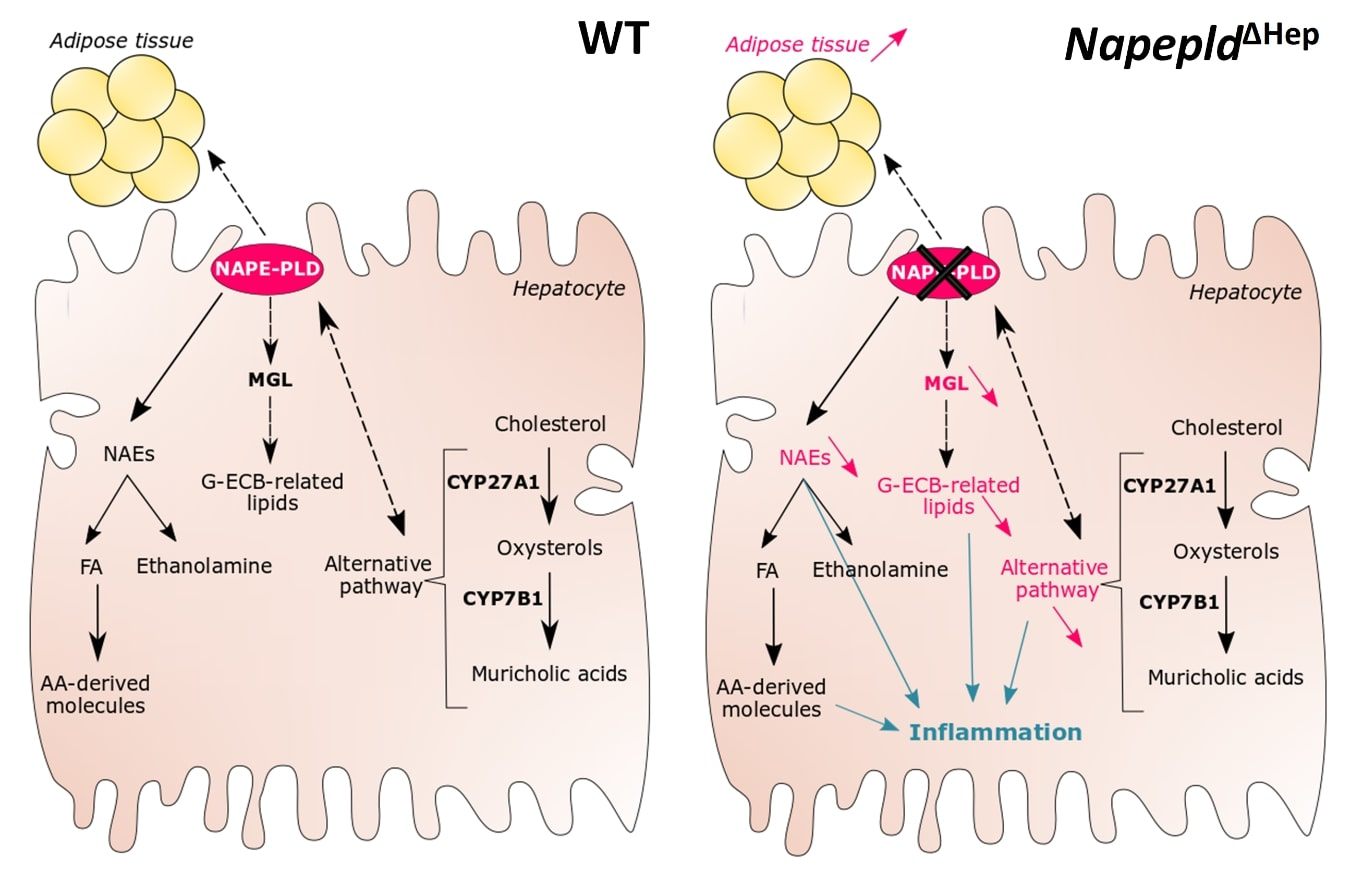
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maccarrone, M.; Bab, I.; Biro, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after thc. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids--at the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef]
- Gatta-Cherifi, B.; Cota, D. New insights on the role of the endocannabinoid system in the regulation of energy balance. Int. J. Obes. 2016, 40, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Van Hul, M.; Lefort, C.; Depommier, C.; Rastelli, M.; Everard, A. Microbial regulation of organismal energy homeostasis. Nat. Metab. 2019, 1, 34–46. [Google Scholar] [CrossRef]
- Jamshidi, N.; Taylor, D.A. Anandamide administration into the ventromedial hypothalamus stimulates appetite in rats. Br. J. Pharmacol. 2001, 134, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.S.; Diep, T.A. N-acylethanolamines, anandamide and food intake. Biochem. Pharmacol. 2009, 78, 553–560. [Google Scholar] [CrossRef]
- Piomelli, D. A fatty gut feeling. Trends Endocrinol. Metab. 2013, 24, 332–341. [Google Scholar] [CrossRef]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- Esposito, G.; Capoccia, E.; Turco, F.; Palumbo, I.; Lu, J.; Steardo, A.; Cuomo, R.; Sarnelli, G.; Steardo, L. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent ppar-alpha activation. Gut 2014, 63, 1300–1312. [Google Scholar] [CrossRef]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Bátkai, S.; Harvey-White, J.; Mackie, K.; Offertáler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic cb1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar] [CrossRef]
- Purohit, V.; Rapaka, R.; Shurtleff, D. Role of cannabinoids in the development of fatty liver (steatosis). AAPS J. 2010, 12, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Morishita, J.; Tsuboi, K.; Tonai, T.; Ueda, N. Molecular characterization of a phospholipase d generating anandamide and its congeners. J. Biol. Chem. 2004, 279, 5298–5305. [Google Scholar] [CrossRef] [PubMed]
- Margheritis, E.; Castellani, B.; Magotti, P.; Peruzzi, S.; Romeo, E.; Natali, F.; Mostarda, S.; Gioiello, A.; Piomelli, D.; Garau, G. Bile acid recognition by nape-pld. ACS Chem. Biol. 2016, 11, 2908–2914. [Google Scholar] [CrossRef] [PubMed]
- Magotti, P.; Bauer, I.; Igarashi, M.; Babagoli, M.; Marotta, R.; Piomelli, D.; Garau, G. Structure of human n-acylphosphatidylethanolamine-hydrolyzing phospholipase d: Regulation of fatty acid ethanolamide biosynthesis by bile acids. Structure 2015, 23, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.; Saghatelian, A.; Simon, G.M.; Cravatt, B.F. Inactivation of n-acyl phosphatidylethanolamine phospholipase d reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry 2006, 45, 4720–4726. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.M.; Cravatt, B.F. Characterization of mice lacking candidate n-acyl ethanolamine biosynthetic enzymes provides evidence for multiple pathways that contribute to endocannabinoid production in vivo. Mol. Biosyst. 2010, 6, 1411–1418. [Google Scholar] [CrossRef]
- Powell, D.R.; Gay, J.P.; Wilganowski, N.; Doree, D.; Savelieva, K.V.; Lanthorn, T.H.; Read, R.; Vogel, P.; Hansen, G.M.; Brommage, R.; et al. Diacylglycerol lipase alpha knockout mice demonstrate metabolic and behavioral phenotypes similar to those of cannabinoid receptor 1 knockout mice. Front. Endocrinol. 2015, 6, 86. [Google Scholar] [CrossRef]
- Inoue, M.; Tsuboi, K.; Okamoto, Y.; Hidaka, M.; Uyama, T.; Tsutsumi, T.; Tanaka, T.; Ueda, N.; Tokumura, A. Peripheral tissue levels and molecular species compositions of n-acyl-phosphatidylethanolamine and its metabolites in mice lacking n-acyl-phosphatidylethanolamine-specific phospholipase d. J. Biochem. 2017, 162, 449–458. [Google Scholar] [CrossRef]
- Leishman, E.; Mackie, K.; Luquet, S.; Bradshaw, H.B. Lipidomics profile of a nape-pld ko mouse provides evidence of a broader role of this enzyme in lipid metabolism in the brain. Biochim. Biophys. Acta 2016, 1861, 491–500. [Google Scholar] [CrossRef]
- Gunschmann, C.; Chiticariu, E.; Garg, B.; Hiz, M.M.; Mostmans, Y.; Wehner, M.; Scharfenberger, L. Transgenic mouse technology in skin biology: Inducible gene knockout in mice. J. Investig. Dermatol. 2014, 134, e22. [Google Scholar] [CrossRef]
- Geurts, L.; Everard, A.; Van Hul, M.; Essaghir, A.; Duparc, T.; Matamoros, S.; Plovier, H.; Castel, J.; Denis, R.G.; Bergiers, M.; et al. Adipose tissue nape-pld controls fat mass development by altering the browning process and gut microbiota. Nat. Commun. 2015, 6, 6495. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Plovier, H.; Rastelli, M.; Van Hul, M.; de Wouters d’Oplinter, A.; Geurts, L.; Druart, C.; Robine, S.; Delzenne, N.M.; Muccioli, G.G.; et al. Intestinal epithelial n-acylphosphatidylethanolamine phospholipase d links dietary fat to metabolic adaptations in obesity and steatosis. Nat. Commun. 2019, 10, 457. [Google Scholar] [CrossRef] [PubMed]
- Wangensteen, T.; Akselsen, H.; Holmen, J.; Undlien, D.; Retterstol, L. A common haplotype in napepld is associated with severe obesity in a norwegian population-based cohort (the hunt study). Obesity 2011, 19, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Collaboration N.C.D.R.F. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar]
- Loomba, R.; Sanyal, A.J. The global nafld epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef]
- Rinella, M.E.; Sanyal, A.J. Nafld in 2014: Genetics, diagnostics and therapeutic advances in nafld. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 65–66. [Google Scholar] [CrossRef]
- Schuler, M.; Dierich, A.; Chambon, P.; Metzger, D. Efficient temporally controlled targeted somatic mutagenesis in hepatocytes of the mouse. Genesis 2004, 39, 167–172. [Google Scholar] [CrossRef]
- Lefort, C.; Van Hul, M.; Delzenne, N.M.; Everard, A.; Cani, P.D. Hepatic myd88 regulates liver inflammation by altering synthesis of oxysterols. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E99–E108. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar]
- Manca, C.; Boubertakh, B.; Leblanc, N.; Deschenes, T.; Lacroix, S.; Martin, C.; Houde, A.; Veilleux, A.; Flamand, N.; Muccioli, G.G.; et al. Germ-free mice exhibit profound gut microbiota-dependent alterations of intestinal endocannabinoidome signaling. J. Lipid Res. 2020, 61, 70–85. [Google Scholar] [CrossRef]
- Mutemberezi, V.; Masquelier, J.; Guillemot-Legris, O.; Muccioli, G.G. Development and validation of an hplc-ms method for the simultaneous quantification of key oxysterols, endocannabinoids, and ceramides: Variations in metabolic syndrome. Anal. Bioanal. Chem. 2016, 408, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Guillemot-Legris, O.; Mutemberezi, V.; Cani, P.D.; Muccioli, G.G. Obesity is associated with changes in oxysterol metabolism and levels in mice liver, hypothalamus, adipose tissue and plasma. Sci. Rep. 2016, 6, 19694. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Harvey-White, J.; Huang, B.X.; Kim, H.Y.; Luquet, S.; Palmiter, R.D.; Krystal, G.; Rai, R.; Mahadevan, A.; et al. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 2008, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Okamoto, Y.; Ikematsu, N.; Inoue, M.; Shimizu, Y.; Uyama, T.; Wang, J.; Deutsch, D.G.; Burns, M.P.; Ulloa, N.M.; et al. Enzymatic formation of n-acylethanolamines from n-acylethanolamine plasmalogen through n-acylphosphatidylethanolamine-hydrolyzing phospholipase d-dependent and -independent pathways. Biochim. Biophys. Acta 2011, 1811, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Muccioli, G.G. Endocannabinoid biosynthesis and inactivation, from simple to complex. Drug Discov. Today 2010, 15, 474–483. [Google Scholar] [CrossRef]
- Movita, D.; Kreefft, K.; Biesta, P.; van Oudenaren, A.; Leenen, P.J.; Janssen, H.L.; Boonstra, A. Kupffer cells express a unique combination of phenotypic and functional characteristics compared with splenic and peritoneal macrophages. J. Leukoc. Biol. 2012, 92, 723–733. [Google Scholar] [CrossRef]
- Elvevold, K.; Smedsrod, B.; Martinez, I. The liver sinusoidal endothelial cell: A cell type of controversial and confusing identity. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G391–G400. [Google Scholar] [CrossRef]
- Carpino, G.; Morini, S.; Ginanni Corradini, S.; Franchitto, A.; Merli, M.; Siciliano, M.; Gentili, F.; Onetti Muda, A.; Berloco, P.; Rossi, M.; et al. Alpha-sma expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig. Liver Dis. 2005, 37, 349–356. [Google Scholar] [CrossRef]
- Manco, R.; Clerbaux, L.A.; Verhulst, S.; Bou Nader, M.; Sempoux, C.; Ambroise, J.; Bearzatto, B.; Gala, J.L.; Horsmans, Y.; van Grunsven, L.; et al. Reactive cholangiocytes differentiate into proliferative hepatocytes with efficient DNA repair in mice with chronic liver injury. J. Hepatol. 2019, 70, 1180–1191. [Google Scholar] [CrossRef]
- Cani, P.D.; Geurts, L.; Matamoros, S.; Plovier, H.; Duparc, T. Glucose metabolism: Focus on gut microbiota, the endocannabinoid system and beyond. Diabetes Metab. 2014, 40, 246–257. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Tonegawa, T.; Nakane, S.; Yamashita, A.; Ishima, Y.; Waku, K. Transacylase-mediated and phosphodiesterase-mediated synthesis of n-arachidonoylethanolamine, an endogenous cannabinoid-receptor ligand, in rat brain microsomes. Comparison with synthesis from free arachidonic acid and ethanolamine. Eur. J. Biochem. 1996, 240, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M. Missing pieces to the endocannabinoid puzzle. Trends Mol. Med. 2020, 26, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef]
- Ueda, N.; Yamanaka, K.; Yamamoto, S. Purification and characterization of an acid amidase selective for n-palmitoylethanolamine, a putative endogenous anti-inflammatory substance. J. Biol. Chem. 2001, 276, 35552–35557. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Maurelli, S.; Melck, D.; De Petrocellis, L.; Di Marzo, V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J. Biol. Chem. 1997, 272, 3315–3323. [Google Scholar] [CrossRef]
- Muccioli, G.G.; Stella, N. Microglia produce and hydrolyze palmitoylethanolamide. Neuropharmacology 2008, 54, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Alhouayek, M.; Muccioli, G.G. Harnessing the anti-inflammatory potential of palmitoylethanolamide. Drug Discov. Today 2014, 19, 1632–1639. [Google Scholar] [CrossRef]
- Zhu, C.; Solorzano, C.; Sahar, S.; Realini, N.; Fung, E.; Sassone-Corsi, P.; Piomelli, D. Proinflammatory stimuli control n-acylphosphatidylethanolamine-specific phospholipase d expression in macrophages. Mol. Pharmacol. 2011, 79, 786–792. [Google Scholar] [CrossRef]
- Ohara, M.; Ohnishi, S.; Hosono, H.; Yamamoto, K.; Fu, Q.; Maehara, O.; Suda, G.; Sakamoto, N. Palmitoylethanolamide ameliorates carbon tetrachloride-induced liver fibrosis in rats. Front. Pharmacol. 2018, 9, 709. [Google Scholar] [CrossRef]
- Petrosino, S.; Di Marzo, V. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br. J. Pharmacol. 2017, 174, 1349–1365. [Google Scholar] [CrossRef]
- Turcotte, C.; Dumais, E.; Archambault, A.S.; Martin, C.; Blanchet, M.R.; Bissonnette, E.; Boulet, L.P.; Laviolette, M.; Di Marzo, V.; Flamand, N. Human leukocytes differentially express endocannabinoid-glycerol lipases and hydrolyze 2-arachidonoyl-glycerol and its metabolites from the 15-lipoxygenase and cyclooxygenase pathways. J. Leukoc. Biol. 2019, 106, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. Fat signals—Lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef]
- Schaap, F.G.; Trauner, M.; Jansen, P.L. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef]
- Dawson, P.A.; Karpen, S.J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. [Google Scholar] [CrossRef]
- Lin, B.C.; Wang, M.; Blackmore, C.; Desnoyers, L.R. Liver-specific activities of fgf19 require klotho beta. J. Biol. Chem. 2007, 282, 27277–27284. [Google Scholar] [CrossRef]
- Kong, B.; Wang, L.; Chiang, J.Y.; Zhang, Y.; Klaassen, C.D.; Guo, G.L. Mechanism of tissue-specific farnesoid x receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 2012, 56, 1034–1043. [Google Scholar] [CrossRef]
- Somm, E.; Henry, H.; Bruce, S.J.; Aeby, S.; Rosikiewicz, M.; Sykiotis, G.P.; Asrih, M.; Jornayvaz, F.R.; Denechaud, P.D.; Albrecht, U.; et al. Beta-klotho deficiency protects against obesity through a crosstalk between liver, microbiota, and brown adipose tissue. JCI Insight. 2017, 2. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring fxr antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Douglass, J.D.; Zhou, Y.X.; Wu, A.; Zadroga, J.A.; Gajda, A.M.; Lackey, A.I.; Lang, W.; Chevalier, K.M.; Sutton, S.W.; Zhang, S.P.; et al. Global deletion of mgl in mice delays lipid absorption and alters energy homeostasis and diet-induced obesity. J. Lipid Res. 2015, 56, 1153–1171. [Google Scholar] [CrossRef] [PubMed]
- Geurts, L.; Muccioli, G.G.; Delzenne, N.M.; Cani, P.D. Chronic endocannabinoid system stimulation induces muscle macrophage and lipid accumulation in type 2 diabetic mice independently of metabolic endotoxaemia. PLoS ONE 2013, 8, e55963. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Schmidt, S.F.; Friedman, J.M. Developmental role for endocannabinoid signaling in regulating glucose metabolism and growth. Diabetes 2013, 62, 2359–2367. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
- Jiang, C.; Xie, C.; Lv, Y.; Li, J.; Krausz, K.W.; Shi, J.; Brocker, C.N.; Desai, D.; Amin, S.G.; Bisson, W.H.; et al. Intestine-selective farnesoid x receptor inhibition improves obesity-related metabolic dysfunction. Nat. Commun. 2015, 6, 10166. [Google Scholar] [CrossRef]
- Ahmad, T.R.; Haeusler, R.A. Bile acids in glucose metabolism and insulin signalling—Mechanisms and research needs. Nat. Rev. Endocrinol. 2019, 15, 701–712. [Google Scholar] [CrossRef]
- Silvestri, C.; Di Marzo, V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 2013, 17, 475–490. [Google Scholar] [CrossRef]
- Guillemot-Legris, O.; Mutemberezi, V.; Muccioli, G.G. Oxysterols in metabolic syndrome: From bystander molecules to bioactive lipids. Trends Mol. Med. 2016, 22, 594–614. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Wahli, W.; Michalik, L. Ppars at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D.; Sasso, O. Peripheral gating of pain signals by endogenous lipid mediators. Nat. Neurosci. 2014, 17, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Alhouayek, M.; Muccioli, G.G. Cox-2-derived endocannabinoid metabolites as novel inflammatory mediators. Trends Pharmacol. Sci. 2014, 35, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Spann, N.J.; Glass, C.K. Sterols and oxysterols in immune cell function. Nat. Immunol. 2013, 14, 893–900. [Google Scholar] [CrossRef]
- Mazuy, C.; Helleboid, A.; Staels, B.; Lefebvre, P. Nuclear bile acid signaling through the farnesoid x receptor. Cell. Mol. Life Sci. 2015, 72, 1631–1650. [Google Scholar] [CrossRef]
- Duparc, T.; Plovier, H.; Marrachelli, V.G.; Van Hul, M.; Essaghir, A.; Stahlman, M.; Matamoros, S.; Geurts, L.; Pardo-Tendero, M.M.; Druart, C.; et al. Hepatocyte myd88 affects bile acids, gut microbiota and metabolome contributing to regulate glucose and lipid metabolism. Gut 2017, 66, 620–632. [Google Scholar] [CrossRef]
- Izzo, A.A.; Piscitelli, F.; Capasso, R.; Marini, P.; Cristino, L.; Petrosino, S.; Di Marzo, V. Basal and fasting/refeeding-regulated tissue levels of endogenous PPAR-alpha ligands in Zucker rats. Obesity (Silver Spring). 2010, 18, 55–62. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Forward Primer Sequence (5’-3’) | Reverse Primer Sequence (5’-3’) |
|---|---|---|---|
| Abcb11 | BSEP | AGATACAACCGAAGGGGACA | TCAACTTCTTCCACAAGCACA |
| Abcb4 | MDR2 | GAGCCCGTGCTGTTCTCTAC | TCTGTTTCTGTCCCCCACTC |
| Abcg5 | ABCG5 | ACCTTACCCACGGTTCCTTT | ACGCATAATCACTGCCTGCT |
| Abcg8 | ABCG8 | CCGTCGTCAGATTTCCAATGA | GGCTTCCGACCCATGAATG |
| Abhd6 | ABHD6 | CTGTCCATAGTGGGGCAAGT | TCAGATGGGTAGTAAGCGGC |
| Acox1 | ACOX1 | CTATGGGATCAGCCAGAAAGG | AGTCAAAGGCATCCACCAAAG |
| Acta2 | αSMA | GGCTGGAGAATTGGATCT | CCAGCAAAGGTCAGAGAAGG |
| Adgre1 | F4/80 | TGACAACCAGACGGCTTGTG | GCAGGCGAGGAAAAGATAGTGT |
| Baat | BAT | GCACAGGCTCATCAACAAGA | TAGAGCACACCACGTTCCTG |
| Ccl2 | MCP1 | GCAGTTAACGCCCCACTCA | TCCAGCCTACTCATTGGGATCA |
| Cd14 | CD14 | CCTGCCCTCTCCACCTTAGAC | TCAGTCCTCTCTCGCCCAAT |
| Cpt1a | CPT1α | AGACCGTGAGGAACTCAAACCTAT | TGAAGAGTCGCTCCCACT |
| Cyp27a1 | CYP27A1 | TCTGGCTACCTGCACTTCCT | GTGTGTTGGATGTCGTGTCC |
| Cyp7a1 | CYP7A1 | GGGATTGCTGTGGTAGTGAGC | GGTATGGAATCAACCCGTTGTC |
| Cyp7b1 | CYP7B1 | TAGGCATGACGATCCTGAAA | TCTCTGGTGAAGTGGACTGAAA |
| Cyp8b1 | CYP8B1 | GATCCGTCGCGGAGATAAGG | CGGGTTGAGGAACCGATCAT |
| Daglb | DAGLβ | CTCCACCAGCAACAAGACAA | GCAGTTCTCCACTTCTGCATC |
| Faah | FAAH | GTGAGGATTTGTTCCGCTTG | GGAGTGGGCATGGTGTAGTT |
| Fabp6 | IBABP | CAAGGCTACCGTGAAGATGGA | CCCACGACCTCCGAAGTCT |
| Fgf15 | FGF15 | GAGGACCAAAACGAACGAAATT | ACGTCCTTGATGGCAATCG |
| Fgfr4 | FGFR4 | CTCGATCCGCTTTGGGAATTC | CAGGTCTGCCAAATCCTTGTC |
| Hmgcr | HMGCR | TGGTGGGACCAACCTTCTAC | GCCATCACAGTGCCACATAC |
| Hnf4a | HNF4α | AAGAGGTCCATGGTGTTTAAGG | ATCGAGGATGCGGATGGA |
| Il1b | IL1β | TCGCTCAGGGTCACAAGAAA | CATCAGAGGCAAGGAGGAAAAC |
| Il6 | IL6 | ACAAGTCGGAGGCTTAATTACACAT | TTGCCATTGCACAACTCTTTTC |
| Itgax | CD11c | ACGTCAGTACAAGGAGATGTTGGA | ATCCTATTGCAGAATGCTTCTTTACC |
| Krt19 | CK19 | AGCGTGATCAGCGGTTTTG | CCTGGTTCTGGCGCTCTATG |
| Lbp | LBP | GTCCTGGGAATCTGTCCTTG | CCGGTAACCTTGCTGTTGTT |
| Mgll | MGL | ATGGTCCTGATTTCACCTCTGGT | TCAACCTCCGACTTGTTCCGAGACA |
| Naaa | NAAA | ATTATGACCATTGGAAGCCTGCA | CGCTCATCACTGTAGTATAAATTGTGTAG |
| Napepld | NAPE-PLD | CTCCATCCCGAATGTGCT | AAGCCAGCCTCTCTCACTCC |
| Npc1l1 | NPC1L1 | GGCTCCATCTGGAGTAGCTG | ATCGCACTACCATCCAGGAC |
| Oatp1b2 | OATP1B2 | ATCCCGTGACTAATCCAACA | ACCAAACTGCTGCTCTATAAACT |
| Pecam1 | CD31 | GGAACGAGAGCCACAGAGAC | TGCACTGCCTTGACTGTCTT |
| Ppara | PPARα | CAACGGCGTCGAAGACAAA | TGACGGTCTCCACGGACAT |
| Rpl19 | RPL19 | GAAGGTCAAAGGGAATGTGTTCA | CCTTGTCTGCCTTCAGCTTGT |
| Saa2 | SAA2 | GGGGTCTGGGCTTCCTATCT | CCATTCTGAAACCCTTGTGG |
| Saa3 | SAA3 | CGCAGCACGAGCAGGAT | CCAGGATCAAGATGCAAAGAATG |
| Slc10a1 | NTCP | GGACAAGGTGCCCTACAAAG | ACAGCCACAGAGAGGGAGAA |
| Slc10a2 | ASBT | TGGGTTTCTTCCTGGCTAGACT | TGTTCTGCATTCCAGTTTCCAA |
| Slc27a5 | BAL | TGTGTGTGAAGGAACCTGGA | ACCCGGACAACTTTGTGAAG |
| Slc51a | OSTα | TACAAGAACACCCTTTGCCC | CGAGGAATCCAGAGACCAAA |
| Slc51b | OSTβ | GTATTTTCGTGCAGAAGATGCG | TTTCTGTTTGCCAGGATGCTC |
| Tnf | TNFα | TCGAGTGACAAGCCTGTAGCC | TTGAGATCCATGCCGTTGG |
| NAEs and ECB-Related Molecules | Bile Acids | ||
|---|---|---|---|
| 1-AG | 1-arachidonoylglycerol | CA | Cholic acid |
| 2-AG | 2-arachidonoylglycerol | CDCA | Chenodeoxycholic acid |
| 1-LG | 1-linoleoylglycerol | DCA | Deoxycholic acid |
| 2-LG | 2-linoleoylglycerol | (α, β or ω) MCA | (α, β or ω) Muricholic acid |
| 2-OG | 2-oleoylglycerol | ||
| 1-PG | 1-palmitoylglycerol | ||
| 2-PG | 2-palmitoylglycerol | ||
| AA | Arachidonic acid | ||
| AEA | N-arachidonoylethanolamine | Oxysterols | |
| DHA | Docosahexaenoic acid | 24(S)-OHC | 24(S)-hydroxycholesterol |
| DHA-1-G | DHA-1-glycerol | 25-OHC | 25-hydroxycholesterol |
| DHA-2-G | DHA-2-glycerol | 27-OHC | 27-hydroxycholesterol |
| DHEA | N-docosahexaenoylethanolamine | 4β-OHC | 4β-hydroxycholesterol |
| DPA-1-G | DPA-1-glycerol | 5α6α-EC | 5α,6α-epoxycholesterol |
| DPA-2-G | DPA-2-glycerol | 5α6β-diOHC | 5α,6β-dihydroxycholesterol |
| DPA-ω3 | DPA-omega3 | 5β6β-EC | 5β,6β-epoxycholesterol |
| EPA | Eicosapentaenoic acid | 7α-OHCone | 7α-hydroxycholestenone |
| EPA-1-G | EPA-1-glycerol | 7-KC | 7-ketocholesterol |
| EPA-2-G | EPA-2-glycerol | 7-OHC | 7-hydroxycholesterol |
| EPEA | N-eicosapentaenoylethanolamine | ||
| LEA | N-linoleylethanolamine | ||
| OEA | N-oleoylethanolamine | ||
| PEA | N-palmitoylethanolamine | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefort, C.; Roumain, M.; Van Hul, M.; Rastelli, M.; Manco, R.; Leclercq, I.; Delzenne, N.M.; Marzo, V.D.; Flamand, N.; Luquet, S.; et al. Hepatic NAPE-PLD Is a Key Regulator of Liver Lipid Metabolism. Cells 2020, 9, 1247. https://doi.org/10.3390/cells9051247
Lefort C, Roumain M, Van Hul M, Rastelli M, Manco R, Leclercq I, Delzenne NM, Marzo VD, Flamand N, Luquet S, et al. Hepatic NAPE-PLD Is a Key Regulator of Liver Lipid Metabolism. Cells. 2020; 9(5):1247. https://doi.org/10.3390/cells9051247
Chicago/Turabian StyleLefort, Charlotte, Martin Roumain, Matthias Van Hul, Marialetizia Rastelli, Rita Manco, Isabelle Leclercq, Nathalie M. Delzenne, Vincenzo Di Marzo, Nicolas Flamand, Serge Luquet, and et al. 2020. "Hepatic NAPE-PLD Is a Key Regulator of Liver Lipid Metabolism" Cells 9, no. 5: 1247. https://doi.org/10.3390/cells9051247
APA StyleLefort, C., Roumain, M., Van Hul, M., Rastelli, M., Manco, R., Leclercq, I., Delzenne, N. M., Marzo, V. D., Flamand, N., Luquet, S., Silvestri, C., Muccioli, G. G., & Cani, P. D. (2020). Hepatic NAPE-PLD Is a Key Regulator of Liver Lipid Metabolism. Cells, 9(5), 1247. https://doi.org/10.3390/cells9051247