Congenital Tufting Enteropathy-Associated Mutant of Epithelial Cell Adhesion Molecule Activates the Unfolded Protein Response in a Murine Model of the Disease

 , and
, and
Abstract
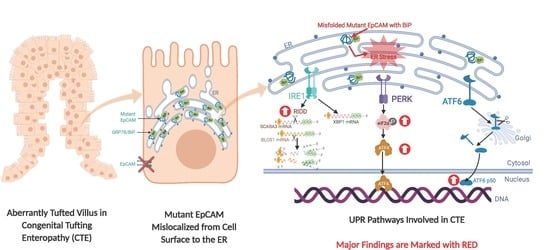
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Culture
2.3. Immunofluorescent (IF) Staining
2.4. Electron Microscopy
2.5. Western Blotting
2.6. RT-PCR and Real-Time qRT-PCR
2.7. Statistical Analysis
3. Results
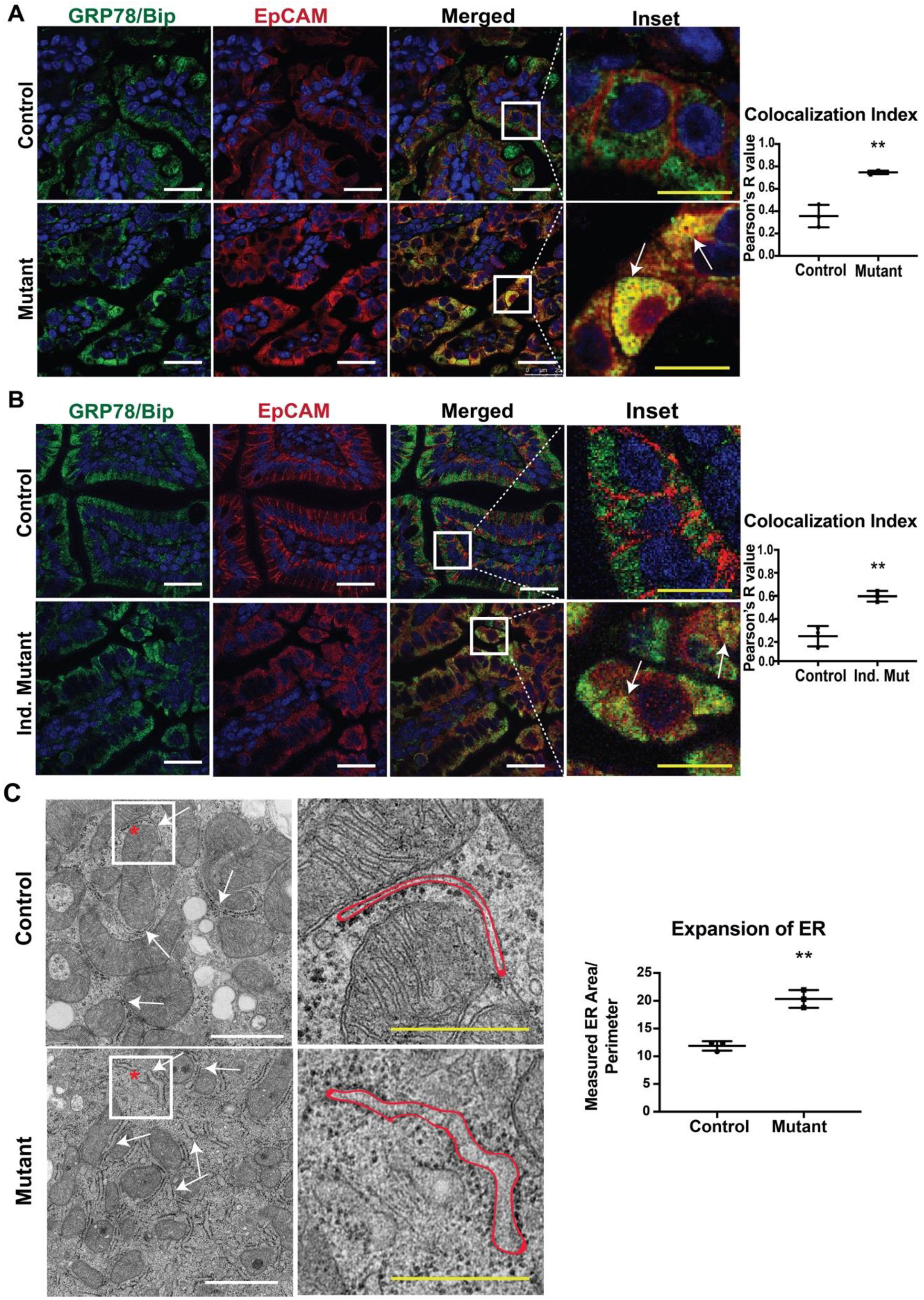
3.1. Mutant EpCAM Accumulates in the ER
3.2. Activation of IRE1-Mediated RIDD Pathway
3.3. PERK-Mediated Pathway Activation
3.4. Increased Nuclear Presence of ATF6
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goulet, O.; Salomon, J.; Ruemmele, F.; de Serres, N.P.; Brousse, N. Intestinal epithelial dysplasia (tufting enteropathy). Orphanet J. Rare Dis. 2007, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Reifen, R.M.; Cutz, E.; Griffiths, A.M.; Ngan, B.Y.; Sherman, P.M. Tufting enteropathy: A newly recognized clinicopathological entity associated with refractory diarrhea in infants. J. Pediatr. Gastroenterol. Nutr. 1994, 18, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Sivagnanam, M.; Mueller, J.L.; Lee, H.; Chen, Z.; Nelson, S.F.; Turner, D.; Zlotkin, S.H.; Pencharz, P.B.; Ngan, B.Y.; Libiger, O.; et al. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology 2008, 135, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Balzar, M.; Winter, M.J.; de Boer, C.J.; Litvinov, S.V. The biology of the 17-1A antigen (Ep-CAM). J. Mol. Med. (Berl.) 1999, 77, 699–712. [Google Scholar] [CrossRef]
- Trzpis, M.; McLaughlin, P.M.; de Leij, L.M.; Harmsen, M.C. Epithelial cell adhesion molecule: More than a carcinoma marker and adhesion molecule. Am. J. Pathol. 2007, 171, 386–395. [Google Scholar] [CrossRef]
- Pathak, S.J.; Mueller, J.L.; Okamoto, K.; Das, B.; Hertecant, J.; Greenhalgh, L.; Cole, T.; Pinsk, V.; Yerushalmi, B.; Gurkan, O.E.; et al. EPCAM mutation update: Variants associated with congenital tufting enteropathy and Lynch syndrome. Hum. Mutat. 2019, 40, 142–161. [Google Scholar] [CrossRef]
- Schnell, U.; Kuipers, J.; Mueller, J.L.; Veenstra-Algra, A.; Sivagnanam, M.; Giepmans, B.N. Absence of cell-surface EpCAM in congenital tufting enteropathy. Hum. Mol. Genet. 2013, 22, 2566–2571. [Google Scholar] [CrossRef]
- Pavsic, M.; Guncar, G.; Djinovic-Carugo, K.; Lenarcic, B. Crystal structure and its bearing towards an understanding of key biological functions of EpCAM. Nat. Commun. 2014, 5, 4764. [Google Scholar] [CrossRef]
- Mueller, J.L.; McGeough, M.D.; Pena, C.A.; Sivagnanam, M. Functional consequences of EpCam mutation in mice and men. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G278–G288. [Google Scholar] [CrossRef]
- Schnell, U.; Kuipers, J.; Giepmans, B.N. EpCAM proteolysis: New fragments with distinct functions? Biosci. Rep. 2013, 33, e00030. [Google Scholar] [CrossRef]
- Anelli, T.; Sitia, R. Protein quality control in the early secretory pathway. EMBO J. 2008, 27, 315–327. [Google Scholar] [CrossRef]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Matus, S.; Glimcher, L.H.; Hetz, C. Protein folding stress in neurodegenerative diseases: A glimpse into the ER. Curr. Opin. Cell Biol. 2011, 23, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 2012, 63, 317–328. [Google Scholar] [CrossRef]
- Blohmke, C.J.; Mayer, M.L.; Tang, A.C.; Hirschfeld, A.F.; Fjell, C.D.; Sze, M.A.; Falsafi, R.; Wang, S.; Hsu, K.; Chilvers, M.A.; et al. Atypical activation of the unfolded protein response in cystic fibrosis airway cells contributes to p38 MAPK-mediated innate immune responses. J. Immunol. 2012, 189, 5467–5475. [Google Scholar] [CrossRef]
- Kaser, A.; Blumberg, R.S. Endoplasmic reticulum stress in the intestinal epithelium and inflammatory bowel disease. Semin. Immunol. 2009, 21, 156–163. [Google Scholar] [CrossRef]
- Negroni, A.; Prete, E.; Vitali, R.; Cesi, V.; Aloi, M.; Civitelli, F.; Cucchiara, S.; Stronati, L. Endoplasmic reticulum stress and unfolded protein response are involved in paediatric inflammatory bowel disease. Dig. Liver Dis. 2014, 46, 788–794. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Sadighi Akha, A.A.; Raden, D.; Kaufman, R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006, 4, e374. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, J.; Prywes, R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 2002, 277, 13045–13052. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Kozan, P.A.; McGeough, M.D.; Peña, C.A.; Mueller, J.L.; Barrett, K.E.; Marchelletta, R.R.; Sivagnanam, M. Mutation of EpCAM leads to intestinal barrier and ion transport dysfunction. J. Mol. Med. 2015, 93, 535–545. [Google Scholar] [CrossRef]
- Dharmsathaphorn, K.; McRoberts, J.A.; Mandel, K.G.; Tisdale, L.D.; Masui, H. A human colonic tumor cell line that maintains vectorial electrolyte transport. Am. J. Physiol. 1984, 246 Pt 1, G204–G208. [Google Scholar] [CrossRef]
- Donato, R.P.; El-Merhibi, A.; Gundsambuu, B.; Mak, K.Y.; Formosa, E.R.; Wang, X.; Abbott, C.A.; Powell, B.C. Studying permeability in a commonly used epithelial cell line: T84 intestinal epithelial cells. Methods Mol. Biol. 2011, 763, 115–137. [Google Scholar]
- Hurley, B.P.; Pirzai, W.; Eaton, A.D.; Harper, M.; Roper, J.; Zimmermann, C.; Ladics, G.S.; Layton, R.J.; Delaney, B. An experimental platform using human intestinal epithelial cell lines to differentiate between hazardous and non-hazardous proteins. Food Chem. Toxicol. 2016, 92, 75–87. [Google Scholar] [CrossRef]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef]
- Foufelle, F.; Fromenty, B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol. Res. Perspect. 2016, 4, e00211. [Google Scholar] [CrossRef]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef] [PubMed]
- Lievremont, J.P.; Rizzuto, R.; Hendershot, L.; Meldolesi, J. BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+. J. Biol. Chem. 1997, 272, 30873–30879. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, T.N.; Sidrauski, C.; Dorfler, S.; Walter, P. Mechanism of non-spliceosomal mRNA splicing in the unfolded protein response pathway. EMBO J. 1999, 18, 3119–3132. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, H.; Nishitoh, H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes (Basel) 2013, 4, 306–333. [Google Scholar] [CrossRef]
- Szabo, R.; Callies, L.K.; Bugge, T.H. Matriptase drives early-onset intestinal failure in a mouse model of congenital tufting enteropathy. Development 2019, 146. [Google Scholar] [CrossRef]
- Wu, C.J.; Feng, X.; Lu, M.; Morimura, S.; Udey, M.C. Matriptase-mediated cleavage of EpCAM destabilizes claudins and dysregulates intestinal epithelial homeostasis. J. Clin. Investig. 2017, 127, 623–634. [Google Scholar] [CrossRef]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef]
- Das, B.; Okamoto, K.; Rabalais, J.; Kozan, P.A.; Marchelletta, R.R.; McGeough, M.D.; Durali, N.; Go, M.; Barrett, K.E.; Das, S.; et al. Enteroids expressing a disease-associated mutant of EpCAM are a model for congenital tufting enteropathy. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G580–G591. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Sen, U.; Vrati, S. Regulated IRE1-dependent decay pathway is activated during Japanese encephalitis virus-induced unfolded protein response and benefits viral replication. J. Gen. Virol. 2014, 95 Pt 1, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Coelho, D.S.; Domingos, P.M. Physiological roles of regulated Ire1 dependent decay. Front. Genet. 2014, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA. IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kashima, M.; Nagano, A.J.; Ishikawa-Fujiwara, T.; Kamei, Y.; Todo, T.; Mori, K. Unfolded protein response transducer IRE1-mediated signaling independent of XBP1 mRNA splicing is not required for growth and development of medaka fish. Elife 2017, 6, e26845. [Google Scholar] [CrossRef]
- Bertolotti, A.; Wang, X.; Novoa, I.; Jungreis, R.; Schlessinger, K.; Cho, J.H.; West, A.B.; Ron, D. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J. Clin. Investig. 2001, 107, 585–593. [Google Scholar] [CrossRef]
- Grey, M.J.; Cloots, E.; Simpson, M.S.; LeDuc, N.; Serebrenik, Y.V.; De Luca, H.; De Sutter, D.; Luong, P.; Thiagarajah, J.R.; Paton, A.W.; et al. IRE1beta negatively regulates IRE1alpha signaling in response to endoplasmic reticulum stress. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef]
- Tam, A.B.; Roberts, L.S.; Chandra, V.; Rivera, I.G.; Nomura, D.K.; Forbes, D.J.; Niwa, M. The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev. Cell 2018, 46, 327–343.e7. [Google Scholar] [CrossRef]
- Prell, T.; Lautenschlager, J.; Witte, O.W.; Carri, M.T.; Grosskreutz, J. The unfolded protein response in models of human mutant G93A amyotrophic lateral sclerosis. Eur. J. Neurosci. 2012, 35, 652–660. [Google Scholar] [CrossRef]
- Lobo, G.P.; Ebke, L.A.; Au, A.; Hagstrom, S.A. TULP1 Missense Mutations Induces the Endoplasmic Reticulum Unfolded Protein Response Stress Complex (ER-UPR). Adv. Exp. Med. Biol. 2016, 854, 223–230. [Google Scholar] [PubMed]
- Jia, Y.; Jucius, T.J.; Cook, S.A.; Ackerman, S.L. Loss of Clcc1 results in ER stress, misfolded protein accumulation, and neurodegeneration. J. Neurosci. 2015, 35, 3001–3009. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| BLOS1 | AGCGTTGGTGGATCACCTC | CACCATTCCAATCCACTGGC |
| SCARA3 (Mouse) | TGCATGGATACTGACCCTGA | GCCGTGTTACCAGCTTCTTC |
| SCARA3 (Human) | CCGCTGCCAGAAGAACCTAT | TGTCTTCGGAGAGAGAGTCCA |
| XBP1 | TTACGGGAGAAAACTCACGGC | GGGTCCAACTTGTCCAGAATGC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, B.; Okamoto, K.; Rabalais, J.; Marchelletta, R.R.; Barrett, K.E.; Das, S.; Niwa, M.; Sivagnanam, M. Congenital Tufting Enteropathy-Associated Mutant of Epithelial Cell Adhesion Molecule Activates the Unfolded Protein Response in a Murine Model of the Disease. Cells 2020, 9, 946. https://doi.org/10.3390/cells9040946
Das B, Okamoto K, Rabalais J, Marchelletta RR, Barrett KE, Das S, Niwa M, Sivagnanam M. Congenital Tufting Enteropathy-Associated Mutant of Epithelial Cell Adhesion Molecule Activates the Unfolded Protein Response in a Murine Model of the Disease. Cells. 2020; 9(4):946. https://doi.org/10.3390/cells9040946
Chicago/Turabian StyleDas, Barun, Kevin Okamoto, John Rabalais, Ronald R. Marchelletta, Kim E. Barrett, Soumita Das, Maho Niwa, and Mamata Sivagnanam. 2020. "Congenital Tufting Enteropathy-Associated Mutant of Epithelial Cell Adhesion Molecule Activates the Unfolded Protein Response in a Murine Model of the Disease" Cells 9, no. 4: 946. https://doi.org/10.3390/cells9040946
APA StyleDas, B., Okamoto, K., Rabalais, J., Marchelletta, R. R., Barrett, K. E., Das, S., Niwa, M., & Sivagnanam, M. (2020). Congenital Tufting Enteropathy-Associated Mutant of Epithelial Cell Adhesion Molecule Activates the Unfolded Protein Response in a Murine Model of the Disease. Cells, 9(4), 946. https://doi.org/10.3390/cells9040946