Genome-Wide Association Study of Diabetogenic Adipose Morphology in the GENetics of Adipocyte Lipolysis (GENiAL) Cohort

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Adipose Tissue Biopsy and Measurement of Adipose Morphology
2.3. Genetic Analysis
2.4. Data Mining
2.5. Adipocyte Cell Culture and Transfection With Small Interfering RNA
2.6. Measurement of Lipid Accumulation and Lipolysis
2.7. Analysis of Cell Proliferation
2.8. Isolation of RNA and Analysis of Gene Expression
2.9. Statistical Analysis of Clinical and In Vitro Data
3. Results
3.1. Clinical Findings
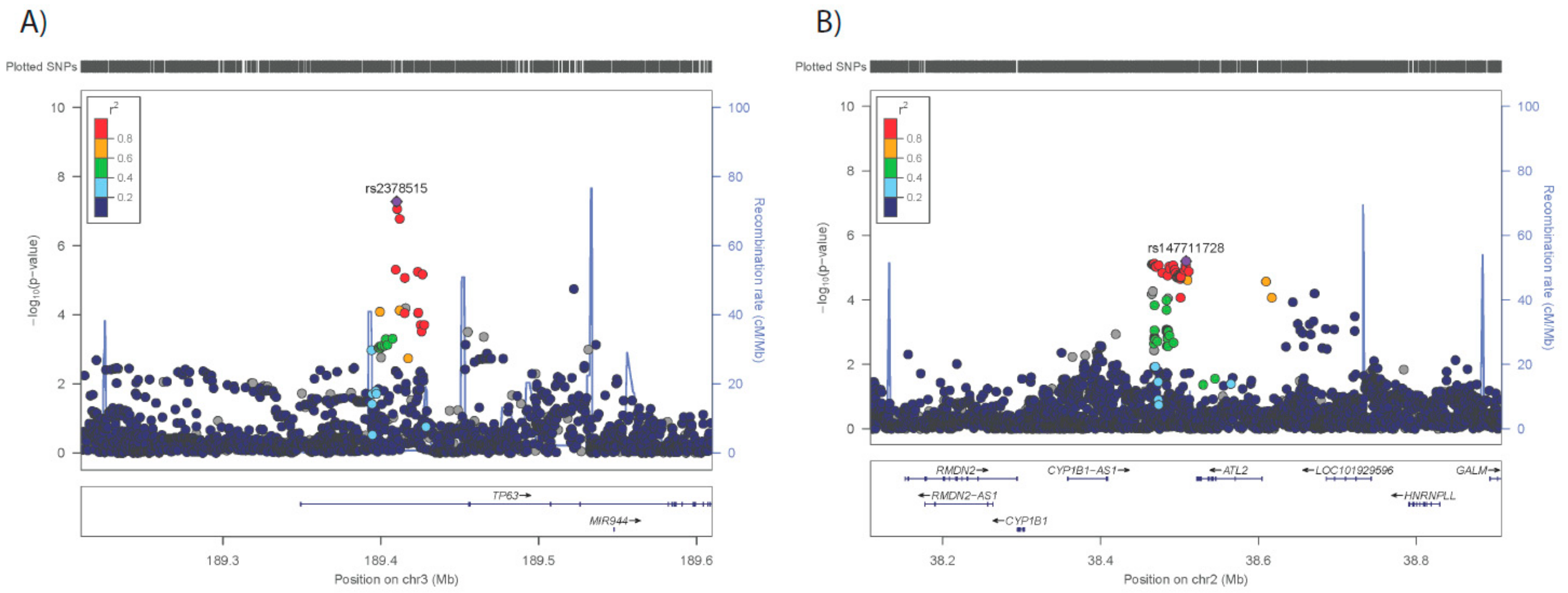
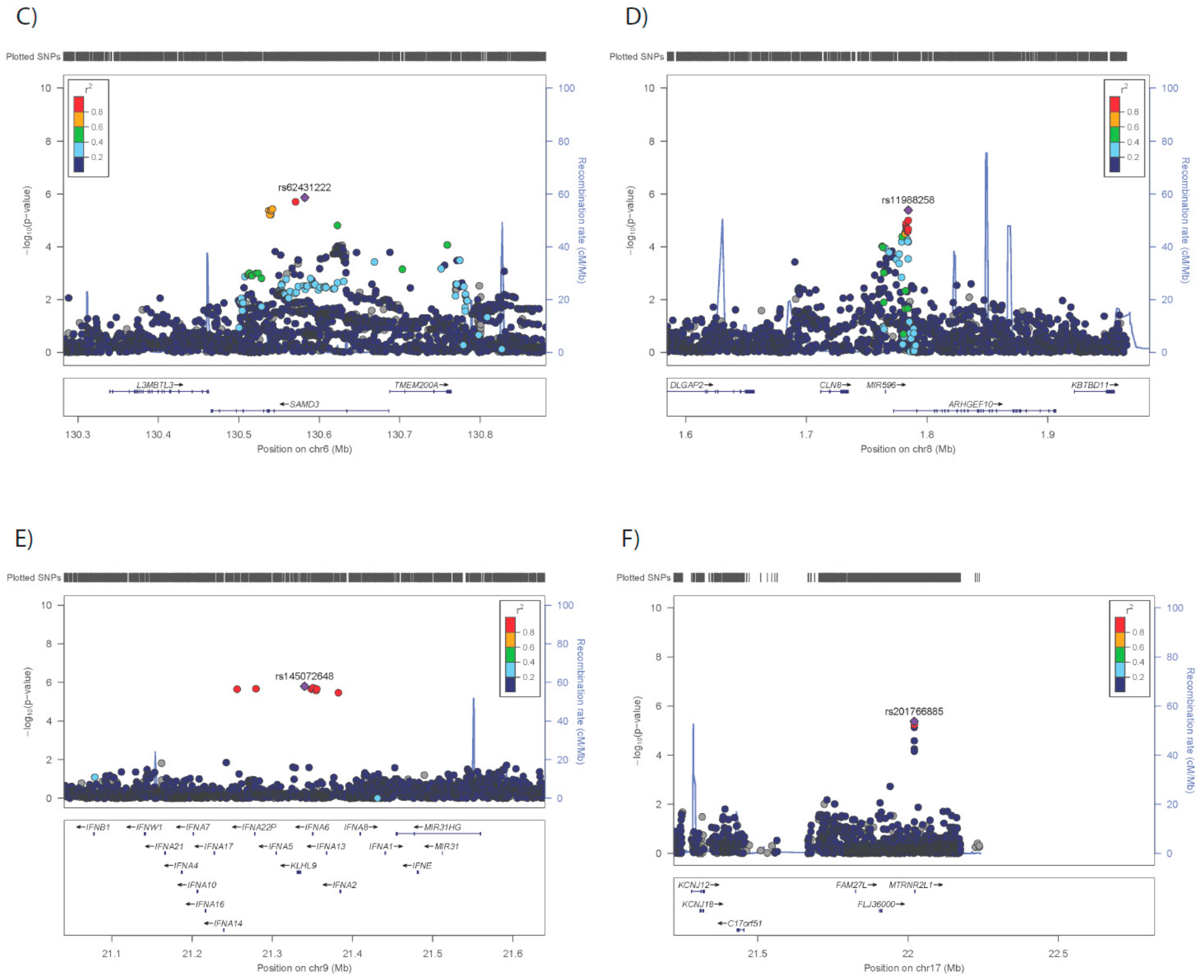
3.2. GWAS for Adipose Morphology
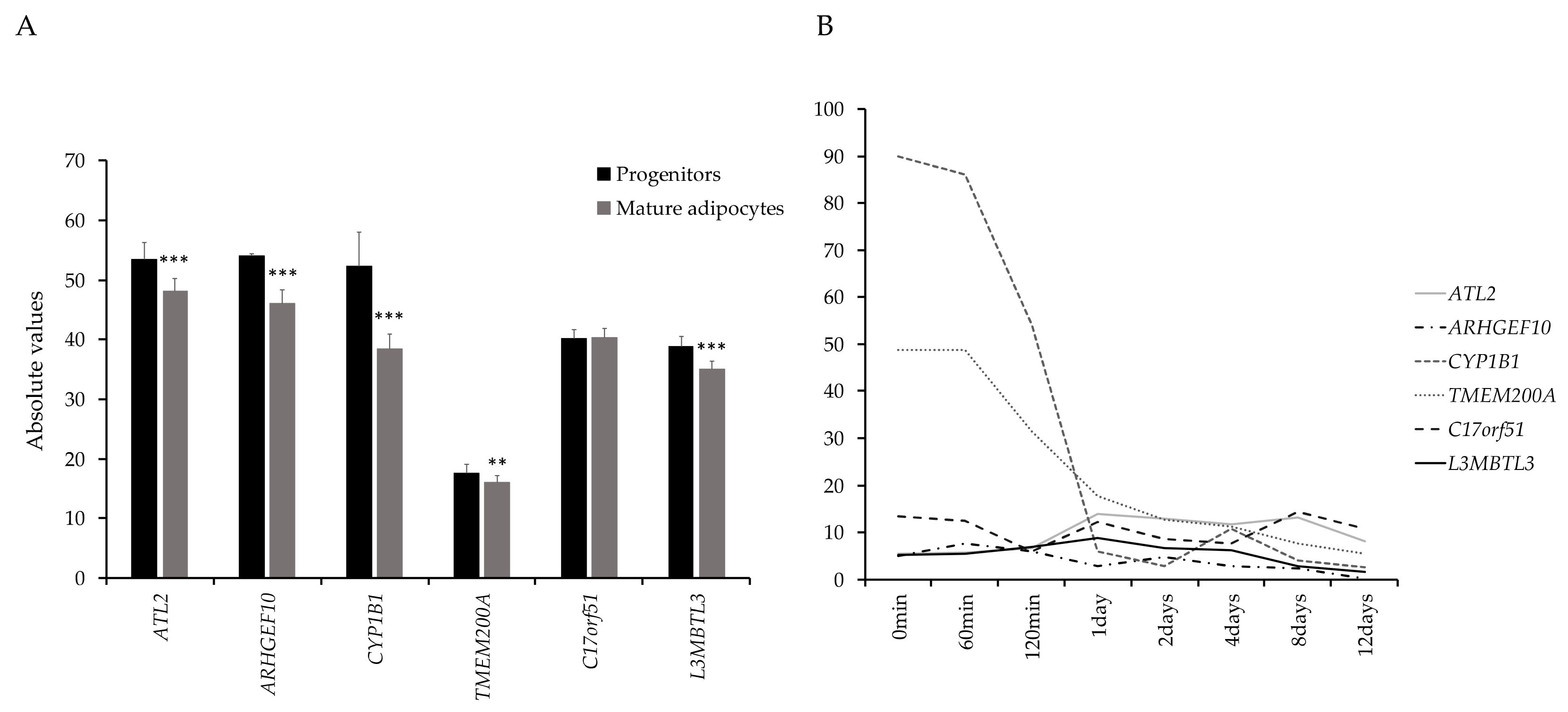
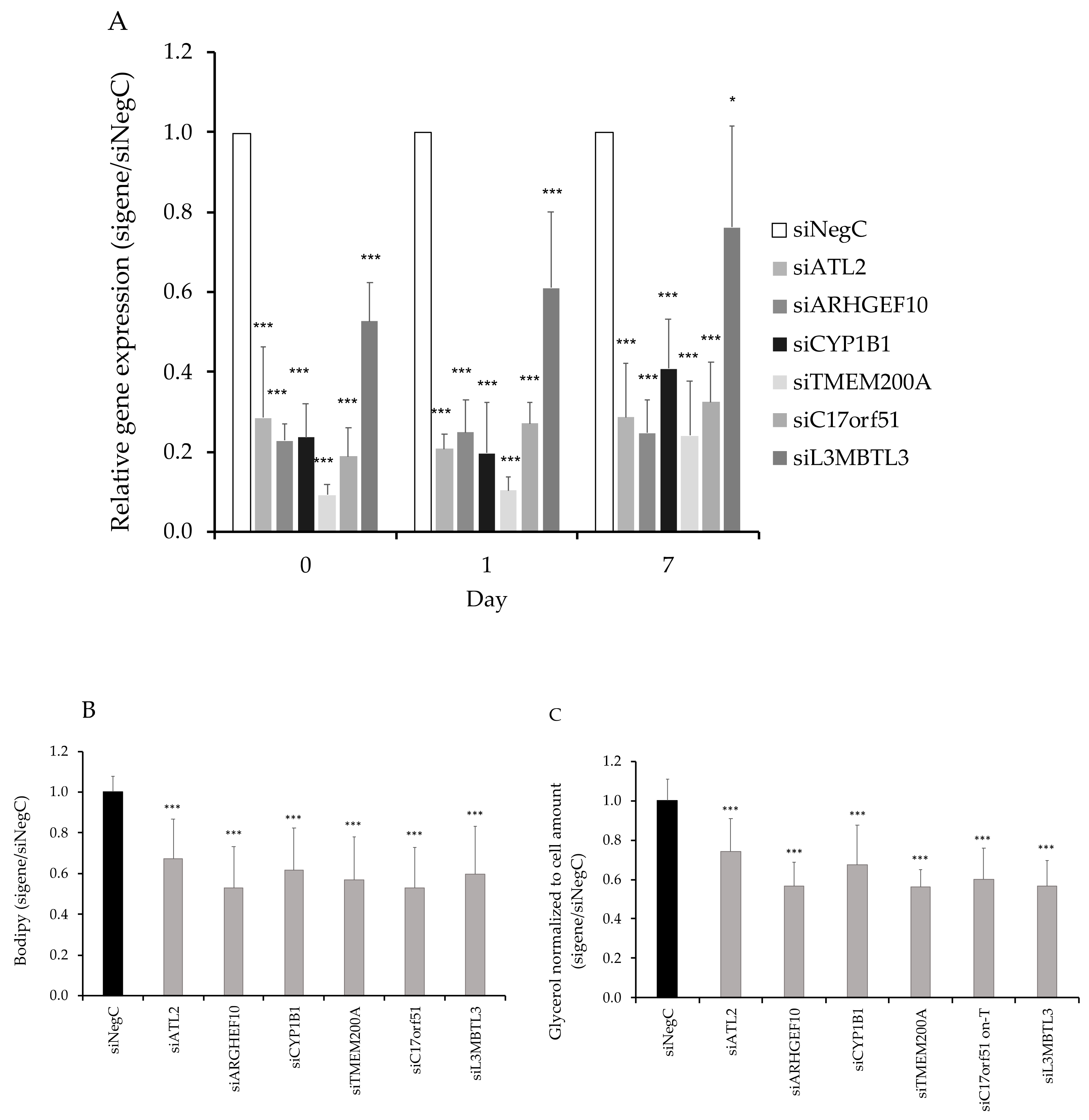
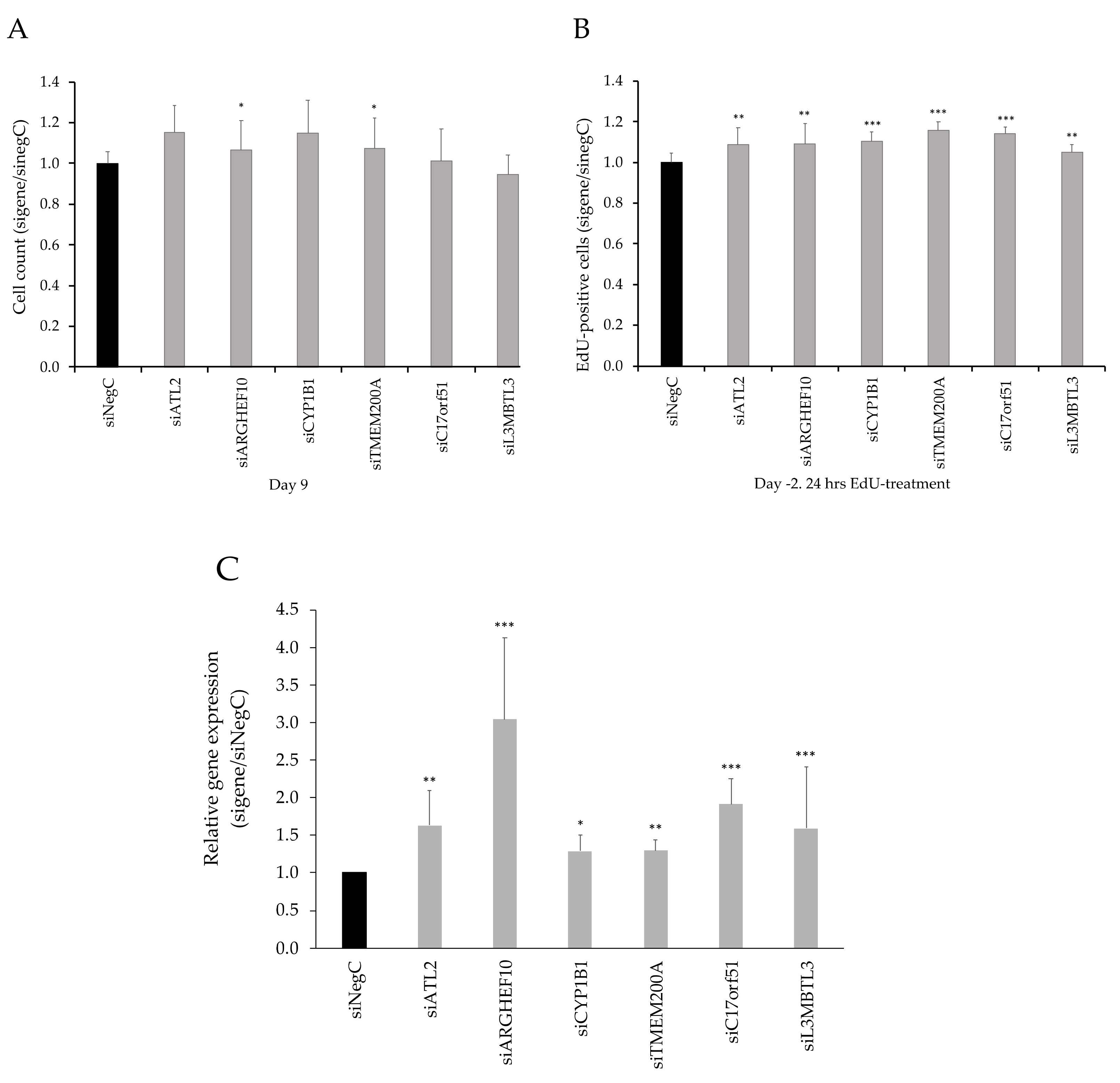
3.3. Functional Evaluation of Candidate Genes
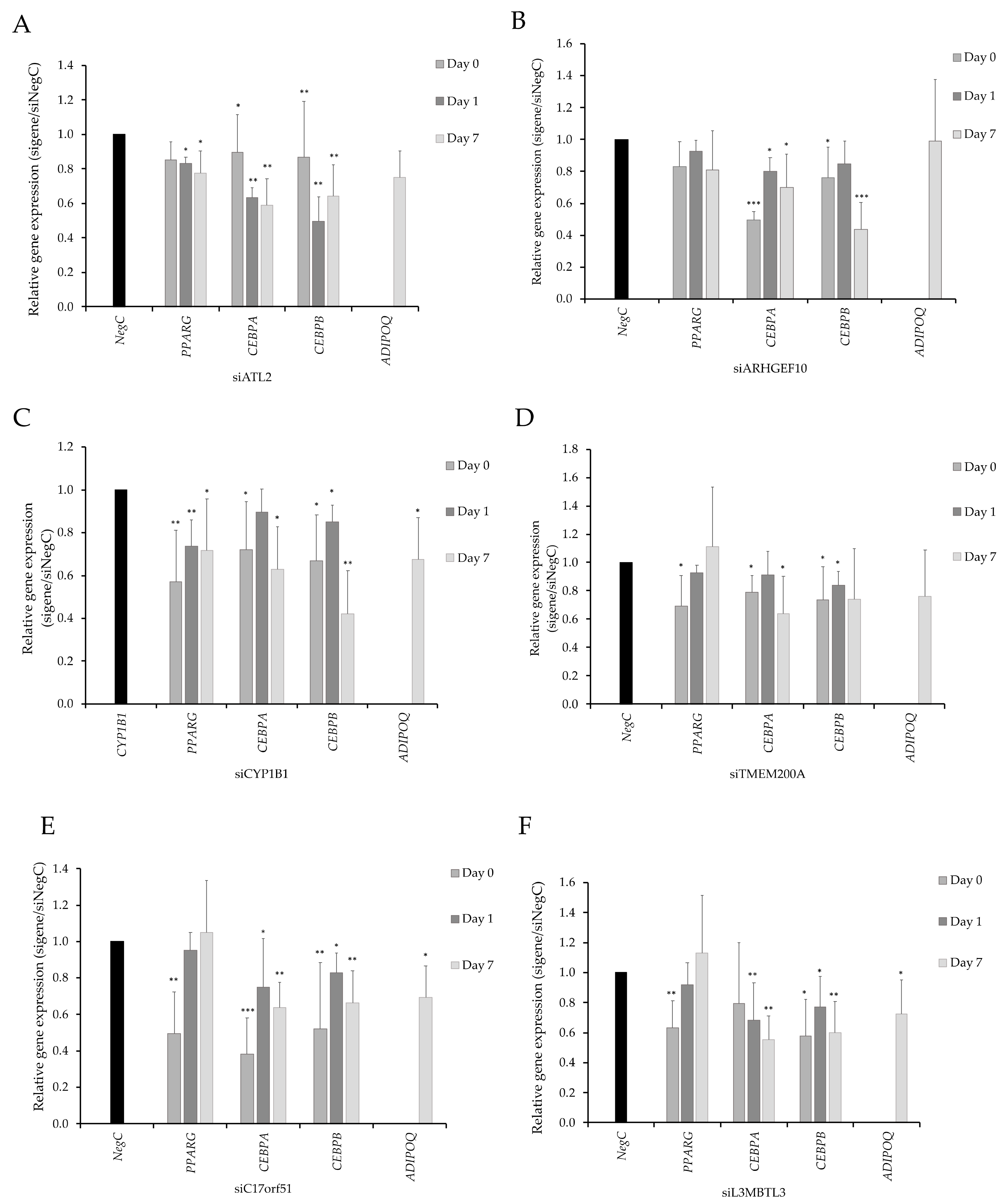
3.4. SAT-Expression of CYP1B1 and ATL2 Correlates With Adipose Morphology
3.5. Overlap Between Result From GWAS Studies of T2D and Adipose Morphology
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability
Conflicts of Interest
References
- Prins, J.B.; O’Rahilly, S. Regulation of adipose cell number in man. Clin. Sci. (Lond) 1997, 92, 3–11. [Google Scholar]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [PubMed]
- Gustafson, B.; Hammarstedt, A.; Hedjazifar, S.; Smith, U. Restricted adipogenesis in hypertrophic obesity: The role of WISP2, WNT, and BMP4. Diabetes 2013, 62, 2997–3004. [Google Scholar] [PubMed]
- Arner, E.; Westermark, P.O.; Spalding, K.L.; Britton, T.; Rydén, M.; Frisén, J.; Bernard, S.; Arner, P. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 2010, 59, 105–109. [Google Scholar] [PubMed]
- Acosta, J.R.; Douagi, I.; Andersson, D.P.; Bäckdahl, J.; Rydén, M.; Arner, P.; Laurencikiene, J. Increased fat cell size: A major phenotype of subcutaneous white adipose tissue in non-obese individuals with type 2 diabetes. Diabetologia 2016, 59, 560–570. [Google Scholar] [PubMed]
- Gao, H.; Mejhert, N.; Fretz, J.A.; Arner, E.; Lorente-Cebrián, S.; Ehrlund, A.; Dahlman-Wright, K.; Gong, X.; Strömblad, S.; Douagi, I. Early B cell factor 1 regulates adipocyte morphology and lipolysis in white adipose tissue. Cell Metab. 2014, 19, 981–992. [Google Scholar]
- Minchin, J.E.; Dahlman, I.; Harvey, C.J.; Mejhert, N.; Singh, M.K.; Epstein, J.A.; Arner, P.; Torres-Vázquez, J.; Rawls, J.F. Plexin D1 determines body fat distribution by regulating the type V collagen microenvironment in visceral adipose tissue. Proc. Natl. Acad. Sci. USA 2015, 112, 4363–4368. [Google Scholar]
- Andersson, D.P.; Dahlman, I.; Eriksson Hogling, D.; Bäckdahl, J.; Toft, E.; Qvisth, V.; Näslund, E.; Thorell, A.; Rydén, M.; Arner, P. Improved metabolism and body composition beyond normal levels following gastric bypass surgery: A longitudinal study. J. Intern. Med. 2019, 285, 92–101. [Google Scholar]
- Arner, P.; Arner, E.; Hammarstedt, A.; Smith, U. Genetic predisposition for Type 2 diabetes, but not for overweight/obesity, is associated with a restricted adipogenesis. PLoS ONE 2011, 6, e18284. [Google Scholar]
- Lotta, L.A.; Gulati, P.; Day, F.R.; Payne, F.; Ongen, H.; van de Bunt, M.; Gaulton, K.J.; Eicher, J.D.; Sharp, S.J.; Luan, J. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet. 2017, 49, 17–26. [Google Scholar]
- Andersson, D.P.; Arner, E.; Hogling, D.E.; Rydén, M.; Arner, P. Abdominal subcutaneous adipose tissue cellularity in men and women. Int. J. Obes. (Lond) 2017, 41, 1564–1569. [Google Scholar]
- Kulyté, A.; Lundbäck, V.; Lindgren, C.M.; Luan, J.; Lotta, A.L.; Langenberg, C.; Arner, P.; Strawbridge, R.J.; Ingrid Dahlman, I. Genome-wide association study of adipocyte lipolysis in the GENetics of adipocyte lipolysis (GENiAL) cohort. Mol. Metab. 2020, 34, 85–96. [Google Scholar] [PubMed]
- Gallagher, D.; Visser, M.; Sepúlveda, D.; Pierson, R.N.; Harris, T.; Heymsfield, S.B. How useful is body mass index for comparison of body fatness across age, sex, and ethnic groups? Am. J. Epidemiol. 1996, 143, 228–239. [Google Scholar] [PubMed]
- Ryden, M.; Arner, P. Subcutaneous Adipocyte Lipolysis Contributes to Circulating Lipid Levels. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1782–1787. [Google Scholar]
- Ryden, M.; Arner, P. Cardiovascular risk score is linked to subcutaneous adipocyte size and lipid metabolism. J. Intern. Med. 2017, 282, 220–228. [Google Scholar]
- Dahlman, I.; Rydén, M.; Brodin, D.; Grallert, H.; Strawbridge, R.J.; Arner, P. Numerous Genes in Loci Associated With Body Fat Distribution Are Linked to Adipose Function. Diabetes 2016, 65, 433–437. [Google Scholar]
- Kolaczynski, J.W.; Morales, L.M.; Moore, J.H., Jr.; Considine, R.V.; Pietrzkowski, Z.; Noto, P.F.; Colberg, J.; Caro, J.F. A new technique for biopsy of human abdominal fat under local anaesthesia with Lidocaine. Int. J. Obes. Relat. Metab. Disord. 1994, 18, 161–166. [Google Scholar]
- Lofgren, P.; Andersson, I.; Wahrenberg, H.; Hoffstedt, J. No difference in lipolysis or glucose transport of subcutaneous fat cells between moderate-fat and low-fat hypocaloric diets in obese women. Horm. Metab. Res. 2005, 37, 734–740. [Google Scholar]
- Hirsch, J.; Gallian, E. Methods for the determination of adipose cell size in man and animals. J. Lipid Res. 1968, 9, 110–119. [Google Scholar]
- Hellmer, J.; Arner, P.; Lundin, A. Automatic luminometric kinetic assay of glycerol for lipolysis studies. Anal. Biochem. 1989, 177, 132–137. [Google Scholar]
- Loh, P.R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; A Reshef, Y.; K Finucane, H.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [PubMed]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [PubMed]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar]
- Pettersson, A.M.; Stenson, B.M.; Lorente-Cebrián, S.; Andersson, D.P.; Mejhert, N.; Krätzel, J.; Aström, G.; Dahlman, I.; Chibalin, A.V.; Arner, P.; et al. LXR is a negative regulator of glucose uptake in human adipocytes. Diabetologia 2013, 56, 2044–2054. [Google Scholar]
- Arner, E.; Daub, C.O.; Vitting-Seerup, K.; Andersson, R.; Lilje, B.; Drabløs, F.; Lennartsson, A.; Rönnerblad, M.; Hrydziuszko, O.; Vitezic, M.; et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 2015, 347, 1010–1014. [Google Scholar]
- Lundback, V.; Kulyte, A.; Strawbridge, R.J.; Ryden, M.; Arner, P.; Marcus, C.; Dahlman, I. FAM13A and POM121C are candidate genes for fasting insulin: Functional follow-up analysis of a genome-wide association study. Diabetologia 2018, 61, 1112–1123. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar]
- Ehrlund, A.; Acosta, J.R.; Björk, C.; Hedén, P.; Douagi, I.; Arner, P.; Laurencikiene, J. The cell-type specific transcriptome in human adipose tissue and influence of obesity on adipocyte progenitors. Sci. Data 2017, 4, 170164. [Google Scholar]
- Li, F.; Zhu, W.; Gonzalez, F.J. Potential role of CYP1B1 in the development and treatment of metabolic diseases. Pharmacol. Ther. 2017, 178, 18–30. [Google Scholar]
- Liu, X.; Huang, T.; Li, L.; Tang, Y.; Tian, Y.; Wang, S.; Fan, C. CYP1B1 deficiency ameliorates obesity and glucose intolerance induced by high fat diet in adult C57BL/6J mice. Am. J. Transl. Res. 2015, 7, 761–771. [Google Scholar] [PubMed]
- Larsen, M.C.; Bushkofsky, J.R.; Gorman, T.; Adhami, V.; Mukhtar, H.; Wang, S.; Reeder, S.B.; Sheibani, N.; Jefcoate, C.R. Cytochrome P450 1B1: An. unexpected modulator of liver fatty acid homeostasis. Arch. Biochem. Biophys. 2015, 571, 21–39. [Google Scholar] [PubMed]
- Li, F.; Jiang, C.; Larsen, M.C.; Bushkofsky, J.; Krausz, K.W.; Wang, T.; Jefcoate, C.R.; Gonzalez, F.J. Lipidomics reveals a link between CYP1B1 and SCD1 in promoting obesity. J. Proteome Res. 2014, 13, 2679–2687. [Google Scholar] [PubMed]
- Le Guillou, S.; Laubier, J.; Péchoux, C.; Aujean, E.; Castille, J.; Leroux, C.; Le Provost, F. Defects of the endoplasmic reticulum and changes to lipid droplet size in mammary epithelial cells due to miR-30b-5p overexpression are correlated to a reduction in Atlastin 2 expression. Biochem. Biophys. Res. Commun. 2019, 512, 283–288. [Google Scholar]
- Akiyama, M.; Okada, Y.; Kanai, M.; Takahashi, A.; Momozawa, Y.; Ikeda, M.; Iwata, N.; Ikegawa, S.; Hirata, M.; Matsuda, K.; et al. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat. Genet. 2017, 49, 1458–1467. [Google Scholar] [PubMed]
- Su, X.; Gi, Y.J.; Chakravarti, D.; Chan, I.L.; Zhang, A.; Xia, X.; Tsai, K.Y.; Flores, E.R. TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell. Metab. 2012, 16, 511–525. [Google Scholar] [PubMed]
- Joseph, J.; Radulovich, N.; Wang, T.; Raghavan, V.; Zhu, C.Q.; Tsao, M.S. Rho guanine nucleotide exchange factor ARHGEF10 is a putative tumor suppressor in pancreatic ductal adenocarcinoma. Oncogene 2020, 39, 308–321. [Google Scholar]
- Hoffstedt, J.; Arner, E.; Wahrenberg, H.; Andersson, D.P.; Qvisth, V.; Löfgren, P.; Rydén, M.; Thörne, A.; Wirén, M.; Palmér, M.; et al. Regional impact of adipose tissue morphology on the metabolic profile in morbid obesity. Diabetologia 2010, 53, 2496–2503. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Men | Women | |||||
|---|---|---|---|---|---|---|
| Non Obese | Obese | P | Non Obese | Obese | P | |
| lean/obese (n) | 155 | 91 | 261 | 441 | ||
| age (years) | 47(14) | 43(12) | 0.01 | 40(13) | 41(10) | 0.38 |
| height (cm) | 179(6) | 181(7) | 0.014 | 167(6) | 166(7) | 0.0044 |
| body weight (kg) | 81(9) | 125(18) | <0.0001 | 67(9) | 108(17) | <0.0001 |
| BMI (kg/m2) | 25(2) | 38(5) | <0.0001 | 24(3) | 39(5) | <0.0001 |
| waist (cm) | 93(8) | 126(13) | <0.0001 | 84(10) | 119(13) | <0.0001 |
| WHR | 0.95(0.06) | 1.05(0.05) | <0.0001 | 0.86(0.07) | 0.96(0.07) | <0.0001 |
| systolic blood pressure (mm Hg) | 129(15) | 140(18) | <0.0001 | 120(16) | 130(16) | <0.0001 |
| diastolic blood pressure (mm Hg) | 79(10) | 86(12) | <0.0001 | 74(10) | 79(10) | <0.0001 |
| plasma total cholesterol (mmol/L) | 5.2(1.3) | 5.3(1.4) | 0.4 | 4.9(1.1) | 5.0(1.0) | 0.13 |
| plasma HDL cholesterol (mmol/L) | 1.3(0.4) | 1.0(0.2) | <0.0001 | 1.6(0.4) | 1.2(0.3) | <0.0001 |
| plasma triacylglycerides (mmol/l) | 1.72(2.1) | 2.36(2.52) | 0.044 | 1.07(0.64) | 1.53(0.87) | <0.0001 |
| plasma non-esterified fatty acids (mmol/L) | 0.49(0.17) | 0.61(0.21) | <0.0001 | 0.62(0.21) | 0.72(0.24) | <0.0001 |
| plasma glycerol (mmol/l) | 60.4(27.7) | 78.5(30.2) | <0.0001 | 81.4(43.0) | 116.8(54.3) | <0.0001 |
| fasting plasma glucose (mmol/L) | 5.5(1.2) | 6.4(2.3) | 0.0012 | 5.0(0.7) | 5.6(1.4) | <0.0001 |
| fasting serum insulin (mU/l) | 7.9(5.3) | 20.8(11.8) | <0.0001 | 6.2(3.3) | 14.8(8.0) | <0.0001 |
| HOMA-IR | 2.0(2.0) | 6.1(4.3) | <0.0001 | 1.4(0.9) | 3.8(2.9) | <0.0001 |
| Cell volumn (pl) | 503(142 | 826(192) | <0.0001 | 474(179) | 862(180) | <0.0001 |
| Spontaneous lipolys (mmol glycerol/2hrs/ESAT) | 0.8(0.7) | 3.8(2.7) | <0.0001 | 1.0(0.9) | 3.4(2.9) | <0.0001 |
| Adipose morphology (pl) | “−6(128)” | 13(171 | 0.36 | “−13(149) | 10(168) | 0.054 |
| Chrom | POS | ID | Effect Allele | BETA | L95 | U95 | P | A1_FREQ |
|---|---|---|---|---|---|---|---|---|
| 1 | 3297459 | rs201839757 | I | 198 | 113 | 283 | 5.75E-06 | 0.0126 |
| 1 | 48004858 | rs12032932 | T | 167 | 96 | 238 | 4.40E-06 | 0.0110 |
| 1 | 76545413 | rs10443175 | T | −40 | −57 | −22 | 8.15E-06 | 0.2247 |
| 1 | 157782519 | rs77346326 | T | 136 | 81 | 191 | 1.28E-06 | 0.0203 |
| 2 | 19739010 | rs3914966 | T | −34 | −48 | −19 | 4.64E-06 | 0.4256 |
| 2* | 38508250 | rs147711728 | T | 164 | 93 | 234 | 6.30E-06 | 0.0108 |
| 2 | 170990253 | rs185528286 | C | 159 | 91 | 226 | 4.45E-06 | 0.0153 |
| 3* | 189410000 | rs2378515 | G | 54 | 34 | 73 | 5.29E-08 | 0.1802 |
| 4 | 170028965 | rs202156267 | A | −164 | −229 | −99 | 9.12E-07 | 0.0313 |
| 6 | 97632917 | rs148707864 | G | 140 | 79 | 200 | 7.01E-06 | 0.0148 |
| 6* | 130581980 | rs62431222 | A | 102 | 61 | 144 | 1.36E-06 | 0.0329 |
| 7 | 52698748 | rs150804725 | I | −112 | −159 | −64 | 4.65E-06 | 0.0297 |
| 7 | 156412814 | rs849073 | G | −34 | −48 | −19 | 9.16E-06 | 0.4351 |
| 8* | 1784364 | rs11988258 | A | −34 | −48 | −20 | 4.06E-06 | 0.4201 |
| 8 | 32613177 | rs75468268 | G | 155 | 87 | 223 | 8.95E-06 | 0.0134 |
| 8 | 69224040 | rs814465 | C | 82 | 49 | 114 | 1.04E-06 | 0.0537 |
| 8 | 141147356 | rs142956251 | A | 155 | 87 | 224 | 8.43E-06 | 0.0139 |
| 9* | 21340235 | rs145072648 | C | 163 | 97 | 230 | 1.59E-06 | 0.0126 |
| 9 | 30859764 | rs138591003 | A | −178 | −255 | −100 | 7.76E-06 | 0.0108 |
| 10 | 97277536 | rs189712177 | G | 186 | 107 | 265 | 4.43E-06 | 0.0115 |
| 10 | 128606691 | rs71490795 | A | 121 | 69 | 174 | 6.52E-06 | 0.0225 |
| 11 | 23509085 | rs148697259 | A | 133 | 75 | 192 | 9.22E-06 | 0.0181 |
| 11 | 99369450 | rs72991567 | G | 67 | 39 | 95 | 2.54E-06 | 0.0804 |
| 12 | 73510344 | rs60148685 | D | 34 | 19 | 48 | 8.09E-06 | 0.4437 |
| 13 | 30571045 | rs200421910 | I | −64 | −92 | −36 | 8.33E-06 | 0.0988 |
| 13 | 109383269 | rs151235076 | T | 188 | 108 | 267 | 4.27E-06 | 0.0097 |
| 16 | 2390565 | rs45501400 | T | 169 | 94 | 243 | 9.89E-06 | 0.0163 |
| 17* | 22020568 | rs201766885 | C | 113 | 65 | 161 | 4.24E-06 | 0.0464 |
| 19 | 53280252 | rs10414169 | T | 131 | 74 | 189 | 8.82E-06 | 0.0183 |
| 22 | 32558640 | rs144964180 | A | 160 | 90 | 229 | 7.28E-06 | 0.0122 |
| 22 | 49801423 | rs9627723 | C | 45 | 26 | 64 | 4.15E-06 | 0.3654 |
| GWAS Adipose Morphology | GWAS Catalogue T2D | Consistent Risk Alleles For | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | CHROM | POS | A1 | A1_FREQ | BETA | P | PUBMED | Reported Genes | Risk Allele | OR BETA | Adipose Hypertrophy and T2D |
| rs12031188 | 1 | 51103268 | C | 0.40 | 15 | 4.50E-02 | 30718926 | FAF1 | C | 1.08 | yes |
| rs58432198 | 1 | 51256091 | T | 0.11 | −24 | 4.58E-02 | 30718926 | FAF1 | C | 1.10 | no |
| rs2088315 | 3 | 162382517 | G | 0.50 | 16 | 3.27E-02 | 30130595 | OTOL1, LINC01192 | ? | 5.26 | not reported |
| rs8192675 | 3 | 170724883 | C | 0.30 | 17 | 4.13E-02 | 28566273 | NR | T | 1.05 | no |
| rs138306797 | 3 | 185545719 | T | 0.02 | 76 | 1.32E-02 | 25760438 | IGF2BP2 | ? | 0.78 | not reported |
| rs9379084 | 6 | 7231843 | A | 0.10 | −48 | 2.94E-04 | 29632382 | RREB1 | G | 1.09 | yes |
| rs9460550 | 6 | 20719561 | A | 0.16 | 26 | 1.01E-02 | 31118516 | CDKAL1 | A | 1.15 | yes |
| rs72892910 | 6 | 50816887 | T | 0.20 | −18 | 4.61E-02 | 30054458 | TFAP2B | T | 0.06 | yes |
| rs9384193 | 6 | 154554249 | C | 0.38 | −16 | 3.97E-02 | 28060188 | OPRM1, CNKSR3 | ? | not reported | |
| rs10231619 | 7 | 43320594 | T | 0.16 | 41 | 7.18E-05 | 25102180 | HECW1 | T | 1.13 | yes |
| rs4729854 | 7 | 102383663 | A | 0.47 | −18 | 2.00E-02 | 30595370 | ? | not reported | ||
| rs896854 | 8 | 95960511 | C | 0.47 | 20 | 8.30E-03 | 20581827 | TP53INP1 | T | 1.06 | no |
| no | |||||||||||
| rs10761745 | 10 | 65101071 | G | 0.14 | −28 | 9.68E-03 | 28406950 | JMJD1C | ? | 1.20 | not reported |
| rs4929965 | 11 | 2197286 | A | 0.37 | 16 | 4.17E-02 | 30718926 | INS, IGF2 | A | 1.12 | yes |
| rs2237892 | 11 | 2839751 | T | 0.07 | −47 | 1.12E-03 | 22961080 | KCNQ1 | C | 1.32 | yes |
| rs2722769 | 11 | 11228374 | G | 0.42 | −16 | 3.48E-02 | 22238593 | GALNTL4, LOC729013 | C | 1.35 | unclear |
| rs7931302 | 11 | 128236058 | C | 0.29 | 19 | 1.92E-02 | 30054458 | ETS1 | C | 0.05 | yes |
| rs11048456 | 12 | 26463082 | C | 0.26 | −17 | 4.66E-02 | 30054458 | ITPR2 | C | 0.05 | yes |
| rs1153188 | 12 | 55098996 | T | 0.26 | 21 | 1.79E-02 | 18372903 | DCD | A | 1.08 | no |
| rs730570 | 14 | 101142890 | G | 0.18 | −20 | 3.74E-02 | 21573907 | C14orf70 | G | 1.14 | no |
| rs1436955 | 15 | 62404382 | T | 0.28 | −17 | 3.62E-02 | 20862305 | C2CD4B | C | 1.13 | yes |
| rs781852 | 17 | 3953102 | G | 0.36 | 17 | 3.43E-02 | 29632382 | ZZEF1 | G | 1.05 | yes |
| rs11873305 | 18 | 58049192 | C | 0.04 | 54 | 1.04E-02 | 22885922 | MC4R | A | 1.18 | no |
| rs7274168 | 20 | 32435978 | C | 0.41 | −20 | 8.28E-03 | 30595370 | ? | not reported | ||
| rs6017317 | 20 | 42946966 | G | 0.23 | 17 | 4.06E-02 | 22158537 | FITM2, R3HDML, HNF4A | G | 1.09 | yes |
| rs16988991 | 20 | 42989777 | A | 0.20 | 17 | 4.63E-02 | 30718926 | HNF4A | A | 1.05 | yes |
| rs2833610 | 21 | 33385186 | A | o | -17 | 2.75E-02 | 21490949 | HUNK | A | 1.17 | no |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lundbäck, V.; Kulyté, A.; Arner, P.; Strawbridge, R.J.; Dahlman, I. Genome-Wide Association Study of Diabetogenic Adipose Morphology in the GENetics of Adipocyte Lipolysis (GENiAL) Cohort. Cells 2020, 9, 1085. https://doi.org/10.3390/cells9051085
Lundbäck V, Kulyté A, Arner P, Strawbridge RJ, Dahlman I. Genome-Wide Association Study of Diabetogenic Adipose Morphology in the GENetics of Adipocyte Lipolysis (GENiAL) Cohort. Cells. 2020; 9(5):1085. https://doi.org/10.3390/cells9051085
Chicago/Turabian StyleLundbäck, Veroniqa, Agné Kulyté, Peter Arner, Rona J. Strawbridge, and Ingrid Dahlman. 2020. "Genome-Wide Association Study of Diabetogenic Adipose Morphology in the GENetics of Adipocyte Lipolysis (GENiAL) Cohort" Cells 9, no. 5: 1085. https://doi.org/10.3390/cells9051085
APA StyleLundbäck, V., Kulyté, A., Arner, P., Strawbridge, R. J., & Dahlman, I. (2020). Genome-Wide Association Study of Diabetogenic Adipose Morphology in the GENetics of Adipocyte Lipolysis (GENiAL) Cohort. Cells, 9(5), 1085. https://doi.org/10.3390/cells9051085