Biomaterials to Neuroprotect the Stroke Brain: A Large Opportunity for Narrow Time Windows

 , ,
, ,
Abstract

1. Introduction
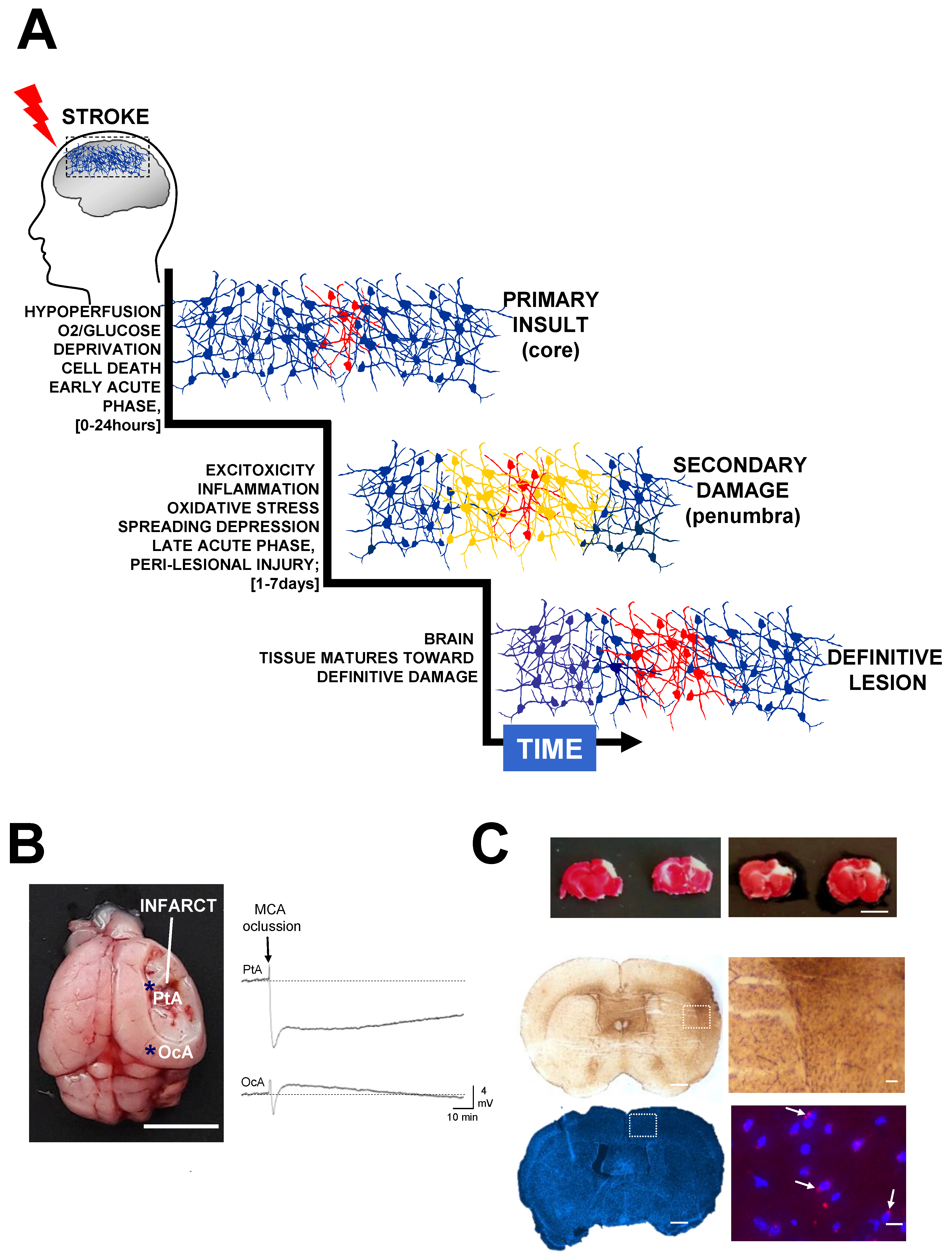
1.1. Lost in Clinical Translation
1.2. The Blood-Brain Barrier
2. Neuroprotective Strategies for Recovery after Ischemic Stroke
2.1. Excitotoxicity
2.2. Oxidative Stress
2.3. Inflammatory Response after Stroke
2.3.1. Central Inflammation
2.3.2. Peripheral Cell Infiltration and Inflammation
2.4. Spreading Depolarization
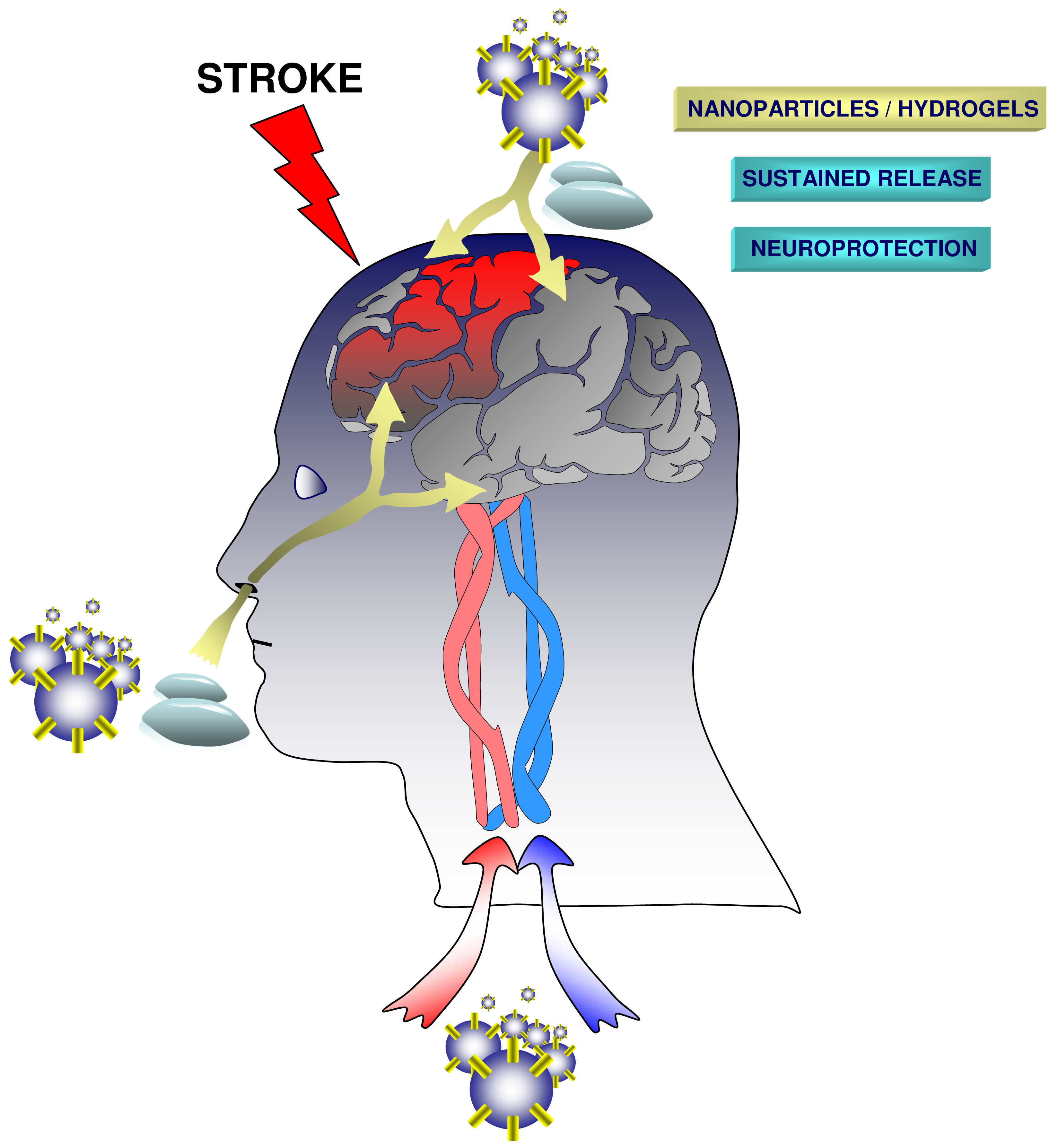
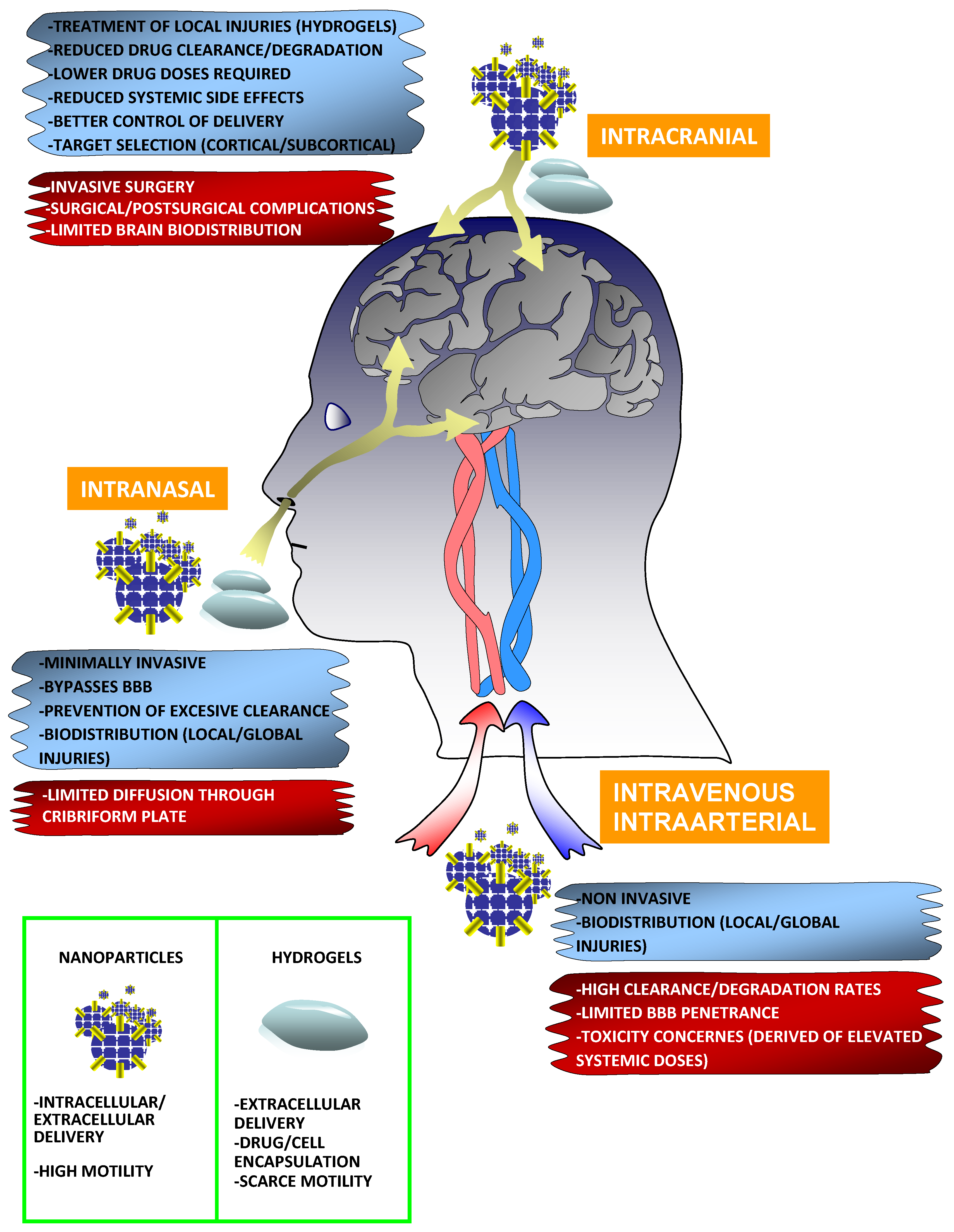
3. Biomaterials and Routes of Administration
3.1. Intracranial Administration
3.2. Intravenous and Intraarterial Administration
3.3. Intranasal Delivery
3.4. Nanoparticles
3.4.1. Functionalization of Nanoparticles
3.4.2. Dendrimers
3.4.3. Liposomes
3.4.4. Micelles
3.5. Hydrogels
Mechanical Properties, Degradation and Dynamic Hydrogels
3.6. Therapeutic Potencial of Biomaterials
4. Neuroprotective Biomaterials for Brain Injury
4.1. Hydrogels and Nanoparticles to Target Angiogenic and Neurogenic Niches
4.2. Hydrogels and Nanoparticles to Target Inflammation
4.3. Antioxidant Strategies
4.4. Biomaterials to Target Excitoxicity
4.5. Other Strategies for Inducing Neuroprotection
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Enderby, P.; Pandyan, A.; Bowen, A.; Hearnden, D.; Ashburn, A.; Conroy, P.; Logan, P.; Thompson, C.; Winter, J. Accessing rehabilitation after stroke—A guessing game? Disabil. Rehabil. 2017, 39, 709–713. [Google Scholar] [CrossRef]
- Moon, S.K.; Alaverdashvili, M.; Cross, A.R.; Whishaw, I.Q. Both compensation and recovery of skilled reaching following small photothrombotic stroke to motor cortex in the rat. Exp. Neurol. 2009, 218, 145–153. [Google Scholar] [CrossRef]
- Broderick, J.P.; Palesch, Y.Y.; Demchuk, A.M.; Yeatts, S.D.; Khatri, P.; Hill, M.D.; Jauch, E.C.; Jovin, T.G.; Yan, B.; Silver, F.L.; et al. Endovascular therapy after intravenous t-PA versus t-PA alone for stroke. N. Engl. J. Med. 2013, 368, 893–903. [Google Scholar] [CrossRef]
- Saver, J.L.; Goyal, M.; Bonafe, A.; Diener, H.C.; Levy, E.I.; Pereira, V.M.; Albers, G.W.; Cognard, C.; Cohen, D.J.; Hacke, W.; et al. Stent-retriever thrombectomy after intravenous t-PA vs. t-PA alone in stroke. N. Engl. J. Med. 2015, 372, 2285–2295. [Google Scholar] [CrossRef]
- Kleindorfer, D.; Kissela, B.; Schneider, A.; Woo, D.; Khoury, J.; Miller, R.; Alwell, K.; Gebel, J.; Szaflarski, J.; Pancioli, A.; et al. Eligibility for recombinant tissue plasminogen activator in acute ischemic stroke: A population-based study. Stroke 2004, 35, e27–e29. [Google Scholar] [CrossRef]
- Pantano, P.; Caramia, F.; Bozzao, L.; Dieler, C.; von Kummer, R. Delayed increase in infarct volume after cerebral ischemia: Correlations with thrombolytic treatment and clinical outcome. Stroke 1999, 30, 502–507. [Google Scholar]
- Shi, K.; Tian, D.C.; Li, Z.G.; Ducruet, A.F.; Lawton, M.T.; Shi, F.D. Global brain inflammation in stroke. Lancet Neurol. 2019, 18, 1058–1066. [Google Scholar] [CrossRef]
- Zhao, S.C.; Ma, L.S.; Chu, Z.H.; Xu, H.; Wu, W.Q.; Liu, F. Regulation of microglial activation in stroke. Acta. Pharmacol. Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef]
- Chamorro, A.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Finsen, B.; Clausen, B.H. Post-stroke inflammation-target or tool for therapy? Acta. Neuropathol. 2019, 137, 693–714. [Google Scholar] [CrossRef]
- Ayata, C. Spreading depression and neurovascular coupling. Stroke 2013, 44, S87–S89. [Google Scholar] [CrossRef]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): A multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- Matsumoto, S.; Murozono, M.; Kanazawa, M.; Nara, T.; Ozawa, T.; Watanabe, Y. Edaravone and cyclosporine A as neuroprotective agents for acute ischemic stroke. Acute Med. Surg. 2018, 5, 213–221. [Google Scholar] [CrossRef]
- Neuroprotection: The end of an era? Lancet 2006, 368, 1548. [CrossRef]
- Shi, L.; Rocha, M.; Leak, R.K.; Zhao, J.; Bhatia, T.N.; Mu, H.; Wei, Z.; Yu, F.; Weiner, S.L.; Ma, F.; et al. A new era for stroke therapy: Integrating neurovascular protection with optimal reperfusion. J. Cereb. Blood Flow Metab. 2018, 38, 2073–2091. [Google Scholar] [CrossRef]
- Louveau, A.; Da Mesquita, S.; Kipnis, J. Lymphatics in Neurological Disorders: A Neuro-Lympho-Vascular Component of Multiple Sclerosis and Alzheimer’s Disease? Neuron 2016, 91, 957–973. [Google Scholar] [CrossRef]
- Cornford, E.M.; Hyman, S. Localization of brain endothelial luminal and abluminal transporters with immunogold electron microscopy. NeuroRx 2005, 2, 27–43. [Google Scholar] [CrossRef]
- De Bock, M.; Van Haver, V.; Vandenbroucke, R.E.; Decrock, E.; Wang, N.; Leybaert, L. Into rather unexplored terrain-transcellular transport across the blood-brain barrier. Glia 2016, 64, 1097–1123. [Google Scholar] [CrossRef]
- Illum, L. Is nose-to-brain transport of drugs in man a reality? J. Pharm. Pharmacol. 2004, 56, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Keum, S.; Marchuk, D.A. A locus mapping to mouse chromosome 7 determines infarct volume in a mouse model of ischemic stroke. Circ. Cardiovasc Genet. 2009, 2, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.J. Strain-Related Differences in the Immune Response: Relevance to Human Stroke. Transl. Stroke Res. 2016, 7, 303–312. [Google Scholar] [CrossRef]
- Upadhyay, R.K. Drug delivery systems, CNS protection, and the blood brain barrier. Biomed. Res. Int. 2014, 2014, 869269. [Google Scholar] [CrossRef] [PubMed]
- Dong, X. Current Strategies for Brain Drug Delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef]
- Begley, D.J.; Brightman, M.W. Structural and functional aspects of the blood-brain barrier. Prog. Drug Res. 2003, 61, 39–78. [Google Scholar] [CrossRef]
- Chen, B.; Friedman, B.; Cheng, Q.; Tsai, P.; Schim, E.; Kleinfeld, D.; Lyden, P.D. Severe blood-brain barrier disruption and surrounding tissue injury. Stroke 2009, 40, e666–e674. [Google Scholar] [CrossRef]
- Neumann-Haefelin, T.; Kastrup, A.; de Crespigny, A.; Yenari, M.A.; Ringer, T.; Sun, G.H.; Moseley, M.E. Serial MRI after transient focal cerebral ischemia in rats: Dynamics of tissue injury, blood-brain barrier damage, and edema formation. Stroke 2000, 31, 1965–1972; discussion 1972–1963. [Google Scholar] [CrossRef]
- Albers, G.W. Late Window Paradox. Stroke 2018, 49, 768–771. [Google Scholar] [CrossRef]
- Bernhardt, J.; Hayward, K.S.; Kwakkel, G.; Ward, N.S.; Wolf, S.L.; Borschmann, K.; Krakauer, J.W.; Boyd, L.A.; Carmichael, S.T.; Corbett, D.; et al. Agreed definitions and a shared vision for new standards in stroke recovery research: The Stroke Recovery and Rehabilitation Roundtable taskforce. Int. J. Stroke 2017, 12, 444–450. [Google Scholar] [CrossRef]
- Dobkin, B.H.; Carmichael, S.T. The Specific Requirements of Neural Repair Trials for Stroke. Neurorehabil. Neural Repair 2016, 30, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, C.R.; Gerigk, M.; Bender, B.; Closhen, D.; Lessmann, V.; Luhmann, H.J. Fluvastatin prevents glutamate-induced blood-brain-barrier disruption in vitro. Life Sci 2008, 82, 1281–1287. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Chan, P.H.; Chen, S.F.; Babuna, O.A.; Simon, R.P.; Weinstein, P.R. Reduction of vasogenic edema and infarction by MK-801 in rats after temporary focal cerebral ischemia. Neurosurgery 1994, 34, 339–345; discussion 345. [Google Scholar] [CrossRef] [PubMed]
- Bordi, F.; Pietra, C.; Ziviani, L.; Reggiani, A. The glycine antagonist GV150526 protects somatosensory evoked potentials and reduces the infarct area in the MCAo model of focal ischemia in the rat. Exp. Neurol. 1997, 145, 425–433. [Google Scholar] [CrossRef]
- Pearlstein, R.D.; Beirne, J.P.; Massey, G.W.; Warner, D.S. Neuroprotective effects of NMDA receptor glycine recognition site antagonism: Dependence on glycine concentration. J. Neurochem 1998, 70, 2012–2019. [Google Scholar] [CrossRef]
- Hoyte, L.; Barber, P.A.; Buchan, A.M.; Hill, M.D. The rise and fall of NMDA antagonists for ischemic stroke. Curr. Mol. Med. 2004, 4, 131–136. [Google Scholar] [CrossRef]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef]
- Neuhaus, A.A.; Couch, Y.; Hadley, G.; Buchan, A.M. Neuroprotection in stroke: The importance of collaboration and reproducibility. Brain 2017, 140, 2079–2092. [Google Scholar] [CrossRef]
- Bratane, B.T.; Cui, H.; Cook, D.J.; Bouley, J.; Tymianski, M.; Fisher, M. Neuroprotection by freezing ischemic penumbra evolution without cerebral blood flow augmentation with a postsynaptic density-95 protein inhibitor. Stroke 2011, 42, 3265–3270. [Google Scholar] [CrossRef]
- Cook, D.J.; Teves, L.; Tymianski, M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 2012, 483, 213–217. [Google Scholar] [CrossRef]
- Hill, M.D.; Martin, R.H.; Mikulis, D.; Wong, J.H.; Silver, F.L.; Terbrugge, K.G.; Milot, G.; Clark, W.M.; Macdonald, R.L.; Kelly, M.E.; et al. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012, 11, 942–950. [Google Scholar] [CrossRef]
- Sims, N.R.; Muyderman, H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta. 2010, 1802, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Teixeira, P.C.; Braunersreuther, V.; Mach, F.; Vuilleumier, N.; Montecucco, F. Pathophysiology and Treatments of Oxidative Injury in Ischemic Stroke: Focus on the Phagocytic NADPH Oxidase 2. Antioxid. Redox Signal. 2015, 23, 460–489. [Google Scholar] [CrossRef] [PubMed]
- Zalba, G.; Fortuno, A.; San Jose, G.; Moreno, M.U.; Beloqui, O.; Diez, J. Oxidative stress, endothelial dysfunction and cerebrovascular disease. Cerebrovasc. Dis. 2007, 24, 24–29. [Google Scholar] [CrossRef]
- Pozo-Rodrigalvarez, A.; Gradillas, A.; Serrano, J.; Fernandez, A.P.; Martinez-Murillo, R.; Perez-Castells, J. New synthesis and promising neuroprotective role in experimental ischemic stroke of ONO-1714. Eur. J. Med. Chem. 2012, 54, 439–446. [Google Scholar] [CrossRef][Green Version]
- Murakami, K.; Kondo, T.; Kawase, M.; Li, Y.; Sato, S.; Chen, S.F.; Chan, P.H. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J. Neurosci 1998, 18, 205–213. [Google Scholar] [CrossRef]
- Moro, M.A.; Almeida, A.; Bolanos, J.P.; Lizasoain, I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic Biol Med. 2005, 39, 1291–1304. [Google Scholar] [CrossRef]
- Kinouchi, H.; Epstein, C.J.; Mizui, T.; Carlson, E.; Chen, S.F.; Chan, P.H. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc. Natl. Acad. Sci. USA 1991, 88, 11158–11162. [Google Scholar] [CrossRef]
- Liu, T.H.; Beckman, J.S.; Freeman, B.A.; Hogan, E.L.; Hsu, C.Y. Polyethylene glycol-conjugated superoxide dismutase and catalase reduce ischemic brain injury. Am. J. Physiol. 1989, 256, H589–H593. [Google Scholar] [CrossRef]
- Ren, Y.; Wei, B.; Song, X.; An, N.; Zhou, Y.; Jin, X.; Zhang, Y. Edaravone’s free radical scavenging mechanisms of neuroprotection against cerebral ischemia: Review of the literature. Int. J. Neurosci. 2015, 125, 555–565. [Google Scholar] [CrossRef]
- Kuroda, S.; Tsuchidate, R.; Smith, M.L.; Maples, K.R.; Siesjo, B.K. Neuroprotective effects of a novel nitrone, NXY-059, after transient focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 1999, 19, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.W.; Cummings, R.M.; Bowes, L.J.; Ridley, R.M.; Green, A.R. Functional and histological evidence for the protective effect of NXY-059 in a primate model of stroke when given 4 h after occlusion. Stroke 2003, 34, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.C.; Ashwood, T.; Wasiewski, W.W.; Emeribe, U. NXY-059 for the treatment of acute ischemic stroke. N. Engl. J. Med. 2007, 357, 562–571. [Google Scholar] [CrossRef]
- Chamorro, A.; Amaro, S.; Castellanos, M.; Segura, T.; Arenillas, J.; Marti-Fabregas, J.; Gallego, J.; Krupinski, J.; Gomis, M.; Canovas, D.; et al. Safety and efficacy of uric acid in patients with acute stroke (URICO-ICTUS): A randomised, double-blind phase 2b/3 trial. Lancet Neurol. 2014, 13, 453–460. [Google Scholar] [CrossRef]
- Turner, R.C.; Naser, Z.J.; Lucke-Wold, B.P.; Logsdon, A.F.; Vangilder, R.L.; Matsumoto, R.R.; Huber, J.D.; Rosen, C.L. Single low-dose lipopolysaccharide preconditioning: Neuroprotective against axonal injury and modulates glial cells. Neuroimmunol. Neuroinflamm. 2017, 4, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef]
- Perego, C.; Fumagalli, S.; De Simoni, M.G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflammation 2011, 8, 174. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Wang, J.; Xing, H.; Wan, L.; Jiang, X.; Wang, C.; Wu, Y. Treatment targets for M2 microglia polarization in ischemic stroke. Biomed. Pharmacother. 2018, 105, 518–525. [Google Scholar] [CrossRef]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef]
- Asahi, M.; Asahi, K.; Jung, J.C.; del Zoppo, G.J.; Fini, M.E.; Lo, E.H. Role for matrix metalloproteinase 9 after focal cerebral ischemia: Effects of gene knockout and enzyme inhibition with BB-94. J. Cereb. Blood Flow Metab. 2000, 20, 1681–1689. [Google Scholar] [CrossRef]
- Asahi, M.; Wang, X.; Mori, T.; Sumii, T.; Jung, J.C.; Moskowitz, M.A.; Fini, M.E.; Lo, E.H. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J. Neurosci. 2001, 21, 7724–7732. [Google Scholar] [CrossRef]
- Chaturvedi, M.; Kaczmarek, L. Mmp-9 inhibition: A therapeutic strategy in ischemic stroke. Mol. Neurobiol 2014, 49, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, G.; Chuang, H.; Chiu, C.; Abdelmaksoud, A.; Ye, Y.; Zhao, L. Portrait of glial scar in neurological diseases. Int. J. Immunopathol. Pharmacol. 2018, 31, 2058738418801406. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Wang, Y.X.; Dou, F.F.; Lu, H.Z.; Ma, Z.W.; Lu, P.H.; Xu, X.M. Glutamine synthetase down-regulation reduces astrocyte protection against glutamate excitotoxicity to neurons. Neurochem. Int. 2010, 56, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Dubbelaar, M.L.; Kracht, L.; Eggen, B.J.L.; Boddeke, E. The Kaleidoscope of Microglial Phenotypes. Front. Immunol. 2018, 9, 1753. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ma, X.; Li, D.; Hao, J. Thiamet G mediates neuroprotection in experimental stroke by modulating microglia/macrophage polarization and inhibiting NF-kappaB p65 signaling. J. Cereb. Blood Flow Metab. 2017, 37, 2938–2951. [Google Scholar] [CrossRef]
- Shao, Y.; Deng, T.; Zhang, T.; Li, P.; Wang, Y. FAM19A3, a novel secreted protein, modulates the microglia/macrophage polarization dynamics and ameliorates cerebral ischemia. FEBS Lett. 2015, 589, 467–475. [Google Scholar] [CrossRef]
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting neutrophils in ischemic stroke: Translational insights from experimental studies. J. Cereb. Blood Flow Metab. 2015, 35, 888–901. [Google Scholar] [CrossRef]
- Lavine, S.D.; Hofman, F.M.; Zlokovic, B.V. Circulating antibody against tumor necrosis factor-alpha protects rat brain from reperfusion injury. J. Cereb. Blood Flow Metab. 1998, 18, 52–58. [Google Scholar] [CrossRef]
- Gary, D.S.; Bruce-Keller, A.J.; Kindy, M.S.; Mattson, M.P. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J. Cereb. Blood Flow Metab. 1998, 18, 1283–1287. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Clausen, B.H.; Babcock, A.A.; Gregersen, R.; Fenger, C.; Nielsen, H.H.; Haugaard, L.S.; Wirenfeldt, M.; Nielsen, M.; Dagnaes-Hansen, F.; et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 2009, 29, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, N. Interleukin-1 and neuronal injury: Mechanisms, modification, and therapeutic potential. Brain Behav. Immun. 2003, 17, 152–157. [Google Scholar] [CrossRef]
- Villa, P.; Triulzi, S.; Cavalieri, B.; Di Bitondo, R.; Bertini, R.; Barbera, S.; Bigini, P.; Mennini, T.; Gelosa, P.; Tremoli, E.; et al. The interleukin-8 (IL-8/CXCL8) receptor inhibitor reparixin improves neurological deficits and reduces long-term inflammation in permanent and transient cerebral ischemia in rats. Mol. Med. 2007, 13, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Brait, V.H.; Rivera, J.; Broughton, B.R.; Lee, S.; Drummond, G.R.; Sobey, C.G. Chemokine-related gene expression in the brain following ischemic stroke: No role for CXCR2 in outcome. Brain Res. 2011, 1372, 169–179. [Google Scholar] [CrossRef]
- Dimitrijevic, O.B.; Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 2007, 38, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.X.; Broughton, B.R.; Kim, H.A.; Lee, S.; Drummond, G.R.; Sobey, C.G. Evidence That Ly6C(hi) Monocytes are Protective in Acute Ischemic Stroke by Promoting M2 Macrophage Polarization. Stroke 2015, 46, 1929–1937. [Google Scholar] [CrossRef]
- Werner, Y.; Mass, E.; Ashok Kumar, P.; Ulas, T.; Handler, K.; Horne, A.; Klee, K.; Lupp, A.; Schutz, D.; Saaber, F.; et al. Cxcr4 distinguishes HSC-derived monocytes from microglia and reveals monocyte immune responses to experimental stroke. Nat. Neurosci. 2020. [Google Scholar] [CrossRef]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; William Shuttleworth, C.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J. Cereb. Blood Flow Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef]
- Nakamura, H.; Strong, A.J.; Dohmen, C.; Sakowitz, O.W.; Vollmar, S.; Sue, M.; Kracht, L.; Hashemi, P.; Bhatia, R.; Yoshimine, T.; et al. Spreading depolarizations cycle around and enlarge focal ischaemic brain lesions. Brain 2010, 133, 1994–2006. [Google Scholar] [CrossRef]
- Klass, A.; Sanchez-Porras, R.; Santos, E. Systematic review of the pharmacological agents that have been tested against spreading depolarizations. J. Cereb. Blood Flow Metab. 2018, 38, 1149–1179. [Google Scholar] [CrossRef] [PubMed]
- Sakowitz, O.W.; Kiening, K.L.; Krajewski, K.L.; Sarrafzadeh, A.S.; Fabricius, M.; Strong, A.J.; Unterberg, A.W.; Dreier, J.P. Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke 2009, 40, e519–e522. [Google Scholar] [CrossRef] [PubMed]
- Barios, J.A.; Pisarchyk, L.; Fernandez-Garcia, L.; Barrio, L.C.; Ramos, M.; Martinez-Murillo, R.; Gonzalez-Nieto, D. Long-term dynamics of somatosensory activity in a stroke model of distal middle cerebral artery oclussion. J. Cereb. Blood Flow Metab. 2016, 36, 606–620. [Google Scholar] [CrossRef]
- Fernandez-Garcia, L.; Perez-Rigueiro, J.; Martinez-Murillo, R.; Panetsos, F.; Ramos, M.; Guinea, G.V.; Gonzalez-Nieto, D. Cortical Reshaping and Functional Recovery Induced by Silk Fibroin Hydrogels-Encapsulated Stem Cells Implanted in Stroke Animals. Front. Cell Neurosci. 2018, 12, 296. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Z.; Rege, S.V.; Wang, M.; Si, G.; Zhou, Y.; Wang, S.; Griffin, J.H.; Goldman, S.A.; Zlokovic, B.V. 3K3A-activated protein C stimulates postischemic neuronal repair by human neural stem cells in mice. Nat. Med. 2016, 22, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Huebsch, N.; Mooney, D.J. Inspiration and application in the evolution of biomaterials. Nature 2009, 462, 426–432. [Google Scholar] [CrossRef]
- Green, J.J.; Elisseeff, J.H. Mimicking biological functionality with polymers for biomedical applications. Nature 2016, 540, 386–394. [Google Scholar] [CrossRef]
- Hubbell, J.A.; Thomas, S.N.; Swartz, M.A. Materials engineering for immunomodulation. Nature 2009, 462, 449–460. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef]
- Khader, B.A.; Towler, M.R. Materials and techniques used in cranioplasty fixation: A review. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 66, 315–322. [Google Scholar] [CrossRef]
- Still, M.; Kane, A.; Roux, A.; Zanello, M.; Dezamis, E.; Parraga, E.; Sauvageon, X.; Meder, J.F.; Pallud, J. Independent Factors Affecting Postoperative Complication Rates After Custom-Made Porous Hydroxyapatite Cranioplasty: A Single-Center Review of 109 Cases. World Neurosurg. 2018, 114, e1232–e1244. [Google Scholar] [CrossRef]
- Taschner, C.A.; Chapot, R.; Costalat, V.; Machi, P.; Courtheoux, P.; Barreau, X.; Berge, J.; Pierot, L.; Kadziolka, K.; Jean, B.; et al. Second-Generation Hydrogel Coils for the Endovascular Treatment of Intracranial Aneurysms: A Randomized Controlled Trial. Stroke 2018, 49, 667–674. [Google Scholar] [CrossRef]
- Mehta, R.I. Polymer-induced central nervous system complications following vascular procedures: Spectrum of iatrogenic injuries and review of outcomes. Hum. Pathol. 2016, 53, 178–190. [Google Scholar] [CrossRef]
- Sugawara, T.; Fujimura, M.; Noshita, N.; Kim, G.W.; Saito, A.; Hayashi, T.; Narasimhan, P.; Maier, C.M.; Chan, P.H. Neuronal death/survival signaling pathways in cerebral ischemia. NeuroRx 2004, 1, 17–25. [Google Scholar] [CrossRef]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef]
- Ruscher, K.; Kuric, E.; Liu, Y.; Walter, H.L.; Issazadeh-Navikas, S.; Englund, E.; Wieloch, T. Inhibition of CXCL12 signaling attenuates the postischemic immune response and improves functional recovery after stroke. J. Cereb. Blood Flow Metab. 2013, 33, 1225–1234. [Google Scholar] [CrossRef]
- Hammarlund-Udenaes, M.; Friden, M.; Syvanen, S.; Gupta, A. On the rate and extent of drug delivery to the brain. Pharm. Res. 2008, 25, 1737–1750. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front. Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Ajay; Bemis, G.W.; Murcko, M.A. Designing libraries with CNS activity. J. Med. Chem. 1999, 42, 4942–4951. [Google Scholar] [CrossRef]
- Fernandez-Garcia, L.; Mari-Buye, N.; Barios, J.A.; Madurga, R.; Elices, M.; Perez-Rigueiro, J.; Ramos, M.; Guinea, G.V.; Gonzalez-Nieto, D. Safety and tolerability of silk fibroin hydrogels implanted into the mouse brain. Acta. Biomater. 2016, 45, 262–275. [Google Scholar] [CrossRef] [PubMed]
- McCracken, P.J.; Manduca, A.; Felmlee, J.; Ehman, R.L. Mechanical transient-based magnetic resonance elastography. Magn. Reson. Med. 2005, 53, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.C.; Curran, G.L.; Glaser, K.J.; Rossman, P.J.; Huston, J., 3rd; Poduslo, J.F.; Jack, C.R., Jr.; Felmlee, J.P.; Ehman, R.L. Magnetic resonance elastography of the brain in a mouse model of Alzheimer’s disease: Initial results. Magn. Reson. Imaging 2012, 30, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Tang-Schomer, M.D.; White, J.D.; Tien, L.W.; Schmitt, L.I.; Valentin, T.M.; Graziano, D.J.; Hopkins, A.M.; Omenetto, F.G.; Haydon, P.G.; Kaplan, D.L. Bioengineered functional brain-like cortical tissue. Proc. Natl. Acad. Sci. USA 2014, 111, 13811–13816. [Google Scholar] [CrossRef] [PubMed]
- Tang-Schomer, M.D.; Kaplan, D.L.; Whalen, M.J. Film interface for drug testing for delivery to cells in culture and in the brain. Acta. Biomater. 2019. [Google Scholar] [CrossRef]
- Moringlane, R.B.; Keric, N.; Freimann, F.B.; Mielke, D.; Burger, R.; Duncker, D.; Rohde, V.; Eckardstein, K.L.V. Efficacy and safety of durotomy after decompressive hemicraniectomy in traumatic brain injury. Neurosurg. Rev. 2017, 40, 655–661. [Google Scholar] [CrossRef]
- Champagne, P.O.; Bojanowski, M.; Fournier-Gosselin, M.P.; Shedid, D. Safety of performing craniotomy in the elderly: The utility of co-morbidity indices. Interdiscip. Neurosurg. 2018, 14, 97–101. [Google Scholar] [CrossRef]
- Baldwin, S.A.; Fugaccia, I.; Brown, D.R.; Brown, L.V.; Scheff, S.W. Blood-brain barrier breach following cortical contusion in the rat. J. Neurosurg. 1996, 85, 476–481. [Google Scholar] [CrossRef]
- Huang, Z.G.; Xue, D.; Preston, E.; Karbalai, H.; Buchan, A.M. Biphasic opening of the blood-brain barrier following transient focal ischemia: Effects of hypothermia. Can. J. Neurol. Sci. 1999, 26, 298–304. [Google Scholar] [CrossRef]
- Hone, E.A.; Hu, H.; Sprowls, S.A.; Farooqi, I.; Grasmick, K.; Lockman, P.R.; Simpkins, J.W.; Ren, X. Blood-Brain Barrier Openings after Stroke. Neurol. Disord. Stroke Int. 2018, 1, 1011. [Google Scholar]
- Kuroiwa, T.; Ting, P.; Martinez, H.; Klatzo, I. The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta. Neuropathol. 1985, 68, 122–129. [Google Scholar] [CrossRef]
- Bharadwaj, V.N.; Lifshitz, J.; Adelson, P.D.; Kodibagkar, V.D.; Stabenfeldt, S.E. Temporal assessment of nanoparticle accumulation after experimental brain injury: Effect of particle size. Sci. Rep. 2016, 6, 29988. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Mathison, S.; Nagilla, R.; Kompella, U.B. Nasal route for direct delivery of solutes to the central nervous system: Fact or fiction? J. Drug Target. 1998, 5, 415–441. [Google Scholar] [CrossRef]
- Kristensson, K.; Olsson, Y. Uptake of exogenous proteins in mouse olfactory cells. Acta Neuropathol 1971, 19, 145–154. [Google Scholar] [CrossRef]
- Illum, L. Nasal drug delivery--possibilities, problems and solutions. J. Control. Release 2003, 87, 187–198. [Google Scholar] [CrossRef]
- Li, R.; Huang, Y.; Chen, L.; Zhou, H.; Zhang, M.; Chang, L.; Shen, H.; Zhou, M.; Su, P.; Zhu, D. Targeted delivery of intranasally administered nanoparticles-mediated neuroprotective peptide NR2B9c to brain and neuron for treatment of ischemic stroke. Nanomedicine 2019, 18, 380–390. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Cai, W.; Chen, X. Nanoplatforms for targeted molecular imaging in living subjects. Small 2007, 3, 1840–1854. [Google Scholar] [CrossRef]
- Krauze, M.T.; McKnight, T.R.; Yamashita, Y.; Bringas, J.; Noble, C.O.; Saito, R.; Geletneky, K.; Forsayeth, J.; Berger, M.S.; Jackson, P.; et al. Real-time visualization and characterization of liposomal delivery into the monkey brain by magnetic resonance imaging. Brain Res. Brain Res. Protoc 2005, 16, 20–26. [Google Scholar] [CrossRef]
- Lee, S.R.; Lee, H.J.; Cha, S.H.; Jeong, K.J.; Lee, Y.; Jeon, C.Y.; Yi, K.S.; Lim, I.; Cho, Z.H.; Chang, K.T.; et al. Long-term survival and differentiation of human neural stem cells in nonhuman primate brain with no immunosuppression. Cell Transplant. 2015, 24, 191–201. [Google Scholar] [CrossRef]
- Lin, B.L.; Zhang, J.Z.; Lu, L.J.; Mao, J.J.; Cao, M.H.; Mao, X.H.; Zhang, F.; Duan, X.H.; Zheng, C.S.; Zhang, L.M.; et al. Superparamagnetic Iron Oxide Nanoparticles-Complexed Cationic Amylose for In Vivo Magnetic Resonance Imaging Tracking of Transplanted Stem Cells in Stroke. Nanomaterials 2017, 7, 107. [Google Scholar] [CrossRef]
- Lu, Y.; Xu, Y.J.; Zhang, G.B.; Ling, D.; Wang, M.Q.; Zhou, Y.; Wu, Y.D.; Wu, T.; Hackett, M.J.; Hyo Kim, B.; et al. Iron oxide nanoclusters for T 1 magnetic resonance imaging of non-human primates. Nat. Biomed. Eng. 2017, 1, 637–643. [Google Scholar] [CrossRef]
- Saito, R.; Krauze, M.T.; Bringas, J.R.; Noble, C.; McKnight, T.R.; Jackson, P.; Wendland, M.F.; Mamot, C.; Drummond, D.C.; Kirpotin, D.B.; et al. Gadolinium-loaded liposomes allow for real-time magnetic resonance imaging of convection-enhanced delivery in the primate brain. Exp. Neurol. 2005, 196, 381–389. [Google Scholar] [CrossRef]
- Bao, G.; Mitragotri, S.; Tong, S. Multifunctional nanoparticles for drug delivery and molecular imaging. Annu Rev. Biomed. Eng 2013, 15, 253–282. [Google Scholar] [CrossRef]
- Feng, X.; Lv, F.; Liu, L.; Tang, H.; Xing, C.; Yang, Q.; Wang, S. Conjugated polymer nanoparticles for drug delivery and imaging. ACS Appl. Mater. Interfaces 2010, 2, 2429–2435. [Google Scholar] [CrossRef]
- Aktas, Y.; Yemisci, M.; Andrieux, K.; Gursoy, R.N.; Alonso, M.J.; Fernandez-Megia, E.; Novoa-Carballal, R.; Quinoa, E.; Riguera, R.; Sargon, M.F.; et al. Development and brain delivery of chitosan-PEG nanoparticles functionalized with the monoclonal antibody OX26. Bioconjugate Chem. 2005, 16, 1503–1511. [Google Scholar] [CrossRef]
- Wohlfart, S.; Gelperina, S.; Kreuter, J. Transport of drugs across the blood-brain barrier by nanoparticles. J. Control. Release 2012, 161, 264–273. [Google Scholar] [CrossRef]
- Poellmann, M.J.; Bu, J.; Hong, S. Would antioxidant-loaded nanoparticles present an effective treatment for ischemic stroke? Nanomedicine 2018, 13, 2327–2340. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Miller, D.W.; Li, S.; Alakhov, V.Y.; Kabanov, A.V.; Elmquist, W.F. Pluronic P85 enhances the delivery of digoxin to the brain: In vitro and in vivo studies. J. Pharm. Exp. 2001, 296, 551–557. [Google Scholar]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Kumar, K.P.; Paramakrishnan, N.; Suresh, B. Targeted delivery of tacrine into the brain with polysorbate 80-coated poly(n-butylcyanoacrylate) nanoparticles. Eur. J. Pharm. Biopharm. 2008, 70, 75–84. [Google Scholar] [CrossRef]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Kumar, K.P.; Paramakrishnan, N.; Suresh, B. Poly(n-butylcyanoacrylate) nanoparticles coated with polysorbate 80 for the targeted delivery of rivastigmine into the brain to treat Alzheimer’s disease. Brain Res. 2008, 1200, 159–168. [Google Scholar] [CrossRef]
- Salvalaio, M.; Rigon, L.; Belletti, D.; D’Avanzo, F.; Pederzoli, F.; Ruozi, B.; Marin, O.; Vandelli, M.A.; Forni, F.; Scarpa, M.; et al. Targeted Polymeric Nanoparticles for Brain Delivery of High Molecular Weight Molecules in Lysosomal Storage Disorders. PLoS ONE 2016, 11, e0156452. [Google Scholar] [CrossRef]
- Harris, N.M.; Ritzel, R.; Mancini, N.S.; Jiang, Y.; Yi, X.; Manickam, D.S.; Banks, W.A.; Kabanov, A.V.; McCullough, L.D.; Verma, R. Nano-particle delivery of brain derived neurotrophic factor after focal cerebral ischemia reduces tissue injury and enhances behavioral recovery. Pharmacol. Biochem. Behav. 2016, 48–56. [Google Scholar] [CrossRef]
- Kurakhmaeva, K.B.; Voronina, T.A.; Kapica, I.G.; Kreuter, J.; Nerobkova, L.N.; Seredenin, S.B.; Balabanian, V.Y.; Alyautdin, R.N. Antiparkinsonian effect of nerve growth factor adsorbed on polybutylcyanoacrylate nanoparticles coated with polysorbate-80. Bull. Exp. Biol. Med. 2008, 145, 259–262. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Crooks, D.R.; Wilson-Ollivierre, H.; Ghosh, M.C.; Sougrat, R.; Lee, J.; Cooperman, S.; Mitchell, J.B.; Beaumont, C.; Rouault, T.A. Iron insufficiency compromises motor neurons and their mitochondrial function in Irp2-null mice. PLoS ONE 2011, 6, e25404. [Google Scholar] [CrossRef]
- Lu, W.L.; Qi, X.R.; Zhang, Q.; Li, R.Y.; Wang, G.L.; Zhang, R.J.; Wei, S.L. A pegylated liposomal platform: Pharmacokinetics, pharmacodynamics, and toxicity in mice using doxorubicin as a model drug. J. Pharmacol. Sci. 2004, 95, 381–389. [Google Scholar] [CrossRef]
- Tiwari, S.B.; Amiji, M.M. A review of nanocarrier-based CNS delivery systems. Curr. Drug Deliv. 2006, 3, 219–232. [Google Scholar] [CrossRef]
- Hong, H.Y.; Choi, J.S.; Kim, Y.J.; Lee, H.Y.; Kwak, W.; Yoo, J.; Lee, J.T.; Kwon, T.H.; Kim, I.S.; Han, H.S.; et al. Detection of apoptosis in a rat model of focal cerebral ischemia using a homing peptide selected from in vivo phage display. J. Control. Release 2008, 131, 167–172. [Google Scholar] [CrossRef]
- Lv, W.; Xu, J.; Wang, X.; Li, X.; Xu, Q.; Xin, H. Bioengineered Boronic Ester Modified Dextran Polymer Nanoparticles as Reactive Oxygen Species Responsive Nanocarrier for Ischemic Stroke Treatment. ACS Nano 2018, 12, 5417–5426. [Google Scholar] [CrossRef]
- Lockman, P.R.; Koziara, J.M.; Mumper, R.J.; Allen, D.D. Nanoparticle surface charges alter blood-brain barrier integrity and permeability. J. Drug Target. 2004, 12, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, V.N.; Nguyen, D.T.; Kodibagkar, V.D.; Stabenfeldt, S.E. Nanoparticle-Based Therapeutics for Brain Injury. Adv. Healthc Mater. 2018, 7. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.G.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef]
- Lai, X.; Zhao, H.; Zhang, Y.; Guo, K.; Xu, Y.; Chen, S.; Zhang, J. Intranasal Delivery of Copper Oxide Nanoparticles Induces Pulmonary Toxicity and Fibrosis in C57BL/6 mice. Sci. Rep. 2018, 8, 4499. [Google Scholar] [CrossRef]
- Santos, S.D.; Xavier, M.; Leite, D.M.; Moreira, D.A.; Custodio, B.; Torrado, M.; Castro, R.; Leiro, V.; Rodrigues, J.; Tomas, H.; et al. PAMAM dendrimers: Blood-brain barrier transport and neuronal uptake after focal brain ischemia. J. Control. Release 2018, 291, 65–79. [Google Scholar] [CrossRef]
- Song, Q.; Song, H.; Xu, J.; Huang, J.; Hu, M.; Gu, X.; Chen, J.; Zheng, G.; Chen, H.; Gao, X. Biomimetic ApoE-Reconstituted High Density Lipoprotein Nanocarrier for Blood-Brain Barrier Penetration and Amyloid Beta-Targeting Drug Delivery. Mol. Pharm. 2016, 13, 3976–3987. [Google Scholar] [CrossRef]
- Godinho, B.; Henninger, N.; Bouley, J.; Alterman, J.F.; Haraszti, R.A.; Gilbert, J.W.; Sapp, E.; Coles, A.H.; Biscans, A.; Nikan, M.; et al. Transvascular Delivery of Hydrophobically Modified siRNAs: Gene Silencing in the Rat Brain upon Disruption of the Blood-Brain Barrier. Mol. Ther. 2018, 26, 2580–2591. [Google Scholar] [CrossRef]
- Dos Santos Rodrigues, B.; Oue, H.; Banerjee, A.; Kanekiyo, T.; Singh, J. Dual functionalized liposome-mediated gene delivery across triple co-culture blood brain barrier model and specific in vivo neuronal transfection. J. Control. Release 2018, 286, 264–278. [Google Scholar] [CrossRef]
- Xing, Y.; Wen, C.Y.; Li, S.T.; Xia, Z.X. Non-viral liposome-mediated transfer of brain-derived neurotrophic factor across the blood-brain barrier. Neural Regen Res. 2016, 11, 617–622. [Google Scholar] [CrossRef]
- Nour, S.A.; Abdelmalak, N.S.; Naguib, M.J.; Rashed, H.M.; Ibrahim, A.B. Intranasal brain-targeted clonazepam polymeric micelles for immediate control of status epilepticus: In vitro optimization, ex vivo determination of cytotoxicity, in vivo biodistribution and pharmacodynamics studies. Drug Deliv. 2016, 23, 3681–3695. [Google Scholar] [CrossRef]
- Basalious, E.B.; Shamma, R.N. Novel self-assembled nano-tubular mixed micelles of Pluronics P123, Pluronic F127 and phosphatidylcholine for oral delivery of nimodipine: In vitro characterization, ex vivo transport and in vivo pharmacokinetic studies. Int. J. Pharm. 2015, 493, 347–356. [Google Scholar] [CrossRef]
- Jin, Q.; Cai, Y.; Li, S.; Liu, H.; Zhou, X.; Lu, C.; Gao, X.; Qian, J.; Zhang, J.; Ju, S.; et al. Edaravone-Encapsulated Agonistic Micelles Rescue Ischemic Brain Tissue by Tuning Blood-Brain Barrier Permeability. Theranostics 2017, 7, 884–898. [Google Scholar] [CrossRef]
- Tuladhar, A.; Morshead, C.M.; Shoichet, M.S. Circumventing the blood-brain barrier: Local delivery of cyclosporin A stimulates stem cells in stroke-injured rat brain. J. Control. Release 2015, 215, 1–11. [Google Scholar] [CrossRef]
- Wang, Y.; Cooke, M.J.; Sachewsky, N.; Morshead, C.M.; Shoichet, M.S. Bioengineered sequential growth factor delivery stimulates brain tissue regeneration after stroke. J. Control. Release 2013, 172, 1–11. [Google Scholar] [CrossRef]
- Bjugstad, K.B.; Redmond, D.E., Jr.; Lampe, K.J.; Kern, D.S.; Sladek, J.R., Jr.; Mahoney, M.J. Biocompatibility of PEG-based hydrogels in primate brain. Cell Transplant. 2008, 17, 409–415. [Google Scholar] [CrossRef]
- Cook, D.J.; Nguyen, C.; Chun, H.N.; Irene, L.L.; Chiu, A.S.; Machnicki, M.; Zarembinski, T.I.; Carmichael, S.T. Hydrogel-delivered brain-derived neurotrophic factor promotes tissue repair and recovery after stroke. J. Cereb. Blood Flow Metab. 2017, 37, 1030–1045. [Google Scholar] [CrossRef]
- Murphy, M.C.; Jones, D.T.; Jack, C.R., Jr.; Glaser, K.J.; Senjem, M.L.; Manduca, A.; Felmlee, J.P.; Carter, R.E.; Ehman, R.L.; Huston, J., 3rd. Regional brain stiffness changes across the Alzheimer’s disease spectrum. Neuroimage Clin. 2016, 10, 283–290. [Google Scholar] [CrossRef]
- Chen, X.; Zhi, F.; Jia, X.; Zhang, X.; Ambardekar, R.; Meng, Z.; Paradkar, A.R.; Hu, Y.; Yang, Y. Enhanced brain targeting of curcumin by intranasal administration of a thermosensitive poloxamer hydrogel. J. Pharm Pharmacol. 2013, 65, 807–816. [Google Scholar] [CrossRef]
- Hoban, D.B.; Newland, B.; Moloney, T.C.; Howard, L.; Pandit, A.; Dowd, E. The reduction in immunogenicity of neurotrophin overexpressing stem cells after intra-striatal transplantation by encapsulation in an in situ gelling collagen hydrogel. Biomaterials 2013, 34, 9420–9429. [Google Scholar] [CrossRef]
- Ballios, B.G.; Cooke, M.J.; Donaldson, L.; Coles, B.L.; Morshead, C.M.; van der Kooy, D.; Shoichet, M.S. A Hyaluronan-Based Injectable Hydrogel Improves the Survival and Integration of Stem Cell Progeny following Transplantation. Stem Cell Rep. 2015, 4, 1031–1045. [Google Scholar] [CrossRef]
- Nih, L.R.; Sideris, E.; Carmichael, S.T.; Segura, T. Injection of Microporous Annealing Particle (MAP) Hydrogels in the Stroke Cavity Reduces Gliosis and Inflammation and Promotes NPC Migration to the Lesion. Adv. Mater. 2017, 29, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Caicco, M.J.; Cooke, M.J.; Wang, Y.; Tuladhar, A.; Morshead, C.M.; Shoichet, M.S. A hydrogel composite system for sustained epi-cortical delivery of Cyclosporin A to the brain for treatment of stroke. J. Control. Release 2013, 166, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Osanai, T.; Kuroda, S.; Yasuda, H.; Chiba, Y.; Maruichi, K.; Hokari, M.; Sugiyama, T.; Shichinohe, H.; Iwasaki, Y. Noninvasive transplantation of bone marrow stromal cells for ischemic stroke: Preliminary study with a thermoreversible gelation polymer hydrogel. Neurosurgery 2010, 66, 1140–1147; discussion 1147. [Google Scholar] [CrossRef] [PubMed]
- Mun, E.A.; Hannell, C.; Rogers, S.E.; Hole, P.; Williams, A.C.; Khutoryanskiy, V.V. On the role of specific interactions in the diffusion of nanoparticles in aqueous polymer solutions. Langmuir 2014, 30, 308–317. [Google Scholar] [CrossRef]
- Nance, E.A.; Woodworth, G.F.; Sailor, K.A.; Shih, T.Y.; Xu, Q.; Swaminathan, G.; Xiang, D.; Eberhart, C.; Hanes, J. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci. Transl. Med. 2012, 4, 149ra119. [Google Scholar] [CrossRef]
- Cruz, L.J.; Stammes, M.A.; Que, I.; van Beek, E.R.; Knol-Blankevoort, V.T.; Snoeks, T.J.A.; Chan, A.; Kaijzel, E.L.; Lowik, C. Effect of PLGA NP size on efficiency to target traumatic brain injury. J. Control. Release 2016, 223, 31–41. [Google Scholar] [CrossRef]
- Potjewyd, G.; Moxon, S.; Wang, T.; Domingos, M.; Hooper, N.M. Tissue Engineering 3D Neurovascular Units: A Biomaterials and Bioprinting Perspective. Trends Biotechnol. 2018, 36, 457–472. [Google Scholar] [CrossRef]
- Emerich, D.F.; Silva, E.; Ali, O.; Mooney, D.; Bell, W.; Yu, S.J.; Kaneko, Y.; Borlongan, C. Injectable VEGF hydrogels produce near complete neurological and anatomical protection following cerebral ischemia in rats. Cell Transplant. 2010, 19, 1063–1071. [Google Scholar] [CrossRef]
- Hao, P.; Duan, H.; Hao, F.; Chen, L.; Sun, M.; Fan, K.S.; Sun, Y.E.; Williams, D.; Yang, Z.; Li, X. Neural repair by NT3-chitosan via enhancement of endogenous neurogenesis after adult focal aspiration brain injury. Biomaterials 2017, 140, 88–102. [Google Scholar] [CrossRef]
- Obermeyer, J.M.; Tuladhar, A.; Payne, S.L.; Ho, E.; Morshead, C.M.; Shoichet, M.S. Local Delivery of Brain-Derived Neurotrophic Factor Enables Behavioral Recovery and Tissue Repair in Stroke-Injured Rats. Tissue Eng. Part. A 2019, 25, 1175–1187. [Google Scholar] [CrossRef]
- Overman, J.J.; Clarkson, A.N.; Wanner, I.B.; Overman, W.T.; Eckstein, I.; Maguire, J.L.; Dinov, I.D.; Toga, A.W.; Carmichael, S.T. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc. Natl. Acad. Sci. USA 2012, 109, E2230–E2239. [Google Scholar] [CrossRef]
- Tian, W.M.; Zhang, C.L.; Hou, S.P.; Yu, X.; Cui, F.Z.; Xu, Q.Y.; Sheng, S.L.; Cui, H.; Li, H.D. Hyaluronic acid hydrogel as Nogo-66 receptor antibody delivery system for the repairing of injured rat brain: In vitro. J. Control. Release 2005, 102, 13–22. [Google Scholar] [CrossRef]
- Martin-Martin, Y.; Fernandez-Garcia, L.; Sanchez-Rebato, M.H.; Mari-Buye, N.; Rojo, F.J.; Perez-Rigueiro, J.; Ramos, M.; Guinea, G.V.; Panetsos, F.; Gonzalez-Nieto, D. Evaluation of Neurosecretome from Mesenchymal Stem Cells Encapsulated in Silk Fibroin Hydrogels. Sci Rep. 2019, 9, 8801. [Google Scholar] [CrossRef]
- Palejwala, A.H.; Fridley, J.S.; Mata, J.A.; Samuel, E.L.; Luerssen, T.G.; Perlaky, L.; Kent, T.A.; Tour, J.M.; Jea, A. Biocompatibility of reduced graphene oxide nanoscaffolds following acute spinal cord injury in rats. Surg. Neurol. Int. 2016, 7, 75. [Google Scholar] [CrossRef]
- Lampe, K.J.; Mooney, R.G.; Bjugstad, K.B.; Mahoney, M.J. Effect of macromer weight percent on neural cell growth in 2D and 3D nondegradable PEG hydrogel culture. J. Biomed. Mater. Res. A 2010, 94, 1162–1171. [Google Scholar] [CrossRef]
- Struzyna, L.A.; Wolf, J.A.; Mietus, C.J.; Adewole, D.O.; Chen, H.I.; Smith, D.H.; Cullen, D.K. Rebuilding Brain Circuitry with Living Micro-Tissue Engineered Neural Networks. Tissue Eng. Part. A 2015, 21, 2744–2756. [Google Scholar] [CrossRef]
- Kloxin, A.M.; Kasko, A.M.; Salinas, C.N.; Anseth, K.S. Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science 2009, 324, 59–63. [Google Scholar] [CrossRef]
- Galarraga, J.H.; Burdick, J.A. Moving hydrogels to the fourth dimension. Nat. Mater. 2019, 18, 914–915. [Google Scholar] [CrossRef]
- Shadish, J.A.; Benuska, G.M.; DeForest, C.A. Bioactive site-specifically modified proteins for 4D patterning of gel biomaterials. Nat. Mater. 2019, 18, 1005–1014. [Google Scholar] [CrossRef]
- Adak, A.; Das, G.; Khan, J.; Mukherjee, N.; Gupta, V.; Mallesh, R.; Ghosh, S. Extracellular Matrix Mimicking (ECM) Neuroprotective Injectable Sulfo-functionalized Peptide Hydrogel for Repairing Brain Injury. ACS Biomater. Sci. Eng. 2020, in press. [Google Scholar] [CrossRef]
- Ghuman, H.; Massensini, A.R.; Donnelly, J.; Kim, S.M.; Medberry, C.J.; Badylak, S.F.; Modo, M. ECM hydrogel for the treatment of stroke: Characterization of the host cell infiltrate. Biomaterials 2016, 91, 166–181. [Google Scholar] [CrossRef]
- Ghuman, H.; Mauney, C.; Donnelly, J.; Massensini, A.R.; Badylak, S.F.; Modo, M. Biodegradation of ECM hydrogel promotes endogenous brain tissue restoration in a rat model of stroke. Acta. Biomater. 2018, 80, 66–84. [Google Scholar] [CrossRef]
- Fabian, R.H.; Derry, P.J.; Rea, H.C.; Dalmeida, W.V.; Nilewski, L.G.; Sikkema, W.K.A.; Mandava, P.; Tsai, A.L.; Mendoza, K.; Berka, V.; et al. Efficacy of Novel Carbon Nanoparticle Antioxidant Therapy in a Severe Model of Reversible Middle Cerebral Artery Stroke in Acutely Hyperglycemic Rats. Front. Neurol. 2018, 9, 199. [Google Scholar] [CrossRef]
- Amani, H.; Habibey, R.; Shokri, F.; Hajmiresmail, S.J.; Akhavan, O.; Mashaghi, A.; Pazoki-Toroudi, H. Selenium nanoparticles for targeted stroke therapy through modulation of inflammatory and metabolic signaling. Sci. Rep. 2019, 9, 6044. [Google Scholar] [CrossRef]
- Korin, N.; Kanapathipillai, M.; Matthews, B.D.; Crescente, M.; Brill, A.; Mammoto, T.; Ghosh, K.; Jurek, S.; Bencherif, S.A.; Bhatta, D.; et al. Shear-activated nanotherapeutics for drug targeting to obstructed blood vessels. Science 2012, 337, 738–742. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Lu, C.H. Effect of human astrocytes on the characteristics of human brain-microvascular endothelial cells in the blood-brain barrier. Colloids Surf. B Biointerfaces 2011, 86, 225–231. [Google Scholar] [CrossRef]
- Thored, P.; Wood, J.; Arvidsson, A.; Cammenga, J.; Kokaia, Z.; Lindvall, O. Long-term neuroblast migration along blood vessels in an area with transient angiogenesis and increased vascularization after stroke. Stroke 2007, 38, 3032–3039. [Google Scholar] [CrossRef]
- Ohab, J.J.; Fleming, S.; Blesch, A.; Carmichael, S.T. A neurovascular niche for neurogenesis after stroke. J. Neurosci. 2006, 26, 13007–13016. [Google Scholar] [CrossRef]
- Ali, Z.; Islam, A.; Sherrell, P.; Le-Moine, M.; Lolas, G.; Syrigos, K.; Rafat, M.; Jensen, L.D. Adjustable delivery of pro-angiogenic FGF-2 by alginate:collagen microspheres. Biol. Open 2018, 7. [Google Scholar] [CrossRef]
- Bible, E.; Qutachi, O.; Chau, D.Y.; Alexander, M.R.; Shakesheff, K.M.; Modo, M. Neo-vascularization of the stroke cavity by implantation of human neural stem cells on VEGF-releasing PLGA microparticles. Biomaterials 2012, 33, 7435–7446. [Google Scholar] [CrossRef]
- Ju, R.; Wen, Y.; Gou, R.; Wang, Y.; Xu, Q. The experimental therapy on brain ischemia by improvement of local angiogenesis with tissue engineering in the mouse. Cell Transplant. 2014, 23, S83–S95. [Google Scholar] [CrossRef]
- Tan, E.Y.; Law, J.W.; Wang, C.H.; Lee, A.Y. Development of a cell transducible RhoA inhibitor TAT-C3 transferase and its encapsulation in biocompatible microspheres to promote survival and enhance regeneration of severed neurons. Pharm. Res. 2007, 24, 2297–2308. [Google Scholar] [CrossRef]
- Deguchi, K.; Tsuru, K.; Hayashi, T.; Takaishi, M.; Nagahara, M.; Nagotani, S.; Sehara, Y.; Jin, G.; Zhang, H.; Hayakawa, S.; et al. Implantation of a new porous gelatin-siloxane hybrid into a brain lesion as a potential scaffold for tissue regeneration. J. Cereb. Blood Flow Metab. 2006, 26, 1263–1273. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, A.; Duan, H.; Zhang, S.; Hao, P.; Ye, K.; Sun, Y.E.; Li, X. NT3-chitosan elicits robust endogenous neurogenesis to enable functional recovery after spinal cord injury. Proc. Natl. Acad. Sci. USA 2015, 112, 13354–13359. [Google Scholar] [CrossRef]
- Chen, H.; Spagnoli, F.; Burris, M.; Rolland, W.B.; Fajilan, A.; Dou, H.; Tang, J.; Zhang, J.H. Nanoerythropoietin is 10-times more effective than regular erythropoietin in neuroprotection in a neonatal rat model of hypoxia and ischemia. Stroke 2012, 43, 884–887. [Google Scholar] [CrossRef]
- Elizarova, O.S.; Litvinova, S.A.; Balaban’ian, V.; Barskov, I.V.; Novikova, S.V.; Stel’mashuk, E.V.; Garibova, T.L.; Voronina, T.A. Neuroprotective effect of recombinant human erythropoietin-loaded poly(lactic-co-glycolic) acid nanoparticles in rats with intracerebral posttraumatic hematoma. Eksp Klin Farm. 2012, 75, 7–10. [Google Scholar]
- Solev, I.N.; Balabanyan, V.Y.; Volchek, I.A.; Elizarova, O.S.; Litvinova, S.A.; Garibova, T.L.; Voronina, T.A. Involvement of BDNF and NGF in the mechanism of neuroprotective effect of human recombinant erythropoietin nanoforms. Bull. Exp. Biol. Med. 2013, 155, 242–244. [Google Scholar] [CrossRef]
- Jin, Y.; Kim, I.Y.; Kim, I.D.; Lee, H.K.; Park, J.Y.; Han, P.L.; Kim, K.K.; Choi, H.; Lee, J.K. Biodegradable gelatin microspheres enhance the neuroprotective potency of osteopontin via quick and sustained release in the post-ischemic brain. Acta. Biomater. 2014, 10, 3126–3135. [Google Scholar] [CrossRef]
- Joachim, E.; Kim, I.D.; Jin, Y.; Kim, K.K.; Lee, J.K.; Choi, H. Gelatin nanoparticles enhance the neuroprotective effects of intranasally administered osteopontin in rat ischemic stroke model. Drug Deliv. Transl. Res. 2014, 4, 395–399. [Google Scholar] [CrossRef]
- Navath, R.S.; Kurtoglu, Y.E.; Wang, B.; Kannan, S.; Romero, R.; Kannan, R.M. Dendrimer-drug conjugates for tailored intracellular drug release based on glutathione levels. Bioconjug. Chem. 2008, 19, 2446–2455. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Reddy, M.K.; Labhasetwar, V. Nanoparticle-mediated delivery of superoxide dismutase to the brain: An effective strategy to reduce ischemia-reperfusion injury. FASEB J. 2009, 23, 1384–1395. [Google Scholar] [CrossRef]
- Singhal, A.; Morris, V.B.; Labhasetwar, V.; Ghorpade, A. Nanoparticle-mediated catalase delivery protects human neurons from oxidative stress. Cell Death Dis. 2013, 4, e903. [Google Scholar] [CrossRef]
- Jiang, Y.; Brynskikh, A.M.; Devika, S.; Kabanov, A.V. SOD1 nanozyme salvages ischemic brain by locally protecting cerebral vasculature. J. Control. Release 2015, 213, 36–44. [Google Scholar] [CrossRef]
- Manickam, D.S.; Brynskikh, A.M.; Kopanic, J.L.; Sorgen, P.L.; Klyachko, N.L.; Batrakova, E.V.; Bronich, T.K.; Kabanov, A.V. Well-defined cross-linked antioxidant nanozymes for treatment of ischemic brain injury. J. Control. Release 2012, 162, 636–645. [Google Scholar] [CrossRef]
- Alkharfy, K.M.; Ahmad, A.; Khan, R.M.; Al-Shagha, W.M. Pharmacokinetic plasma behaviors of intravenous and oral bioavailability of thymoquinone in a rabbit model. Eur. J. Drug Metab. Pharmacokinet. 2015, 40, 319–323. [Google Scholar] [CrossRef]
- Xiao, X.Y.; Zhu, Y.X.; Bu, J.Y.; Li, G.W.; Zhou, J.H.; Zhou, S.P. Evaluation of Neuroprotective Effect of Thymoquinone Nanoformulation in the Rodent Cerebral Ischemia-Reperfusion Model. Biomed. Res. Int. 2016, 2016, 2571060. [Google Scholar] [CrossRef]
- Haque, S.; Md, S.; Fazil, M.; Kumar, M.; Sahni, J.K.; Ali, J.; Baboota, S. Venlafaxine loaded chitosan NPs for brain targeting: Pharmacokinetic and pharmacodynamic evaluation. Carbohydr. Polym. 2012, 89, 72–79. [Google Scholar] [CrossRef]
- Ding, Y.; Qiao, Y.; Wang, M.; Zhang, H.; Li, L.; Zhang, Y.; Ge, J.; Song, Y.; Li, Y.; Wen, A. Enhanced Neuroprotection of Acetyl-11-Keto-beta-Boswellic Acid (AKBA)-Loaded O-Carboxymethyl Chitosan Nanoparticles Through Antioxidant and Anti-Inflammatory Pathways. Mol. Neurobiol. 2016, 53, 3842–3853. [Google Scholar] [CrossRef]
- Klose, D.; Laprais, M.; Leroux, V.; Siepmann, F.; Deprez, B.; Bordet, R.; Siepmann, J. Fenofibrate-loaded PLGA microparticles: Effects on ischemic stroke. Eur. J. Pharm. Sci. 2009, 37, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Kopke, R.D.; Choi, C.H.; Foster, S.B.; Doblas, S.; Towner, R.A. Nitrones as therapeutics. Free Radic. Biol. Med. 2008, 45, 1361–1374. [Google Scholar] [CrossRef]
- Pinarbasli, O.; Aktas, Y.; Dalkara, T.; Andrieux, K.; Alonso, M.J.; Fernandez-Megia, E.; Novoa-Carballal, R.; Riguera, R.; Couvreur, P.; Capan, Y. Preparation and evaluation of alpha-phenyl-n-tert-butyl nitrone (PBN)-encapsulated chitosan and PEGylated chitosan nanoparticles. Pharmazie 2009, 64, 436–439. [Google Scholar]
- Hazekawa, M.; Sakai, Y.; Yoshida, M.; Haraguchi, T.; Uchida, T. Single injection of ONO-1301-loaded PLGA microspheres directly after ischaemia reduces ischaemic damage in rats subjected to middle cerebral artery occlusion. J. Pharm. Pharmacol. 2012, 64, 353–359. [Google Scholar] [CrossRef]
- Kakkar, V.; Muppu, S.K.; Chopra, K.; Kaur, I.P. Curcumin loaded solid lipid nanoparticles: An efficient formulation approach for cerebral ischemic reperfusion injury in rats. Eur. J. Pharm. Biopharm. 2013, 85, 339–345. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, Y.; Lv, W.; Wang, Z.; Lv, L.; Wang, B.; Liu, X.; Liu, Y.; Hu, Q.; Sun, W.; et al. Dual targeted nanocarrier for brain ischemic stroke treatment. J. Control. Release 2016, 233, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, M.; Figiel, I.; Sreedhar, B.; Kaczmarek, L. Neuroprotection from tissue inhibitor of metalloproteinase-1 and its nanoparticles. Neurochem. Int. 2012, 61, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, M.; Molino, Y.; Sreedhar, B.; Khrestchatisky, M.; Kaczmarek, L. Tissue inhibitor of matrix metalloproteinases-1 loaded poly(lactic-co-glycolic acid) nanoparticles for delivery across the blood-brain barrier. Int. J. Nanomed. 2014, 9, 575–588. [Google Scholar] [CrossRef]
- Verma, S.K.; Arora, I.; Javed, K.; Akhtar, M.; Samim, M. Enhancement in the Neuroprotective Power of Riluzole Against Cerebral Ischemia Using a Brain Targeted Drug Delivery Vehicle. ACS Appl. Mater. Interfaces 2016, 8, 19716–19723. [Google Scholar] [CrossRef]
- Wahl, A.S.; Omlor, W.; Rubio, J.C.; Chen, J.L.; Zheng, H.; Schroter, A.; Gullo, M.; Weinmann, O.; Kobayashi, K.; Helmchen, F.; et al. Neuronal repair. Asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science 2014, 344, 1250–1255. [Google Scholar] [CrossRef]
- Schwab, M.E. Functions of Nogo proteins and their receptors in the nervous system. Nat. Rev. Neurosci. 2010, 11, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Cai, Q.; Tian, D.; Kong, D.K.; Gou, X.; Chen, Z.; Strittmatter, S.M.; Wang, Z.; Sheth, K.N.; Zhou, J. Targeted drug delivery to ischemic stroke via chlorotoxin-anchored, lexiscan-loaded nanoparticles. Nanomedicine 2016, 12, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; Yoshioka, C.; Ito, Y.; Funakami, Y.; Nishikawa, H.; Kawabata, A. Intravenous Administration of Cilostazol Nanoparticles Ameliorates Acute Ischemic Stroke in a Cerebral Ischemia/Reperfusion-Induced Injury Model. Int. J. Mol. Sci. 2015, 16, 29329–29344. [Google Scholar] [CrossRef]
- Chen, C.; Mei, H.; Shi, W.; Deng, J.; Zhang, B.; Guo, T.; Wang, H.; Hu, Y. EGFP-EGF1-conjugated PLGA nanoparticles for targeted delivery of siRNA into injured brain microvascular endothelial cells for efficient RNA interference. PLoS ONE 2013, 8, e60860. [Google Scholar] [CrossRef]
- Lee, J.; Hyun, H.; Kim, J.; Ryu, J.H.; Kim, H.A.; Park, J.H.; Lee, M. Dexamethasone-loaded peptide micelles for delivery of the heme oxygenase-1 gene to ischemic brain. J. Control. Release 2012, 158, 131–138. [Google Scholar] [CrossRef]
- Kim, I.D.; Lim, C.M.; Kim, J.B.; Nam, H.Y.; Nam, K.; Kim, S.W.; Park, J.S.; Lee, J.K. Neuroprotection by biodegradable PAMAM ester (e-PAM-R)-mediated HMGB1 siRNA delivery in primary cortical cultures and in the postischemic brain. J. Control. Release 2010, 142, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lin, G.; Luan, Y.; Ding, J.; Li, P.C.; Zhao, Z.; Qian, C.; Liu, G.; Ju, S.; Teng, G.J. HIF-prolyl hydroxylase 2 silencing using siRNA delivered by MRI-visible nanoparticles improves therapy efficacy of transplanted EPCs for ischemic stroke. Biomaterials 2019, 197, 229–243. [Google Scholar] [CrossRef]
- Gonzalez-Nieto, D.; Li, L.; Kohler, A.; Ghiaur, G.; Ishikawa, E.; Sengupta, A.; Madhu, M.; Arnett, J.L.; Santho, R.A.; Dunn, S.K.; et al. Connexin-43 in the osteogenic BM niche regulates its cellular composition and the bidirectional traffic of hematopoietic stem cells and progenitors. Blood 2012, 119, 5144–5154. [Google Scholar] [CrossRef]
- Kim, D.H.; Seo, Y.K.; Thambi, T.; Moon, G.J.; Son, J.P.; Li, G.; Park, J.H.; Lee, J.H.; Kim, H.H.; Lee, D.S.; et al. Enhancing neurogenesis and angiogenesis with target delivery of stromal cell derived factor-1alpha using a dual ionic pH-sensitive copolymer. Biomaterials 2015, 61, 115–125. [Google Scholar] [CrossRef]
- Boisserand, L.S.; Kodama, T.; Papassin, J.; Auzely, R.; Moisan, A.; Rome, C.; Detante, O. Biomaterial Applications in Cell-Based Therapy in Experimental Stroke. Stem Cells Int. 2016, 2016, 6810562. [Google Scholar] [CrossRef]
- Gonzalez-Nieto, D.; Fernández-García, L.; Pérez-Rigueiro, J.; Guinea, G.V.; Panetsos, F. Hydrogels-Assisted Cell Engraftment for Repairing the Stroke-Damaged Brain: Chimera or Reality. Polymers 2018, 10, 184. [Google Scholar] [CrossRef]
- Hwang, B.W.; Kim, S.J.; Park, K.M.; Kim, H.; Yeom, J.; Yang, J.A.; Jeong, H.; Jung, H.; Kim, K.; Sung, Y.C.; et al. Genetically engineered mesenchymal stem cell therapy using self-assembling supramolecular hydrogels. J. Control. Release 2015, 220, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Nih, L.R.; Carmichael, S.T.; Segura, T. Hydrogels for brain repair after stroke: An emerging treatment option. Curr. Opin. Biotechnol. 2016, 40, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Sanche, L. Convection-Enhanced Delivery in Malignant Gliomas: A Review of Toxicity and Efficacy. J. Oncol. 2019, 2019, 9342796. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, A.T.; Haida, M.; Ohba, H.; Yamano, M.; Fukumoto, D.; Tsukada, H. Liposome-encapsulated hemoglobin ameliorates ischemic stroke in nonhuman primates: Longitudinal observation. Artif. Organs 2013, 37, 904–912. [Google Scholar] [CrossRef]
- Kawaguchi, A.T.; Haida, M.; Yamano, M.; Fukumoto, D.; Ogata, Y.; Tsukada, H. Liposome-encapsulated hemoglobin ameliorates ischemic stroke in nonhuman primates: An acute study. J. Pharmacol. Exp. Ther. 2010, 332, 429–436. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Stroke Model, Species | Therapeutic Molecule, Main Properties | Biomaterial and Format, Administration Route, Time Window of Application | Main Therapeutic Effects | References |
|---|---|---|---|---|
| MCAO (permanent), rat | Superoxide dismutase/Catalase, anti-oxidant enzymes | PGA NPs, intravenous injection, ~ 1 min post-stroke | Reduction of infarct volume | [49] |
| MCAO (transient), mouse | BDNF, neuroprotective, neurogenic, and regenerative factor | PEG/PGA NPs, intravenous injection, 3–24 h post-stroke | Reduction in cerebral tissue loss | [134] |
| MCAO (transient), rat | Osteopontin, anti-inflammatory, anti-apoptotic, and neurogenic roles | Gelatin microspheres, intrastriatal delivery, 1–12 h post-stroke | Reduced infarct volume | [199] |
| Photothrombotic stroke model (permanent), mouse | Edaravone, antioxidant compound/ROS scavenger | PEG-PLA micelles, intravenous injection, 6 h post-stroke | Higher efficiency of Edaravone delivery. Reduction of ROS, decreasing of ischemic area that leads to behavioural improvement | [152] |
| Endothelin-1 model, rat | Cyclosporin-A, immunosuppressant and anti-inflammatory compound. Neurogenic and regenerative properties | PLGA microparticles encapsulated in hyaluronan and methylcellulose hydrogels, epicortical delivery, immediately after entothelin-1 injection | Sustained delivery of Cyclosporin-A for 14 days. Decreasing infarct volume. Neuroprotective effect even with the hydrogel formulation alone. | [153] |
| Endothelin-1 model, mouse | EGF/erythropoietin, stimulators of neural precursors formation and neuroprotective properties | PLGA-PEG NPs encapsulated in hyaluronan and methylcellulose hydrogels, epicortical administration, 4 days post-stroke | Attenuation of injury response and cell death. Reduction of infarct cavity. Increasing content of mature neurons in damage and peri-lesional areas | [154] |
| Focal aspiration brain injury model, rat | Neurotrophin-3, neurogenic, angiogenic, and anti-inflammatory factor | Chitosan particles, intracerebral delivery (lesion cavity, cortex), immediately after tissue aspiration | Reduction of inflammation (peripheral leukocytes and microglia) | [169] |
| Hypoxia-ischemia exposure model, neonatal rat | Erythropoietin, enhances oxygen delivery, neurogenesis, anti-oxidant properties (decreases ROS formation) | PLGA NPs, intraperitoneal, 1, 24, and 48 h after hypoxia | Reduction of infarct volume and improvement of behavioral deficits with lower doses than the administration of free Erythropoietin | [196] |
| MCAO (transient), rat | Osteopontin, anti-inflammatory, anti-apoptotic, and neurogenic roles | Gelatin microspheres, intranasal, 6 h post-stroke | Reduction of infarct volume | [200] |
| MCAO (transient), rat | Superoxide dismutase, anti-oxidant enzyme | PLGA NPs, arterial (intracarotid), immediately after reperfusion (1 h post-ischemia) | Reduction of infarct volume leading to functional recovery | [203] |
| MCAO (transient), mouse | Superoxide dismutase, anti-oxidant enzyme | Polyion-cationic complexes-PEG, intravenous injection, immediately after reperfusion (1 h post-ischemia) | Reduction of infarct volume (no effect with superoxide dismutase alone) | [205] |
| MCAO (transient), rat | Thymoquinone, anti-inflammatory and anti-oxidant factor | PLGA-Chitosan NPs, intranasal, administration for 12 days: from -5 (pre-stroke) to 7 (post-stroke) days | Reduction of infarct volume and significant motor improvement associated with stronger scavenging and anti-oxidant capacity | [208] |
| Control (healthy), rat | Venlafaxine, antidepressant molecule | Chitosan NPs, intranasal delivery | Efficient delivery of Venlafaxine into the brain via the intranasal route in comparison with the intranasal or intravenous delivery of free Venlafaxine | [209] |
| MCAO (transient), rat | Acetyl-11-keto-β-boswellic acid, anti-oxidant and anti-inflammatory effects | Chitosan NPs, intravenous injection, 1 h after reperfusion (2 h post-ischemia) | Higher rates of delivery in the brain with respect to the administration of the free molecule. Reduction of infarct volume that leads to behavioral improvements. Positive outcomes related with decreasing oxidative stress/inflammation and reduced neuronal mortality post-ischemia | [210] |
| MCAO (transient), rat | Fenofibrate, activator of Peroxisome Proliferator Activated Receptors (anti-inflammatory and anti-oxidative receptors) | PLGA microparticles, intracerebral administration, 24 h before ischemia | Moderate (non-significant) decrease of infarct volume with respect to the effect of free Fenofibrate. Evolution of infarct volume on untreated MCAO animals is non-determined | [211] |
| MCAO (transient), rat | ONO-1301, prostacyclin agonist with thromboxane synthase inhibitory activity | PLGA microspheres, subcutaneous injection, immediately after MCAO | Significant reduction in the infarct volume and cerebral edema. Oral administration of the free molecule produced similar neuroprotective efficacy, although repetitive doses were needed. | [214] |
| Global cerebral (transient), rat | Curcumin, antioxidant and anti-inflammatory effects | Polysorbate 80/lecithin-based NPs, oral administration, starting 5 days before ischemia for 3 days | Motor and cognitive improvement. No positive effect with the free molecule | [215] |
| MCAO (transient), rat | NR2B9, prevents the excitotoxocity and hyperproduction of NO by disrupting NMDA-PSD-95 signaling | PLGA-PEG nanoparticles decorated with wheat germ agglutinin (WGA) to target olfactory epithelium receptors, intranasal delivery, immediately after reperfusion (2 h post-ischemia) | Increasing content of NR2B9 in the brain with respect to the intranasal administration of free molecule. Significant reduction in infarct size and better neurological improvement | [117] |
| MCAO (transient), rat | NR2B9, prevents the excitotoxocity and hyperproduction of NO by disrupting NMDA-PSD-95 signaling | Dextran NPs decorated with ROS-responsive boronic ester and red blood shell membrane, intravenous injection, immediately after reperfusion (2 h post-ischemia) | NR2B9: Increasing time in circulation and highly efficient to target NR2B9 in the brain. Significant reduction in infarct size and better neurological outcome | [140] |
| MCAO (transient), rat | ZL006, prevents the excitotoxocity and hyperproduction of NO by disrupting NMDA-PSD-95 signaling | Soybean-lecithin-cholesterol-based liposome-PEG decorated with peptides against transferring receptor of the brain endothelium, intravenous injection | Reduction of infarct volume. Behavioural improvement | [216] |
| MCAO (transient), rat | Riluzole, variable effects antagonizing NMDA receptors or/and blocking voltage-gated Calcium/sodium channels | Chitosan NPs formulated with N-isopropyl acrylamide/tween80, intraperitoneal injection, 1 h post-ischemia | Decreasing content of lipid peroxide and increasing levels of glutathione and ROS detoxification enzymes. Reduction of lesion and improvement of behavioral deficits | [219] |
| MCAO (transient), mouse | Cilostazol, vasodilatating, anti-inflammatory with anti-oxidant properties | Zirconia/Methylcellulose NPs, intravenous delivery, 3 h after reperfusion (5 h post-ischemia) | Reduction of infarct size | [223] |
| MCAO (transient), mouse | NEP1-40, antagonist of the Nogo-66 receptor,. enhances neural plasticity and suppresses cell death after injury | PLGA NPs decorated with chlorotoxin to target MMP-2 and lexiscan to transiently increase the BBB permeability, intravenous injection, 3 doses (0, 24, and 48 h after MCAO) | Reduction of infarct volume and better neurological outcome | [222] |
| MCAO (transient), rat | Heme oxygenase-1, anti-oxidant enzyme | R3V6-peptide -based micelles stabilized with Dexamethasone, intracerebral injection (striatum), 1 h before ischemia | Reduction of infarct volume | [225] |
| MCAO (transient), rat | siRNA against HMGB1, (HMGB1 is a pro-inflammatory signal) | PAMAM-based dendrimers, intracerebral injection (cortex). 6–24 h before ischemia | Reduction of neuronal mortality and infarct volume | [226] |
| Photothrombotic stroke model (permanent), mouse | siRNA to silence PHID2, (PHD2 is a factor involved in the up-regulation of genes related to a cellular response to hypoxia) | Polyethylenimine superparamagnetic iron oxide NPs delivered from endothelial progenitor cells, intra-cardiac injection, 24 h after stroke | Significant reduction of infarct volume at 7 days after treatment. Stimulation of endogenous vascularization and neurogenesis. Significant Improvement of behavioral deficits (at 2 weeks after treatment) | [227] |
| MCAO (permanent), rat | Cxcl12, a chemoattractant and neuroprotective molecule | pH-responsive copolymer poly (urethane amino sulfamethazine) (PUASM) micelles, intracerebral delivery, 24 h after ischemia | Significant angiogenesis and neurogenesis, lack of a neuroprotective effect | [229] |
| MCAO (transient), rat | VEGF, an angiogenic and neuroprotectant molecule | Alginate-based hydrogels, intracerebral injection (striatum), 15 min before ischemia | Higher content of VEGF administrated in the striatum than delivery of the free molecule. Behavioral recovery and significant reduction of infarct volume | [168] |
| Focal aspiration brain injury model, rat | Neurotrophin-3, neuroprotective, neurogenic, and anti-inflammatory factor | Chitosan particles, intracerebral administration (cortex), immediately after injury | Increasing neurogenesis and synaptogenesis, anti-inflammatory effects, and behavioral improvement | [169] |
| Endothelin-1 model, rat | BDNF, a neuroprotector molecule and inductor of plasticity | PLGA NPs dispersed on the hyaluronan and methylcellulose hydrogels, epi-cortical administration (brain surface), immediately after stroke | Significant behavioral recovery and reduced cortical lesions (these positive effects were seen even with the biomaterial alone) | [170] |
| Endothelin-1 model, mouse | Dual delivery of EGF and EPO, neurogenic, anti-inflammatory, and neuroprotective factors | PLGA NPs dispersed on the hyaluronan and methylcellulose hydrogels, epi-cortical administration (brain surface), 4 days after stroke | Increasing neurogenesis and reduction of injury response, inflammation and cell death (attenuation of inflammation and injury were also observed, even with the biomaterial alone) | [154] |
| MCAO (transient), hyperglycemic rat | Carbon-HCCs, antioxidant potential | Carbon-PEG NPs, intravenous administration, two doses (immediately before reperfusion and 2 h after reperfusion) | Reduction in infarct size and behavioral improvement | [183] |
| MCAO (transient), rat | Selenium, modulator of neurogenesis with anti-oxidant properties | Selenium-PEG NPs decorated with Anti-transferrin receptor antibody, intraperitoneal injection, 1 h before stroke | Reduction in infarct size, high levels of myelination, neural, and axonal density. Behavioral improvement. No toxicity concerns at therapeutic doses | [184] |
| MCAO (permanent), mouse | Hyaluronic acid hydrogel (porous), promotes cell neural stem cell infiltration and reduces inflammation | MAP-HA-based hydrogels, intracerebral delivery (stroke cavity), 5 days post-stroke | Reduction of inflammation and astrogliosis, increasing neurogenesis and angiogenesis. Remarkable neural progenitor cell migration to lesional and peri-lesional areas | [161] |
| MCAO (transient), rat | Porcine urinary bladder ECM, promotes cell neural stem cell infiltration and reduces inflammation | Porcine urinary bladder ECM hydrogels, intracerebral administration (stroke cavity), 14 days post-stroke | Increasing microglia polarization towards anti-inflammatory phenotypes | [181] |
| MCAO (transient), macaque monkey | Hemoglobin, oxygen carrier supporting tissue oxygenation | Phosphatidylcholine-cholesterol-liposome decorated with PEG, intravenous injection, 5 min after ischemia | Reduction of infarct area, behavioral improvement | [235,236] |
| MCAO (transient), macaque monkey | BDNF, neuroprotective, neurogenic, and regenerative factor | HA-PEG based hydrogel, intracerebral administration (stroke cavity), 3 months post-stroke | Significant increase of BDNF content in peri-infarct tissue in relation to the delivery of free BDNF | [156] |
| Control (healthy), St. Kitts green monkey | No therapeutic compound | PEG-PLA-based hydrogel, intracerebral injection (cortex and striatum) | Moderate inflammation and astrogliosis. Full degradation of material four months after implantation | [155] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Nieto, D.; Fernández-Serra, R.; Pérez-Rigueiro, J.; Panetsos, F.; Martinez-Murillo, R.; Guinea, G.V. Biomaterials to Neuroprotect the Stroke Brain: A Large Opportunity for Narrow Time Windows. Cells 2020, 9, 1074. https://doi.org/10.3390/cells9051074
González-Nieto D, Fernández-Serra R, Pérez-Rigueiro J, Panetsos F, Martinez-Murillo R, Guinea GV. Biomaterials to Neuroprotect the Stroke Brain: A Large Opportunity for Narrow Time Windows. Cells. 2020; 9(5):1074. https://doi.org/10.3390/cells9051074
Chicago/Turabian StyleGonzález-Nieto, Daniel, Rocío Fernández-Serra, José Pérez-Rigueiro, Fivos Panetsos, Ricardo Martinez-Murillo, and Gustavo V. Guinea. 2020. "Biomaterials to Neuroprotect the Stroke Brain: A Large Opportunity for Narrow Time Windows" Cells 9, no. 5: 1074. https://doi.org/10.3390/cells9051074
APA StyleGonzález-Nieto, D., Fernández-Serra, R., Pérez-Rigueiro, J., Panetsos, F., Martinez-Murillo, R., & Guinea, G. V. (2020). Biomaterials to Neuroprotect the Stroke Brain: A Large Opportunity for Narrow Time Windows. Cells, 9(5), 1074. https://doi.org/10.3390/cells9051074