Cardiomyocyte-Specific Deletion of Orai1 Reveals Its Protective Role in Angiotensin-II-Induced Pathological Cardiac Remodeling

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Cardiomyocyte Isolation
2.3. Analysis of Orai1 Expression and Cellular Hypertrophy by Immunocytochemistry
2.4. RNA Isolation and qPCR Analysis
2.5. Histological Analysis
2.6. Statistical Analysis
3. Results
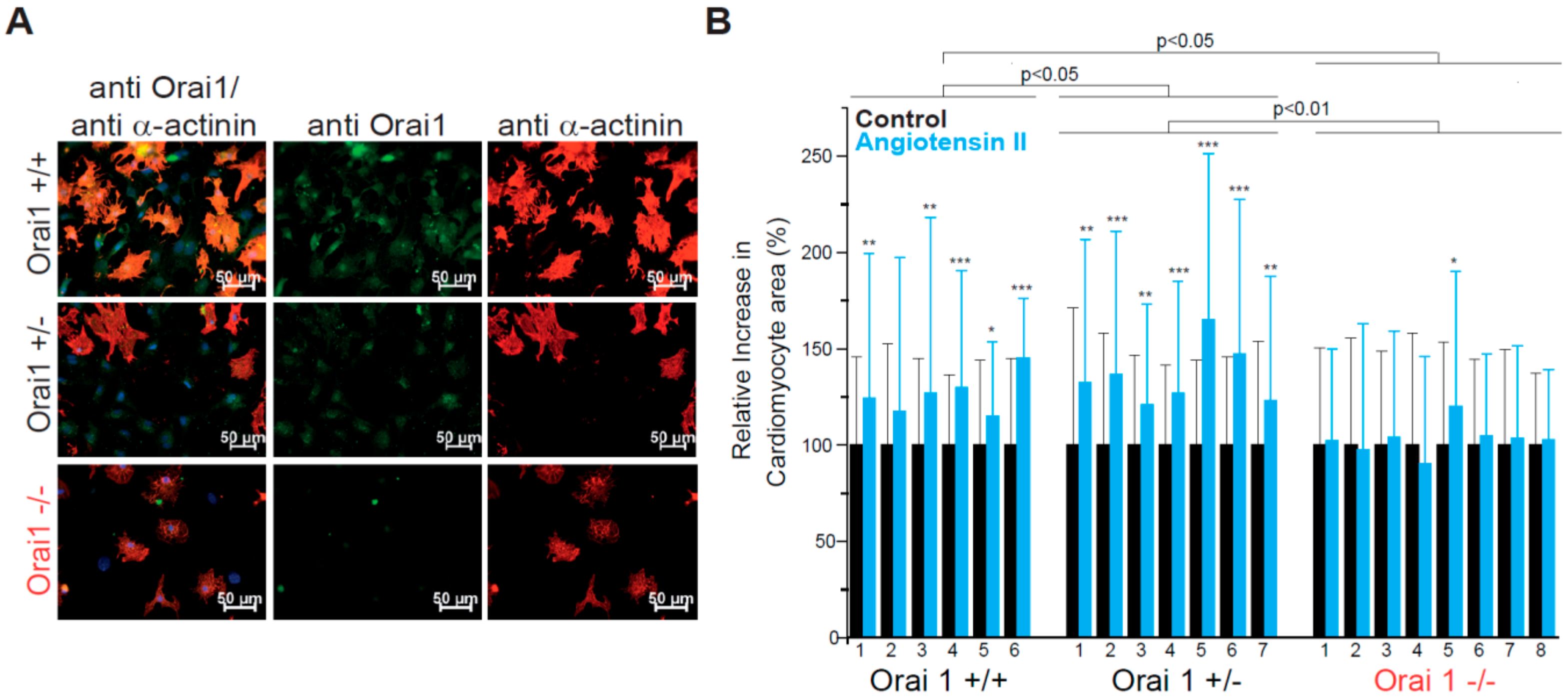
3.1. Orai1 Deletion Protects from Neurohumorally Induced Cellular Hypertrophy in Embryonic Cardiomyocytes
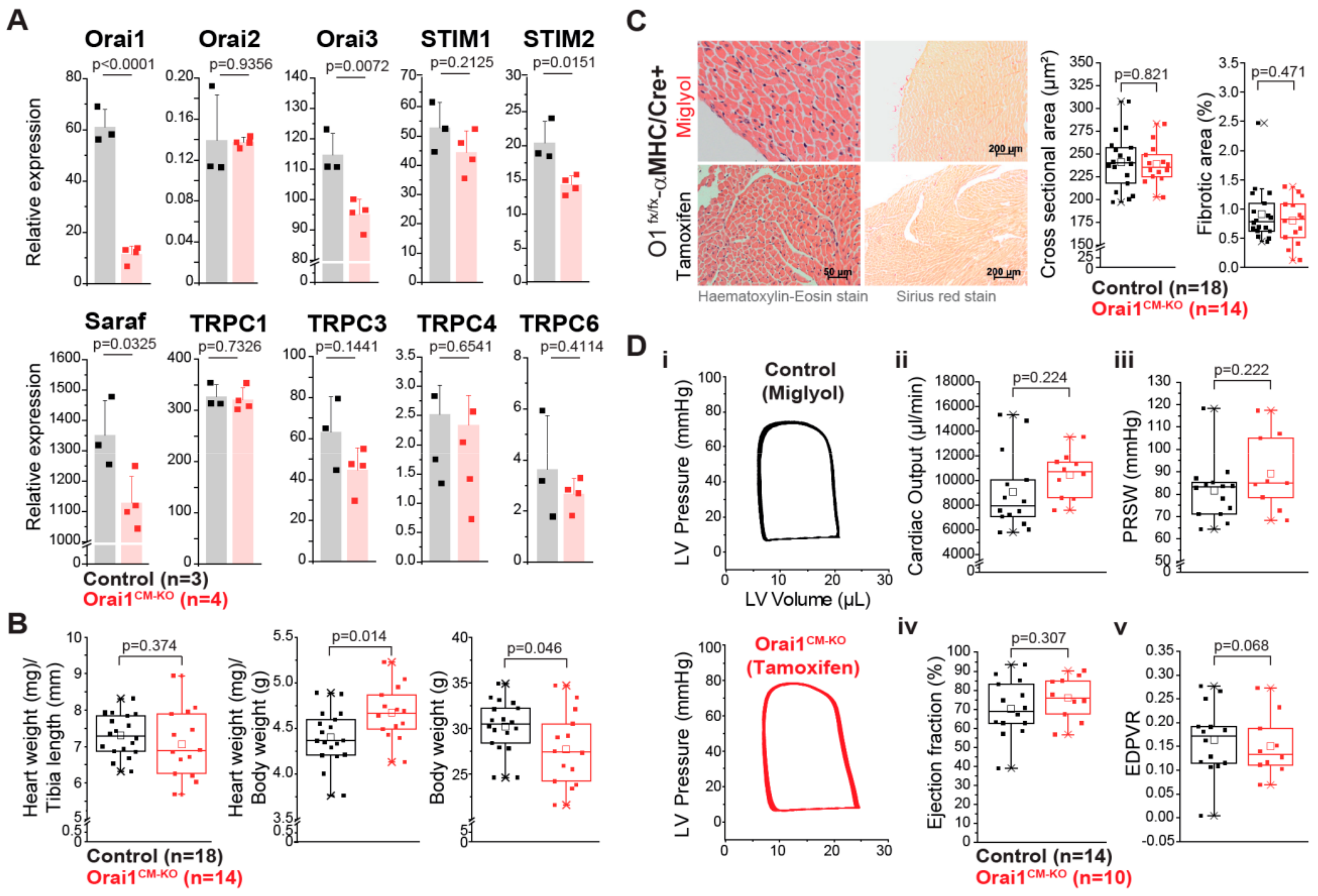
3.2. Unaltered Cardiac Function in Cardiomyocyte-Specific Orai1-Deficient Mice
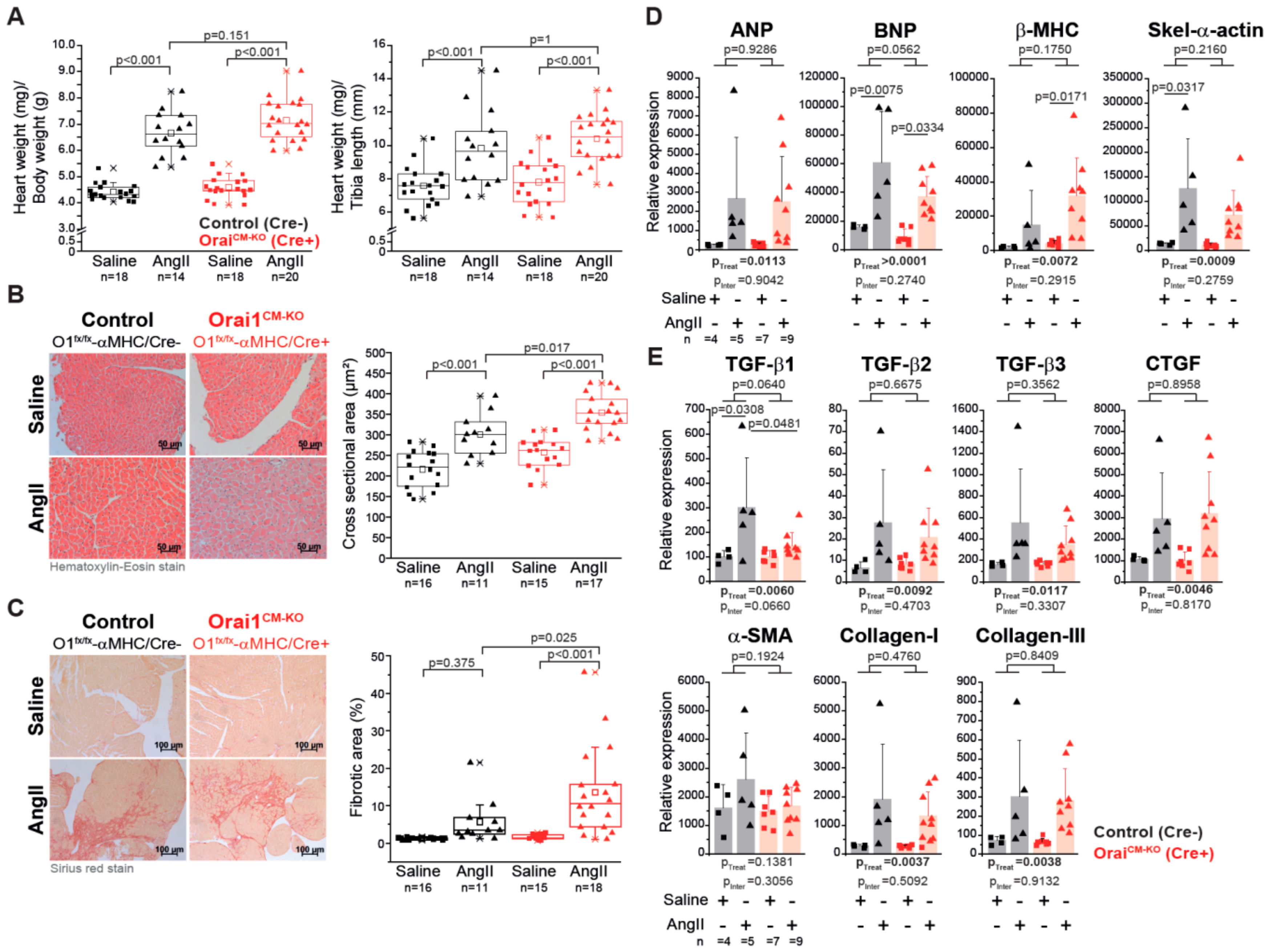
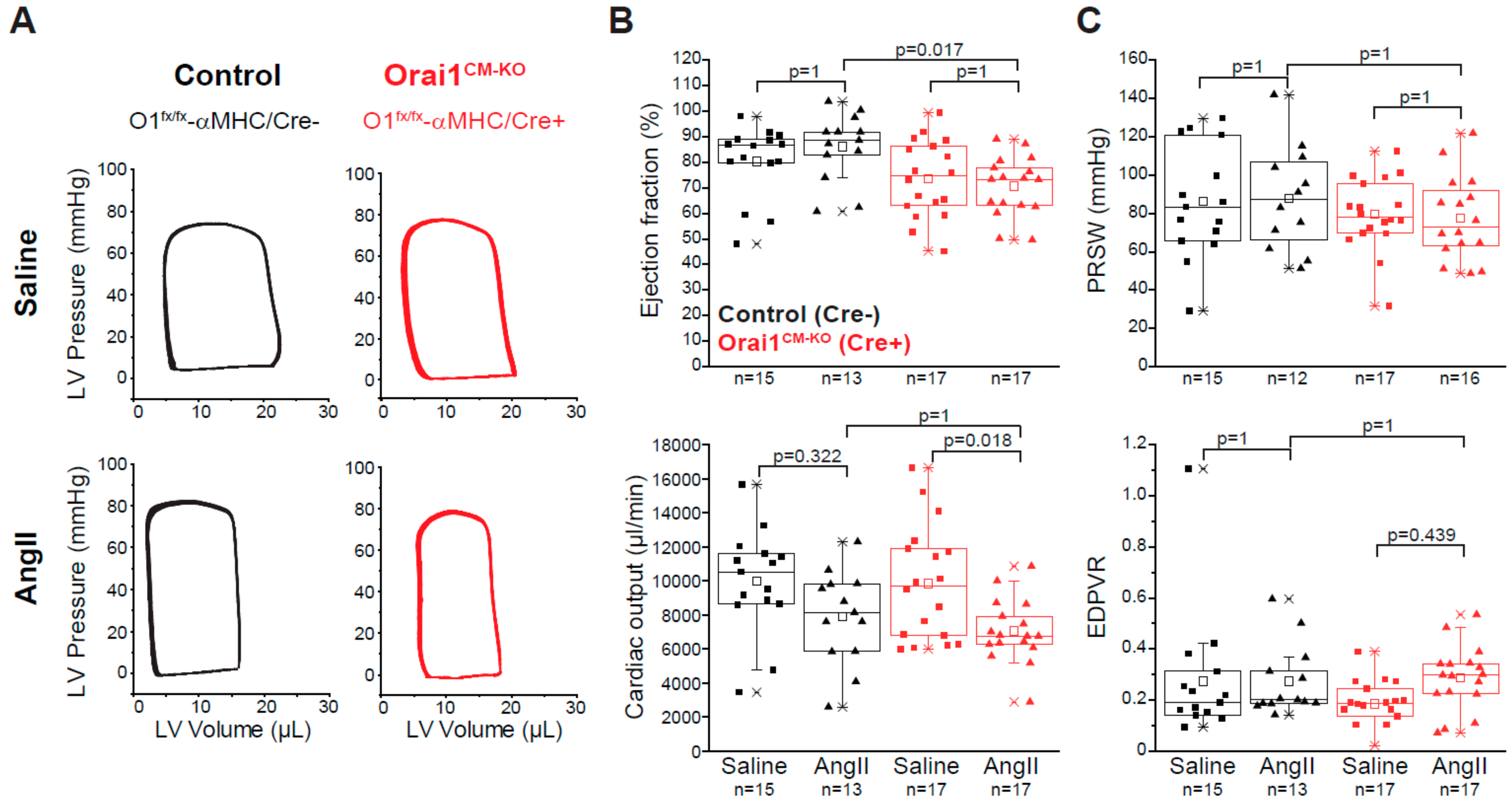
3.3. Deletion of Orai1 in Cardiomyocytes Aggravates the Outcome of Morphometric and Functional Parameters after Angiotensin-II-Induced Cardiac Remodeling
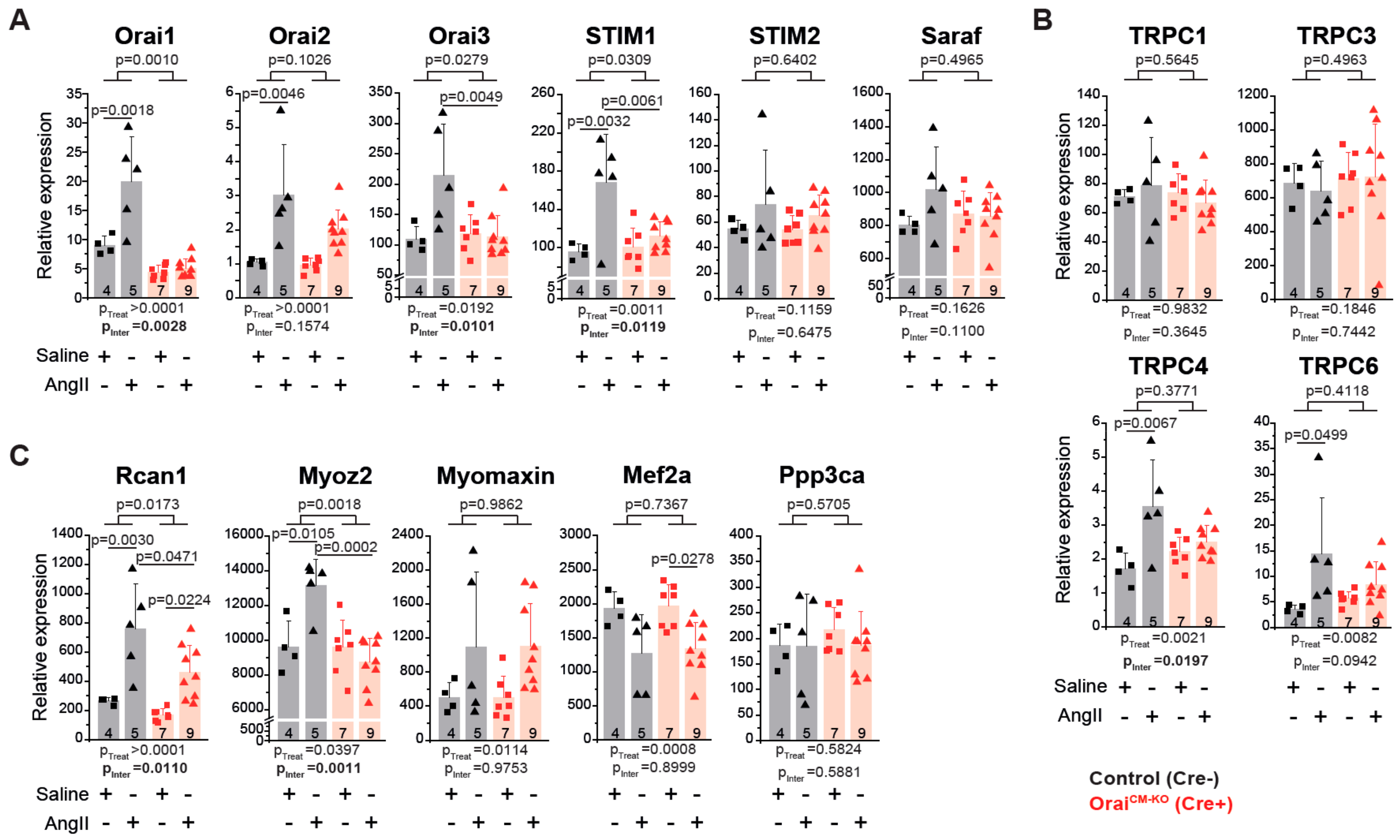
3.4. Differential Gene Expression in Orai1CM-KO Hearts after Neurohumorally Induced Cardiac Remodelling
4. Discussion
4.1. In Vitro Analysis of Orai1 in Embryonic Cardiomyocytes
4.2. Impact of Specific Cardiomyocyte Deletion of Orai1 on the Adult Heart
4.3. Protective Function of Orai1 Proteins in Adult Cardiomyocytes During Neurohumorally Induced Cardiac Hypertrophy
4.4. Cardiac Transcriptional Changes in the Absence of Orai1 in Cardiomyocytes after AngII-Evoked Cardiac Remodelling
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2016, 14, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Wettschureck, N.; Offermanns, S. Mammalian G Proteins and Their Cell Type Specific Functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [PubMed]
- Goonasekera, S.A.; Molkentin, J.D. Unraveling the secrets of a double life: Contractile versus signaling Ca2+ in a cardiac myocyte. J. Mol. Cell. Cardiol. 2012, 52, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Anderson, M.E. Mechanisms of altered Ca2+ handling in heart failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef] [PubMed]
- Eder, P. Cardiac Remodeling and Disease: SOCE and TRPC Signaling in Cardiac Pathology. Advances in Experimental Medicine and Biology 2017, 993, 505–521. [Google Scholar] [CrossRef]
- Freichel, M.; Berlin, M.; Schürger, A.; Mathar, I.; Bacmeister, L.; Medert, R.; Frede, W.; Marx, A.; Segin, S.; Londoño, J.E.C.; et al. TRP Channels in the Heart. In Methods for Neural Ensemble Recordings; Informa UK Limited: Colchester, UK, 2017; pp. 149–185. [Google Scholar]
- Numaga-Tomita, T.; Oda, S.; Shimauchi, T.; Nishimura, A.; Mangmool, S.; Nishida, M. TRPC3 Channels in Cardiac Fibrosis. Front. Cardiovasc. Med. 2017, 4, 56. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Iribe, G.; Nishida, M.; Naruse, K. Role of TRPC3 and TRPC6 channels in the myocardial response to stretch: Linking physiology and pathophysiology. Prog. Biophys. Mol. Boil. 2017, 130, 264–272. [Google Scholar] [CrossRef]
- Londoño, J.E.C.; Tian, Q.; Hammer, K.; Schröder, L.; Londoño, J.C.; Reil, J.C.; He, T.; Oberhofer, M.; Mannebach, S.; Mathar, I.; et al. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur. Hear. J. 2015, 36, 2257–2266. [Google Scholar] [CrossRef]
- Parekh, A.B.; Penner, R. Store depletion and calcium influx. Physiol. Rev. 1997, 77, 901–930. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J. Store-Operated Calcium Channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 Is a Plasma Membrane Protein Essential for Store-Operated Ca2+ Entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.-F.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release–activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef]
- Gross, S.A.; Wissenbach, U.; Philipp, S.E.; Freichel, M.; Cavalié, A.; Flockerzi, V.; Guy, J.E.; Whittle, E.; Kumaran, D.; Lindqvist, Y.; et al. Murine ORAI2 Splice Variants Form Functional Ca2+Release-activated Ca2+(CRAC) Channels. J. Biol. Chem. 2007, 282, 19375–19384. [Google Scholar] [CrossRef] [PubMed]
- Feske, S. CRAC channels and disease – From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 2019, 80, 112–116. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Li, G.-Y.; Plevin, M.; Ames, J.B.; Ikura, M. Stored Ca2+ Depletion-induced Oligomerization of Stromal Interaction Molecule 1 (STIM1) via the EF-SAM Region: An Initiation Mechanism for Capacitive Ca2+ Entry. J. Biol. Chem. 2006, 281, 35855–35862. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T. STIM Is a Ca2+ Sensor Essential for Ca2+-Store-Depletion-Triggered Ca2+ Influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef]
- Roos, J.; Digregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Hojayev, B.; Jiang, N.; Wang, Z.V.; Tandan, S.; Rakalin, A.; Rothermel, B.A.; Gillette, T.G.; Hill, J. STIM1-dependent store-operated Ca2+ entry is required for pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2011, 52, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Hulot, J.-S.; Fauconnier, J.; Ramanujam, D.; Chaanine, A.; Aubart, F.; Sassi, Y.; Merkle, S.; Cazorla, O.; Ouillé, A.; Dupuis, M.; et al. Critical role for stromal interaction molecule 1 in cardiac hypertrophy. Circulation 2011, 124, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Völkers, M.; Salz, M.; Herzog, N.; Frank, D.; Dolatabadi, N.; Frey, N.; Gude, N.; Friedrich, O.; Koch, W.J.; Katus, H.A.; et al. Orai1 and Stim1 regulate normal and hypertrophic growth in cardiomyocytes. J. Mol. Cell. Cardiol. 2010, 48, 1329–1334. [Google Scholar] [CrossRef]
- Hunton, D.L.; Zou, L.; Pang, Y.; Marchase, R.B. Adult rat cardiomyocytes exhibit capacitative calcium entry. Am. J. Physiol. Circ. Physiol. 2004, 286, H1124–H1132. [Google Scholar] [CrossRef]
- Touchberry, C.; Elmore, C.J.; Nguyen, T.M.; Andresen, J.J.; Zhao, X.; Orange, M.; Weisleder, N.; Brotto, M.; Claycomb, W.C.; Wacker, M.J. Store-operated calcium entry is present in HL-1 cardiomyocytes and contributes to resting calcium. Biochem. Biophys. Res. Commun. 2011, 416, 45–50. [Google Scholar] [CrossRef]
- Hunton, D.L.; A Lucchesi, P.; Pang, Y.; Cheng, X.; Dell’Italia, L.J.; Marchase, R.B. Capacitative Calcium Entry Contributes to Nuclear Factor of Activated T-cells Nuclear Translocation and Hypertrophy in Cardiomyocytes. J. Biol. Chem. 2002, 277, 14266–14273. [Google Scholar] [CrossRef]
- Nakayama, H.; Wilkin, B.J.; Bodi, I.; Molkentin, J.D. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 2006, 20, 1660–1670. [Google Scholar] [CrossRef]
- Ju, Y.-K.; Chu, Y.; Chaulet, H.; Lai, D.; Gervasio, O.L.; Graham, R.M.; Cannell, M.B.; Allen, D.G. Store-Operated Ca2+Influx and Expression of TRPC Genes in Mouse Sinoatrial Node. Circ. Res. 2007, 100, 1605–1614. [Google Scholar] [CrossRef]
- Völkers, M.; Dolatabadi, N.; Gude, N.; Most, P.; Sussman, M.A.; Hassel, D. Orai1 deficiency leads to heart failure and skeletal myopathy in zebrafish. J. Cell Sci. 2012, 125, 287–294. [Google Scholar] [CrossRef]
- Dai, F.; Zhang, Y.; Wang, Q.; Li, D.; Yang, Y.; Ma, S.; Yang, D. Overexpression of SARAF Ameliorates Pressure Overload–Induced Cardiac Hypertrophy Through Suppressing STIM1-Orai1 in Mice. Cell. Physiol. Biochem. 2018, 47, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Benard, L.; Lompré, A.-M. STIM1 and Orai in cardiac hypertrophy and vascular proliferative diseases. Front. Biosci. 2013, 5, 766–773. [Google Scholar] [CrossRef]
- Collins, H.E.; Zhu-Mauldin, X.; Marchase, R.B.; Chatham, J.C. STIM1/Orai1-mediated SOCE: current perspectives and potential roles in cardiac function and pathology. Am. J. Physiol. Circ. Physiol. 2013, 305, H446–H458. [Google Scholar] [CrossRef] [PubMed]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Domínguez-Rodríguez, A.; Gallardo-Castillo, I.; Ribas, J.; Ordóñez, A.; Rosado, J.A.; Smani, T. The Complex Role of Store Operated Calcium Entry Pathways and Related Proteins in the Function of Cardiac, Skeletal and Vascular Smooth Muscle Cells. Front. Physiol. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.; Katz, D.; Bryson, V. SOCE and STIM1 signaling in the heart: Timing and location matter. Cell Calcium 2019, 77, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Srikanth, S.; Oh-Hora, M.; Hogan, P.G.; Lamperti, E.D.; Yamashita, M.; Gelinas, C.; Neems, D.S.; Sasaki, Y.; Feske, S.; et al. Hair Loss and Defective T- and B-Cell Function in Mice Lacking ORAI1. Mol. Cell. Biol. 2008, 28, 5209–5222. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.E.; Wolf, M.J.; Smyth, J.T. Suppression of store-operated calcium entry causes dilated cardiomyopathy of the Drosophila heart. Biol. Open 2020, 9, bio049999. [Google Scholar] [CrossRef]
- Horton, J.S.; Buckley, C.L.; Alvarez, E.M.; Schorlemmer, A.; Stokes, A. The calcium release-activated calcium channel Orai1 represents a crucial component in hypertrophic compensation and the development of dilated cardiomyopathy. Channels 2013, 8, 35–43. [Google Scholar] [CrossRef]
- Sabourin, J.; Bartoli, F.; Antigny, F.; Gomez, A.-M.; Benitah, J.-P. Transient Receptor Potential Canonical (TRPC)/Orai1-dependent Store-operated Ca2+Channels. J. Biol. Chem. 2016, 291, 13394–13409. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Z.C.; Zhang, P.; Poon, E.; Kong, C.W.; Boheler, K.R.; Huang, Y.; Li, R.A.; Yao, X. Nitric Oxide-cGMP-PKG Pathway Acts on Orai1 to Inhibit the Hypertrophy of Human Embryonic Stem Cell-Derived Cardiomyocytes. Stem Cells 2015, 33, 2973–2984. [Google Scholar] [CrossRef]
- Zheng, C.; Lo, C.-Y.; Meng, Z.; Li, Z.; Zhong, M.; Zhang, P.; Lu, J.; Yang, Z.; Yan, F.; Zhang, Y.; et al. Gastrodin Inhibits Store-Operated Ca2+ Entry and Alleviates Cardiac Hypertrophy. Front. Pharmacol. 2017, 8, 534. [Google Scholar] [CrossRef]
- McCarl, C.-A.; Picard, C.; Khalil, S.; Kawasaki, T.; Röther, J.; Papolos, A.; Kutok, J.; Hivroz, C.; LeDeist, F.; Plogmann, K.; et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy Clin. Immunol. 2009, 124, 1311–1318.e7. [Google Scholar] [CrossRef]
- Ahuja, M.; Schwartz, D.M.; Tandon, M.; Son, A.; Zeng, M.; Swaim, W.; Eckhaus, M.; Hoffman, V.; Cui, Y.; Xiao, B.; et al. Orai1-Mediated Antimicrobial Secretion from Pancreatic Acini Shapes the Gut Microbiome and Regulates Gut Innate Immunity. Cell Metab. 2017, 25, 635–646. [Google Scholar] [CrossRef]
- Schwenk, F.; Baron, U.; Rajewsky, K. A cre -transgenic mouse strain for the ubiquitous deletion of loxP -flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995, 23, 5080–5081. [Google Scholar] [CrossRef] [PubMed]
- Takefuji, M.; Wirth, A.; Lukasova, M.; Takefuji, S.; Boettger, T.; Braun, T.; Althoff, T.; Offermanns, S.; Wettschureck, N. G13-Mediated Signaling Pathway Is Required for Pressure Overload-Induced Cardiac Remodeling and Heart Failure. Circulation 2012, 126, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Muzumdar, M.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genes 2007, 45, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Nagayama, T.; Mukhopadhyay, P.; Bátkai, S.; Kass, D.A. Measurement of cardiac function using pressure–volume conductance catheter technique in mice and rats. Nat. Protoc. 2008, 3, 1422–1434. [Google Scholar] [CrossRef]
- Bacmeister, L.; Segin, S.; Medert, R.; Lindner, D.; Freichel, M.; Londoño, J.E.C. Assessment of PEEP-Ventilation and the Time Point of Parallel-Conductance Determination for Pressure-Volume Analysis Under β-Adrenergic Stimulation in Mice. Front. Cardiovasc. Med. 2019, 6, 36. [Google Scholar] [CrossRef]
- Wissenbach, U.; Philipp, S.E.; Gross, S.A.; Cavalié, A.; Flockerzi, V. Primary structure, chromosomal localization and expression in immune cells of the murine ORAI and STIM genes. Cell Calcium 2007, 42, 439–446. [Google Scholar] [CrossRef]
- Sachdeva, R.; Fleming, T.; Schumacher, D.; Homberg, S.; Stilz, K.; Mohr, F.; Wagner, A.H.; Tsvilovskyy, V.; Mathar, I.; Freichel, M. Methylglyoxal evokes acute Ca2+ transients in distinct cell types and increases agonist-evoked Ca2+ entry in endothelial cells via CRAC channels. Cell Calcium 2019, 78, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. STIM and Orai in platelet function. Cell Calcium 2011, 50, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Swaim, W.; Ambudkar, I.S. Local Ca²+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca²+ signals required for specific cell functions. PLoS Biol. 2011, 9, e1001025. [Google Scholar] [CrossRef]
- Kim, H.J.; Lv, P.; Sihn, C.-R.; Yamoah, E.N. Cellular and Molecular Mechanisms of Autosomal Dominant Form of Progressive Hearing Loss, DFNA2*. J. Biol. Chem. 2010, 286, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Koitabashi, N.; Bedja, D.; Zaiman, A.L.; Pinto, Y.M.; Zhang, M.; Gabrielson, K.L.; Takimoto, E.; Kass, D.A. Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circ. Res. 2009, 105, 12–15. [Google Scholar] [CrossRef]
- Parks, C.; Alam, M.A.; Sullivan, R.; Mancarella, S. STIM1-dependent Ca2+ microdomains are required for myofilament remodeling and signaling in the heart. Sci. Rep. 2016, 6, 25372. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Shcheglovitov, A.; Dolmetsch, R. The CRAC Channel Activator STIM1 Binds and Inhibits L-Type Voltage-Gated Calcium Channels. Science 2010, 330, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, X.; Mancarella, S.; Hendron, E.; Eguchi, S.; Soboloff, J.; Tang, X.D.; Gill, D.L. The Calcium Store Sensor, STIM1, Reciprocally Controls Orai and CaV1.2 Channels. Science 2010, 330, 105–109. [Google Scholar] [CrossRef]
- Collins, H.E.; He, L.; Zou, L.; Qu, J.; Zhou, L.; Litovsky, S.H.; Yang, Q.; Young, M.E.; Marchase, R.B.; Chatham, J.C. Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function. Am. J. Physiol. Circ. Physiol. 2014, 306, H1231–H1239. [Google Scholar] [CrossRef]
- Collins, H.E.; Pat, B.M.; Zou, L.; Litovsky, S.H.; Wende, A.R.; Young, M.E.; Chatham, J.C. Novel role of the ER/SR Ca2+ sensor STIM1 in the regulation of cardiac metabolism. Am. J. Physiol. Circ. Physiol. 2019, 316, H1014–H1026. [Google Scholar] [CrossRef]
- Saliba, Y.; Keck, M.; Marchand, A.; Atassi, F.; Ouillé, A.; Cazorla, O.; Trebak, M.; Pavoine, C.; Lacampagne, A.; Hulot, J.-S.; et al. Emergence of Orai3 activity during cardiac hypertrophy. Cardiovasc. Res. 2014, 105, 248–259. [Google Scholar] [CrossRef]
- Mancarella, S.; Kamatham, S. Abstract 922: Cardiac-specific Deletion of Orai3 Channel Causes Dilated Cardiomyopathy. Circ. Res. 2019, 125, 125. [Google Scholar] [CrossRef]
- Tsvilovskyy, V.; Solís-López, A.; Schumacher, D.; Medert, R.; Roers, A.; Kriebs, U.; Freichel, M. Deletion of Orai2 augments endogenous CRAC currents and degranulation in mast cells leading to enhanced anaphylaxis. Cell Calcium 2018, 71, 24–33. [Google Scholar] [CrossRef]
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Jani, P.K.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; DeHaven, W.I.; Bird, G.S.; Billingsley, J.M.; Wang, H.; E Rao, P.; Hutchings, A.B.; Jouvin, M.-H.; Putney, J.; Kinet, J.-P. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat. Immunol. 2007, 9, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, F.; Bailey, M.A.; Rode, B.; Mateo, P.; Antigny, F.; Bedouet, K.; Gerbaud, P.; Gosain, R.; Plante, J.; Norman, K.; et al. Orai1 Channel Inhibition Preserves Left Ventricular Systolic Function and Normal Ca 2+ Handling After Pressure Overload. Circ. 2020, 141, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Komuro, I.; Shiojima, I.; Hayashi, D.; Kudoh, S.; Mizuno, T.; Kijima, K.; Matsubara, H.; Sugaya, T.; Murakami, K.; et al. Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation 1998, 97, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Dewenter, M.; Von Der Lieth, A.; Katus, H.A.; Backs, J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circ. Res. 2017, 121, 1000–1020. [Google Scholar] [CrossRef]
- Feske, S.; Skolnik, E.Y.; Prakriya, M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 2012, 12, 532–547. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Lu, J.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A Calcineurin-Dependent Transcriptional Pathway for Cardiac Hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef]
- Huang, H.-T.; Brand, O.M.; Mathew, M.; Ignatiou, C.; Ewen, E.P.; McCalmon, S.A.; Naya, F.J. Myomaxin Is a Novel Transcriptional Target of MEF2A That Encodes a Xin-related α-Actinin-interacting Protein. J. Biol. Chem. 2006, 281, 39370–39379. [Google Scholar] [CrossRef]
- Yang, J.; Rothermel, B.; Vega, R.B.; Frey, N.; McKinsey, T.A.; Olson, E.N.; Bassel-Duby, R.; Williams, R.S. Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circ. Res. 2000, 87, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Rothermel, B.A.; Weinheimer, C.J.; Kovacs, A.; Naseem, R.H.; Bassel-Duby, R.; Williams, R.S.; Olson, E.N. Dual roles of modulatory calcineurin-interacting protein 1 in cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2003, 100, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tadross, M.R.; Tsien, R.W. Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 2016, 351, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Farmer, L.K.; Rollason, R.; Whitcomb, D.J.; Ni, L.; Goodliff, A.; Lay, A.; Birnbaumer, L.; Heesom, K.J.; Xu, S.-Z.; Saleem, M.A.; et al. TRPC6 Binds to and Activates Calpain, Independent of Its Channel Activity, and Regulates Podocyte Cytoskeleton, Cell Adhesion, and Motility. J. Am. Soc. Nephrol. 2019, 30, 1910–1924. [Google Scholar] [CrossRef]
- Frey, N.; Barrientos, T.; Shelton, J.M.; Frank, D.; Rütten, H.; Gehring, D.; Kuhn, C.; Lutz, M.; Rothermel, B.; Bassel-Duby, R.; et al. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat. Med. 2004, 10, 1336–1343. [Google Scholar] [CrossRef]
- Frank, D.; Kuhn, C.; Van Eickels, M.; Gehring, D.; Hanselmann, C.; Lippl, S.; Will, R.; Katus, H.A.; Frey, N. Calsarcin-1 Protects Against Angiotensin-II–Induced Cardiac Hypertrophy. Circulation 2007, 116, 2587–2596. [Google Scholar] [CrossRef][Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Orai1-WT | Orai1CM-KO | p-Value | |
|---|---|---|---|
| n = 14 | n = 10 | ||
| Body temperature (°C) | 37.19 (±0.56) | 37.34 (±0.71) | 0.534 |
| HR (bpm) | 597.2 (±35.5) | 603.1 (±27.0) | 0.665 |
| ESP (mmHg) | 67.3 (±6.4) | 64.4 (±7.7) | 0.324 |
| EDP (mmHg) | 8.4 (±3.0) | 7.4 (±3.3) | 0.451 |
| ESV (µL) | 8.1 (±5.7) | 6.8 (±4.0) | 0.530 |
| EDV (µL) | 21.8 (±7.8) | 22.9 (±5.3) | 0.702 |
| SV (µL) | 15.1 (±4.8) | 17.3 (±2.8) | 0.216 |
| CO (µL/min) | 9071.6 (±3101.2) | 10442.5 (±1798.1) | 0.224 |
| Systolic parameters | |||
| EF (%) | 71.3 (±16.7) | 76.1 (±11.2) | 0.442 |
| Stroke Work (mmHg x µL) | 883.5 (±317.1) | 962.1 (±162.3) | 0.482 |
| dP/dt max. (mmHg/s) | 6591.9 (±1143.2) | 6492.4 (±690.8) | 0.809 |
| PRSW | 81.4 (±13.2) | 89.3 (±16.7) | 0.222 |
| ESPVR | 17.8 (±18.9) | 12.7 (±4.5) | 0.534 |
| Diastolic parameters | |||
| Tau (ms) | 7.0 (±2.6) | 6.3 (±1.1) | 0.407 |
| EDPVR | 0.16 (±0.07) | 0.15 (±0.06) | 0.677 |
| dP/dt min. (mmHg/s) | −6679.5 (±1295.3) | −6522.3 (±622.8) | 0.727 |
| Orai1-WT | p-1 | Orai1CM-KO | p-2 | p-3 | |||
|---|---|---|---|---|---|---|---|
| NaCl 0.9% | AngII | NaCl 0.9% | AngII | ||||
| n = 15 | n = 13/12 | n = 17 | n = 17/16 | ||||
| Body temp.(°C) | 36.57 (±0.45) | 36.95 (±0.77) | 0.803 | 36.76 (±0.86) | 37.12 (±0.52) | 0.701 | 1 |
| HF (bpm) | 607.42 (±31.99) | 583.5 (±37.78) | 0.323 | 620.58 (±29.65) | 614.93 (±29.33) | 1 | 0.060 |
| ESP (mmHg) | 63.71 (±9.20) | 63.19 (±13.57) | 1 | 64.40 (±10.75) | 66.33 (±10.52) | 1 | 1 |
| EDP (mmHg) | 6.53 (±3.49) | 3.14 (±2.99) | 0.052 | 3.87 (±2.99) | 3.11 (±3.61) | 1 | 1 |
| ESV (µL) | 4.43 (±2.77) | 2.87 (±2.35) | 1 | 6.97 (±4.18) | 5.79 (±4.04) | 1 | 0.164 |
| EDV (µL) | 19.83 (±5.61) | 15.63 (±4.97) | 0.319 | 21.85 (±6.24) | 16.11 (±5.42) | 0.025 | 1 |
| SV (µL) | 16.46 (±5.16) | 13.57 (±4.84) | 0.361 | 16.16 (±5.35) | 11.41 (±2.88) | 0.021 | 0.589 |
| CO (µL/min) | 9989.75 (±3052.44) | 7897.54 (±2726.28) | 0.211 | 10024.73 (±3361.74) | 7044.08 (±1856.75) | 0.015 | 0.8417 |
| Systolic parameters | |||||||
| EF (%) | 80.36 (±14.30) | 86.02 (±13.32) | 1 | 72.10 (±14.01) | 70.69 (±11.55) | 1 | 0.016 |
| Stroke Work (mmHg x µL) | 990.61 (±334.67) | 863.20 (±447.33) | 1 | 935.46 (±391.33) | 669.18 (±250.88) | 0.205 | 0.879 |
| dP/dt max. (mmHg/s) | 7738.79 (±885.73) | 6650.58 (±1510.60) | 0.337 | 6933.16 (±1810.65) | 6231 (±1473.95) | 1 | 1 |
| PRSW | 86.26 (±28.95) | 87.90 (±27.04) | 1 | 79.32 (±19.44) | 77.48 (±21.78) | 1 | 1 |
| ESPVR | 14.77 (±8.36) | 19.11 (±8.44) | 1 | 15.76 (±11.01) | 14.88 (±7.66) | 1 | 1 |
| Diastolic parameters | |||||||
| Tau (ms) | 5.44 (±1.08) | 5.42 (±1.24) | 1 | 5.21 (±1.11) | 5.66 (±1.80) | 1 | 1 |
| EDPVR | 0.27 (±0.25) | 0.27 (±0.14) | 1 | 0.19 (±0.09) | 0.29 (±0.13) | 0.439 | 1 |
| dP/dt min. (mmHg/s) | −7361.30 (±941.17) | −6643.33 (±1613.47) | 1 | −6961.90 (±1515.91) | −6545.75 (±1483.59) | 1 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segin, S.; Berlin, M.; Richter, C.; Medert, R.; Flockerzi, V.; Worley, P.; Freichel, M.; Camacho Londoño, J.E. Cardiomyocyte-Specific Deletion of Orai1 Reveals Its Protective Role in Angiotensin-II-Induced Pathological Cardiac Remodeling. Cells 2020, 9, 1092. https://doi.org/10.3390/cells9051092
Segin S, Berlin M, Richter C, Medert R, Flockerzi V, Worley P, Freichel M, Camacho Londoño JE. Cardiomyocyte-Specific Deletion of Orai1 Reveals Its Protective Role in Angiotensin-II-Induced Pathological Cardiac Remodeling. Cells. 2020; 9(5):1092. https://doi.org/10.3390/cells9051092
Chicago/Turabian StyleSegin, Sebastian, Michael Berlin, Christin Richter, Rebekka Medert, Veit Flockerzi, Paul Worley, Marc Freichel, and Juan E. Camacho Londoño. 2020. "Cardiomyocyte-Specific Deletion of Orai1 Reveals Its Protective Role in Angiotensin-II-Induced Pathological Cardiac Remodeling" Cells 9, no. 5: 1092. https://doi.org/10.3390/cells9051092
APA StyleSegin, S., Berlin, M., Richter, C., Medert, R., Flockerzi, V., Worley, P., Freichel, M., & Camacho Londoño, J. E. (2020). Cardiomyocyte-Specific Deletion of Orai1 Reveals Its Protective Role in Angiotensin-II-Induced Pathological Cardiac Remodeling. Cells, 9(5), 1092. https://doi.org/10.3390/cells9051092