Ciliary Genes in Renal Cystic Diseases


Abstract
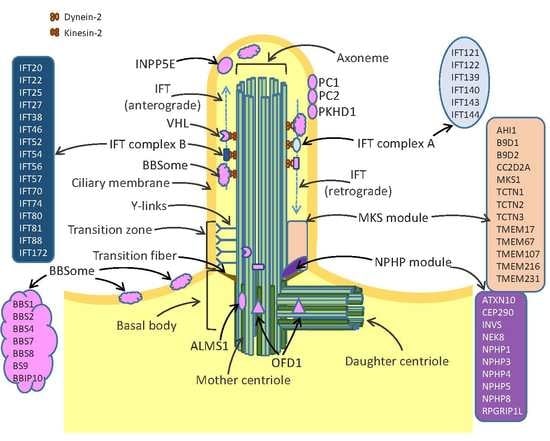
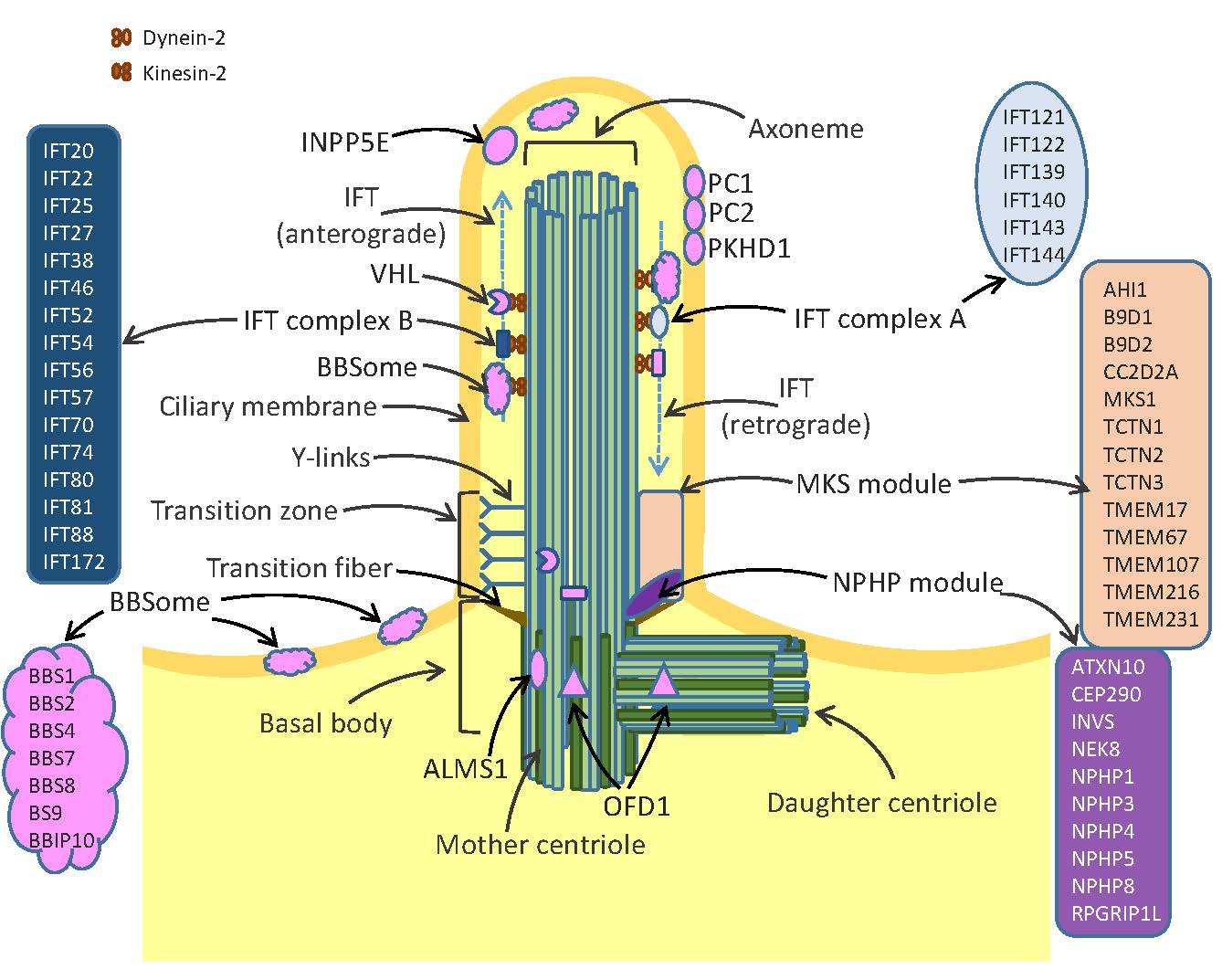
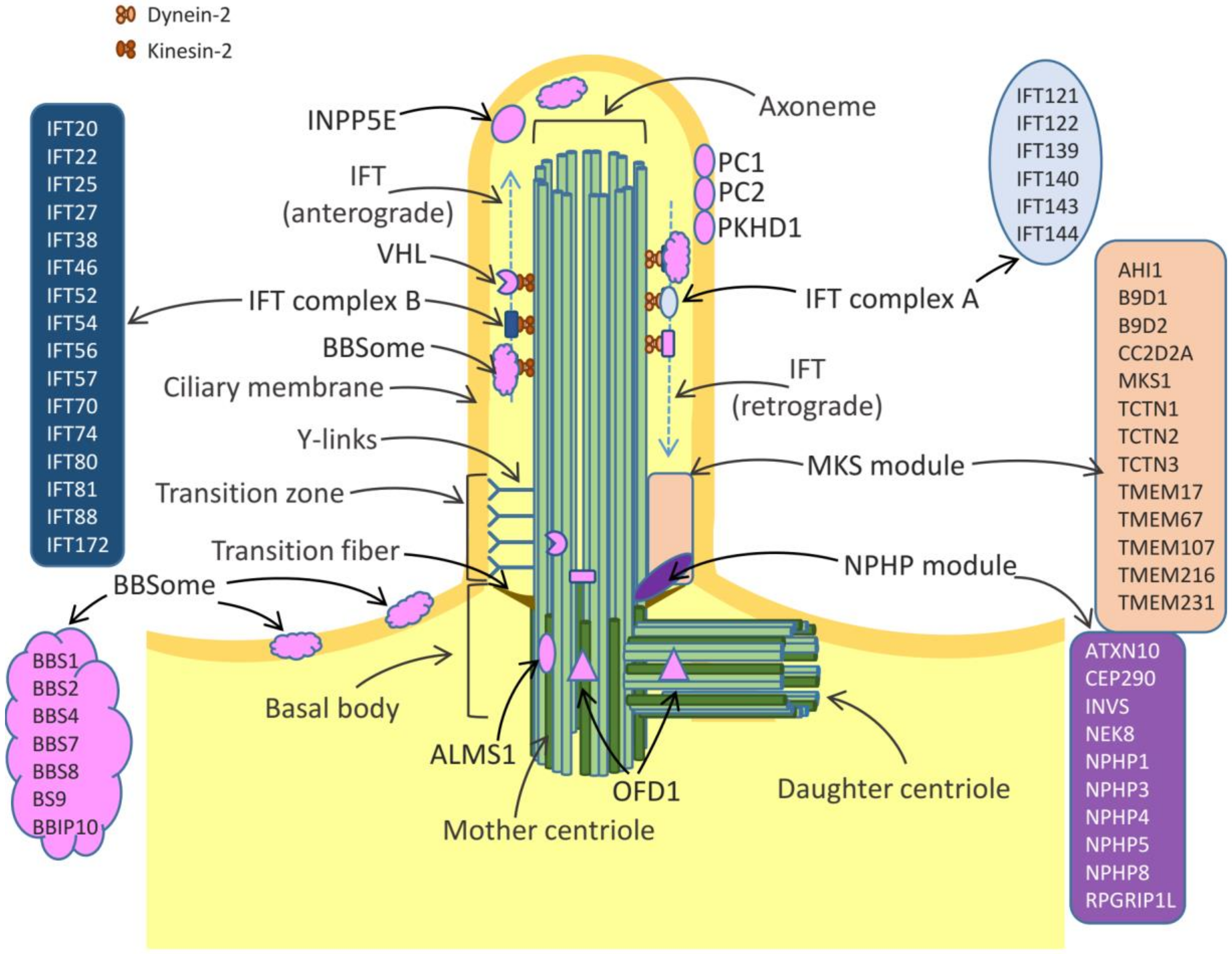
1. Structure and Function of Cilia
2. Role of Cilia in Renal Diseases
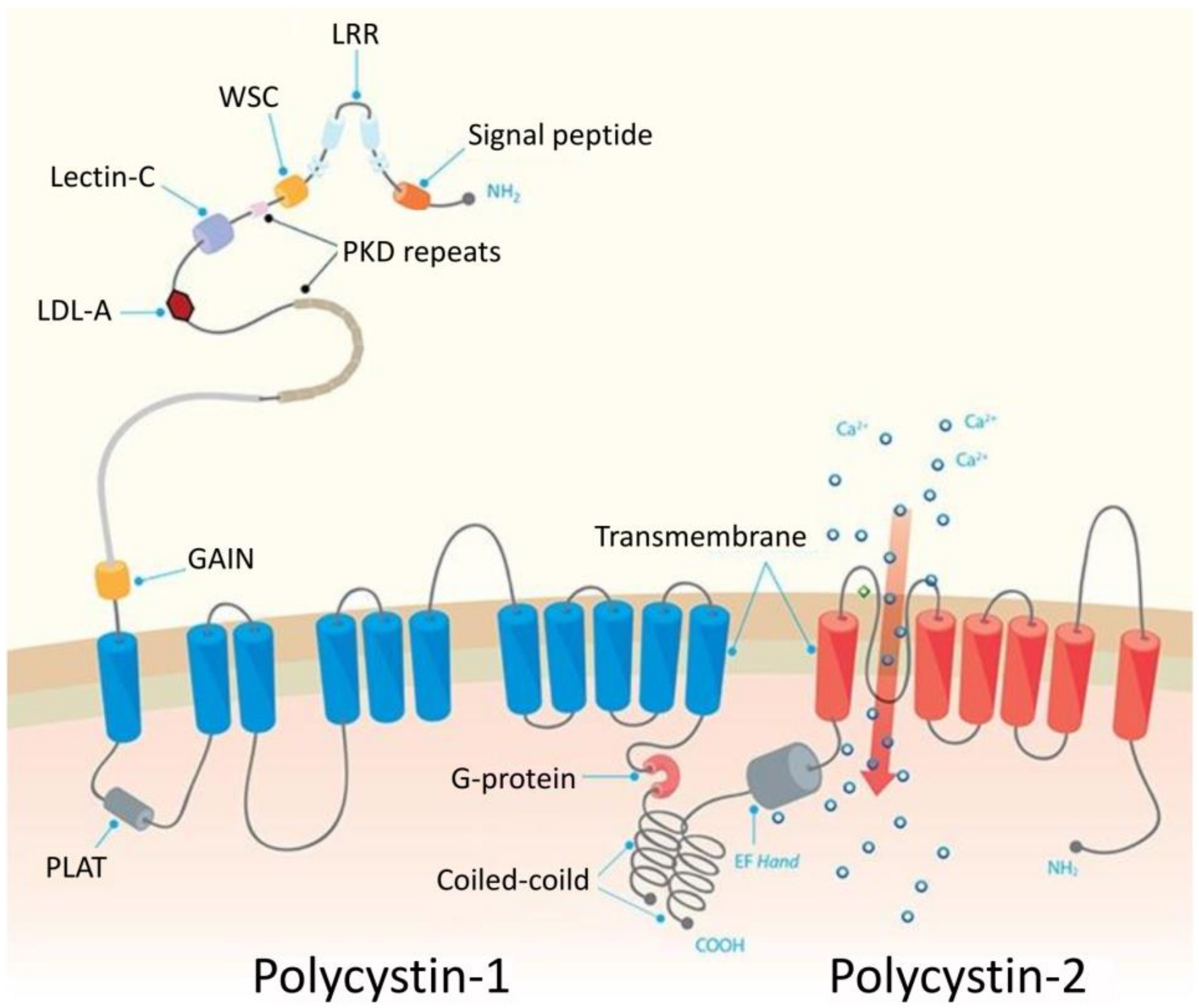
2.1. Polycystic Kidney Disease
2.2. Nephronophthisis
2.3. Joubert Syndrome
2.4. Meckel-Gruber Syndrome
2.5. Bardet-Biedl Syndrome
2.6. Senior-Loken Syndrome
2.7. Alström Syndrome
2.8. Orofaciodigital Syndrome
2.9. Sensenbrenner Syndrome
3. Relevance of Ciliary Genes in Renal Cancer
3.1. The Role of VHL in Ciliogenesis and Renal Cancer
3.2. Other Ciliary Genes Involved in Renal Cancer
4. Ciliary Genes as Therapeutic Targets
5. Conclusions
Funding
Conflicts of Interest
References
- Davis, E.E.; Brueckner, M.; Katsanis, N. The emerging complexity of the vertebrate cilium: New functional roles for an ancient organelle. Dev. Cell 2006, 11, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Haimo, L.T.; Rosenbaum, J.L. Cilia, Flagella, and Microtubules. J. Cell Biol. 1981, 91, S125–S130. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.; Dynlacht, B.D. Cilium assembly and disassembly. Nat. Cell Biol. 2016, 18, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.B.; Schroder, J.M.; Satir, P.; Christensen, S.T. The ciliary cytoskeleton. Compr. Physiol. 2012, 2, 779–803. [Google Scholar] [CrossRef]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Praetorius, H.A.; Spring, K.R. The renal cell primary cilium functions as a flow sensor. Curr. Opin. Nephrol. Hypertens. 2003, 12, 517–520. [Google Scholar] [CrossRef]
- Ernstrom, G.G.; Chalfie, M. Genetics of sensory mechanotransduction. Annu. Rev. Genet. 2002, 36, 411–453. [Google Scholar] [CrossRef]
- Kulaga, H.M.; Leitch, C.C.; Eichers, E.R.; Badano, J.L.; Lesemann, A.; Hoskins, B.E.; Lupski, J.R.; Beales, P.L.; Reed, R.R.; Katsanis, N. Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat. Genet. 2004, 36, 994–998. [Google Scholar] [CrossRef]
- Perantoni, A.O. Renal development: Perspectives on a Wnt-dependent process. Semin. Cell Dev. Biol. 2003, 14, 201–208. [Google Scholar] [CrossRef]
- Simons, M.; Gloy, J.; Ganner, A.; Bullerkotte, A.; Bashkurov, M.; Kronig, C.; Schermer, B.; Benzing, T.; Cabello, O.A.; Jenny, A.; et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005, 37, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Bisgrove, B.W.; Yost, H.J. The roles of cilia in developmental disorders and disease. Development 2006, 133, 4131–4143. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Christensen, S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.S.; Ben-Shahar, Y.; Moninger, T.O.; Kline, J.N.; Welsh, M.J. Motile cilia of human airway epithelia are chemosensory. Science 2009, 325, 1131–1134. [Google Scholar] [CrossRef]
- Babu, D.; Roy, S. Left-right asymmetry: Cilia stir up new surprises in the node. Open Biol. 2013, 3, 130052. [Google Scholar] [CrossRef]
- Bartoloni, L.; Blouin, J.L.; Pan, Y.Z.; Gehrig, C.; Maiti, A.K.; Scamuffa, N.; Rossier, C.; Jorissen, M.; Armengot, M.; Meeks, M.; et al. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA 2002, 99, 10282–10286. [Google Scholar] [CrossRef]
- Avasthi, P.; Maser, R.L.; Tran, P.V. Primary Cilia in Cystic Kidney Disease. Results Probl. Cell Differ. 2017, 60, 281–321. [Google Scholar] [CrossRef]
- Lambacher, N.J.; Bruel, A.L.; van Dam, T.J.; Szymanska, K.; Slaats, G.G.; Kuhns, S.; McManus, G.J.; Kennedy, J.E.; Gaff, K.; Wu, K.M.; et al. TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat. Cell Biol. 2016, 18, 122–131. [Google Scholar] [CrossRef]
- Pazour, G.J.; Dickert, B.L.; Vucica, Y.; Seeley, E.S.; Rosenbaum, J.L.; Witman, G.B.; Cole, D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J. Cell Biol. 2000, 151, 709–718. [Google Scholar] [CrossRef]
- Schrick, J.J.; Onuchic, L.F.; Reeders, S.T.; Korenberg, J.; Chen, X.N.; Moyer, J.H.; Wilkinson, J.E.; Woychik, R.P. Characterization of the Human Homolog of the Mouse Tg737 Candidate Polycystic Kidney-Disease Gene. Hum. Mol. Genet. 1995, 4, 559–567. [Google Scholar] [CrossRef]
- Plotnikova, O.V.; Golemis, E.A.; Pugacheva, E.N. Cell cycle-dependent ciliogenesis and cancer. Cancer Res. 2008, 68, 2058–2061. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.W. The primary cilium as a multiple cellular signaling scaffold in development and disease. BMB Rep. 2012, 45, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Berbari, N.F.; Yoder, B.K. Ciliary Dysfunction in Developmental Abnormalities and Diseases. Curr. Top. Dev. Biol. 2008, 85, 371–427. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, W.; Song, L.; Zhu, W. Cilia, adenomatous polyposis coli and associated diseases. Oncogene 2012, 31, 1475–1483. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thi-Kim Vu, H.; Rink, J.C.; McKinney, S.A.; McClain, M.; Lakshmanaperumal, N.; Alexander, R.; Sanchez Alvarado, A. Stem cells and fluid flow drive cyst formation in an invertebrate excretory organ. Elife 2015, 4, e07405. [Google Scholar] [CrossRef]
- Bergmann, C. Genetics of Autosomal Recessive Polycystic Kidney Disease and Its Differential Diagnoses. Front. Pediatr. 2017, 5, 221. [Google Scholar] [CrossRef]
- Malekshahabi, T.; Rad, N.K.; Serra, A.L.; Moghadasali, R. Autosomal dominant polycystic kidney disease: Disrupted pathways and potential therapeutic interventions. J. Cell. Physiol. 2019, 234, 12451–12470. [Google Scholar] [CrossRef]
- Saigusa, T.; Dang, Y.; Bunni, M.A.; Amria, M.Y.; Steele, S.L.; Fitzgibbon, W.R.; Bell, P.D. Activation of the intrarenal renin-angiotensin-system in murine polycystic kidney disease. Physiol. Rep. 2015, 3, e12405. [Google Scholar] [CrossRef]
- Wang, Z.; Ng, C.; Liu, X.; Wang, Y.; Li, B.; Kashyap, P.; Chaudhry, H.A.; Castro, A.; Kalontar, E.M.; Ilyayev, L.; et al. The ion channel function of polycystin-1 in the polycystin-1/polycystin-2 complex. EMBO Rep. 2019, 20, e48336. [Google Scholar] [CrossRef]
- Sun, K.; Xu, D.; Mei, C. The association between autosomal dominant polycystic kidney disease and cancer. Int. Urol. Nephrol. 2019, 51, 93–100. [Google Scholar] [CrossRef]
- Ghata, J.; Cowley, B.D., Jr. Polycystic Kidney Disease. Compr. Physiol. 2017, 7, 945–975. [Google Scholar] [CrossRef] [PubMed]
- Trudel, M.; Yao, Q.; Qian, F. The Role of G-Protein-Coupled Receptor Proteolysis Site Cleavage of Polycystin-1 in Renal Physiology and Polycystic Kidney Disease. Cells 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- International Polycystic Kidney Disease Consortium. Polycystic kidney disease: The complete structure of the PKD1 gene and its protein. Cell 1995, 81, 289–298. [Google Scholar] [CrossRef]
- Merrick, D.; Bertuccio, C.A.; Chapin, H.C.; Lal, M.; Chauvet, V.; Caplan, M.J. Polycystin-1 cleavage and the regulation of transcriptional pathways. Pediatr. Nephrol. 2014, 29, 505–511. [Google Scholar] [CrossRef]
- Ward, C.J.; Turley, H.; Ong, A.C.M.; Comley, M.; Biddolph, S.; Chetty, R.; Ratcliffe, P.J.; Gatter, K.; Harris, P.C. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc. Natl. Acad. Sci. USA 1996, 93, 1524–1528. [Google Scholar] [CrossRef]
- Rodova, M.; Islam, M.R.; Maser, R.L.; Calvet, J.P. The polycystic kidney disease-1 promoter is a target of the beta-catenin/T-cell factor pathway. J. Biol. Chem. 2002, 277, 29577–29583. [Google Scholar] [CrossRef]
- Bhunia, A.K.; Piontek, K.; Boletta, A.; Liu, L.J.; Qian, F.; Xu, P.N.; Germino, F.J.; Germino, G.G. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 2002, 109, 157–168. [Google Scholar] [CrossRef]
- Jeon, J.O.; Yoo, K.H.; Park, J.H. Expression of the Pkd1 gene is momentously regulated by Sp1. Nephron Exp. Nephrol. 2007, 107, 57–64. [Google Scholar] [CrossRef]
- Wang, Q.; Han, G.; Ye, J.; Gao, X.; Niu, H.; Zhao, J.; Chai, Y.; Li, N.; Yin, H. Characterization of the polycystic kidney disease 2 gene promoter. Genomics 2014, 104, 512–519. [Google Scholar] [CrossRef]
- Van Bodegom, D.; Saifudeen, Z.; Dipp, S.; Puri, S.; Magenheimer, B.S.; Calvet, J.P.; El-Dahr, S.S. The polycystic kidney disease-1 gene is a target for p53-mediated transcriptional repression. J. Biol. Chem. 2006, 281, 31234–31244. [Google Scholar] [CrossRef]
- Wang, Q.; Zheng, W.; Wang, Z.; Yang, J.; Hussein, S.; Tang, J.; Chen, X.Z. Filamin-a increases the stability and plasma membrane expression of polycystin-2. PLoS ONE 2015, 10, e0123018. [Google Scholar] [CrossRef]
- Kim, S.; Nie, H.; Nesin, V.; Tran, U.; Outeda, P.; Bai, C.X.; Keeling, J.; Maskey, D.; Watnick, T.; Wessely, O.; et al. The polycystin complex mediates Wnt/Ca(2+) signalling. Nat. Cell Biol. 2016, 18, 752–764. [Google Scholar] [CrossRef]
- Su, Q.; Hu, F.; Ge, X.; Lei, J.; Yu, S.; Wang, T.; Zhou, Q.; Mei, C.; Shi, Y. Structure of the human PKD1-PKD2 complex. Science 2018, 361, eaat9819. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.M.; Watanabe, E.H.; Onuchic, L.F. Polycystins and Molecular Basis of Autosomal Dominant Polycystic Kidney Disease. In Polycystic Kidney Disease; Li, X., Ed.; Codon Publications: Brisbane, Australia, 2015. [Google Scholar]
- Hiesberger, T.; Bai, Y.; Shao, X.; McNally, B.T.; Sinclair, A.M.; Tian, X.; Somlo, S.; Igarashi, P. Mutation of hepatocyte nuclear factor-1beta inhibits Pkhd1 gene expression and produces renal cysts in mice. J. Clin. Investig. 2004, 113, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Gresh, L.; Fischer, E.; Reimann, A.; Tanguy, M.; Garbay, S.; Shao, X.; Hiesberger, T.; Fiette, L.; Igarashi, P.; Yaniv, M.; et al. A transcriptional network in polycystic kidney disease. EMBO J. 2004, 23, 1657–1668. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef]
- Lu, H.; Galeano, M.C.R.; Ott, E.; Kaeslin, G.; Kausalya, P.J.; Kramer, C.; Ortiz-Bruchle, N.; Hilger, N.; Metzis, V.; Hiersche, M.; et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 2017, 49, 1025–1034. [Google Scholar] [CrossRef]
- Simms, R.J.; Eley, L.; Sayer, J.A. Nephronophthisis. Eur. J. Hum. Genet. 2009, 17, 406–416. [Google Scholar] [CrossRef]
- Ala-Mello, S.; Sankila, E.M.; Koskimies, O.; de la Chapelle, A.; Kaariainen, H. Molecular studies in Finnish patients with familial juvenile nephronophthisis exclude a founder effect and support a common mutation causing mechanism. J. Med. Genet. 1998, 35, 279–283. [Google Scholar] [CrossRef][Green Version]
- Stokman, M.; Lilien, M.; Knoers, N. Nephronophthisis. In Gene Reviews(R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Wolf, M.T. Nephronophthisis and related syndromes. Curr. Opin. Pediatr. 2015, 27, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Zhou, W.B. Nephronophthisis-associated ciliopathies. J. Am. Soc. Nephrol. 2007, 18, 1855–1871. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Tao, Y.H. Nephronophthisis: A review of genotype-phenotype correlation. Nephrology 2018, 23, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Szymanska, K.; Johnson, C.A. The transition zone: An essential functional compartment of cilia. Cilia 2012, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Delous, M.; Hellman, N.E.; Gaude, H.M.; Silbermann, F.; Le Bivic, A.; Salomon, R.; Antignac, C.; Saunier, S. Nephrocystin-1 and nephrocystin-4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum. Mol. Genet. 2009, 18, 4711–4723. [Google Scholar] [CrossRef]
- Seeger-Nukpezah, T.; Little, J.L.; Serzhanova, V.; Golemis, E.A. Cilia and cilia-associated proteins in cancer. Drug Discov. Today Dis. Mech. 2013, 10, e135–e142. [Google Scholar] [CrossRef]
- Phillips, C.L.; Miller, K.J.; Filson, A.J.; Nurnberger, J.; Clendenon, J.L.; Cook, G.W.; Dunn, K.W.; Overbeek, P.A.; Gattone, V.H., II; Bacallao, R.L. Renal cysts of inv/inv mice resemble early infantile nephronophthisis. J. Am. Soc. Nephrol. 2004, 15, 1744–1755. [Google Scholar] [CrossRef]
- Olbrich, H.; Fliegauf, M.; Hoefele, J.; Kispert, A.; Otto, E.; Volz, A.; Wolf, M.T.; Sasmaz, G.; Trauer, U.; Reinhardt, R.; et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet. 2003, 34, 455–459. [Google Scholar] [CrossRef]
- Bergmann, C.; Fliegauf, M.; Bruchle, N.O.; Frank, V.; Olbrich, H.; Kirschner, J.; Schermer, B.; Schmedding, I.; Kispert, A.; Kranzlin, B.; et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am. J. Hum. Genet. 2008, 82, 959–970. [Google Scholar] [CrossRef]
- Mollet, G.; Salomon, R.; Gribouval, O.; Silbermann, F.; Bacq, D.; Landthaler, G.; Milford, D.; Nayir, A.; Rizzoni, G.; Antignac, C.; et al. The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat. Genet. 2002, 32, 300–305. [Google Scholar] [CrossRef]
- Roepman, R.; Letteboer, S.J.; Arts, H.H.; van Beersum, S.E.; Lu, X.; Krieger, E.; Ferreira, P.A.; Cremers, F.P. Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 18520–18525. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.L.; Winkelbauer, M.E.; Schafer, J.C.; Michaud, E.J.; Yoder, B.K. Functional redundancy of the B9 proteins and nephrocystins in Caenorhabditis elegans ciliogenesis. Mol. Biol. Cell 2008, 19, 2154–2168. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.L.; Li, C.; Kida, K.; Inglis, P.N.; Mohan, S.; Semenec, L.; Bialas, N.J.; Stupay, R.M.; Chen, N.; Blacque, O.E.; et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J. Cell Biol. 2011, 192, 1023–1041. [Google Scholar] [CrossRef]
- Otto, E.A.; Loeys, B.; Khanna, H.; Hellemans, J.; Sudbrak, R.; Fan, S.L.; Muerb, U.; O’Toole, J.F.; Helou, J.; Attanasio, M.; et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 2005, 37, 282–288. [Google Scholar] [CrossRef]
- Tsang, W.Y.; Bossard, C.; Khanna, H.; Peranen, J.; Swaroop, A.; Malhotra, V.; Dynlacht, B.D. CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev. Cell 2008, 15, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Rachel, R.A.; May-Simera, H.L.; Veleri, S.; Gotoh, N.; Choi, B.Y.; Murga-Zamalloa, C.; McIntyre, J.C.; Marek, J.; Lopez, I.; Hackett, A.N.; et al. Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis. J. Clin. Investig. 2012, 122, 1233–1245. [Google Scholar] [CrossRef]
- Stowe, T.R.; Wilkinson, C.J.; Iqbal, A.; Stearns, T. The centriolar satellite proteins Cep72 and Cep290 interact and are required for recruitment of BBS proteins to the cilium. Mol. Biol. Cell 2012, 23, 3322–3335. [Google Scholar] [CrossRef]
- Schafer, T.; Putz, M.; Lienkamp, S.; Ganner, A.; Bergbreiter, A.; Ramachandran, H.; Gieloff, V.; Gerner, M.; Mattonet, C.; Czarnecki, P.G.; et al. Genetic and physical interaction between the NPHP5 and NPHP6 gene products. Hum. Mol. Genet. 2008, 17, 3655–3662. [Google Scholar] [CrossRef]
- Attanasio, M.; Uhlenhaut, N.H.; Sousa, V.H.; O’Toole, J.F.; Otto, E.; Anlag, K.; Klugmann, C.; Treier, A.C.; Helou, J.; Sayer, J.A.; et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet. 2007, 39, 1018–1024. [Google Scholar] [CrossRef]
- Delous, M.; Baala, L.; Salomon, R.; Laclef, C.; Vierkotten, J.; Tory, K.; Golzio, C.; Lacoste, T.; Besse, L.; Ozilou, C.; et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007, 39, 875–881. [Google Scholar] [CrossRef]
- Arts, H.H.; Doherty, D.; van Beersum, S.E.; Parisi, M.A.; Letteboer, S.J.; Gorden, N.T.; Peters, T.A.; Marker, T.; Voesenek, K.; Kartono, A.; et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 2007, 39, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Laclef, C.; Anselme, I.; Besse, L.; Catala, M.; Palmyre, A.; Baas, D.; Paschaki, M.; Pedraza, M.; Metin, C.; Durand, B.; et al. The role of primary cilia in corpus callosum formation is mediated by production of the Gli3 repressor. Hum. Mol. Genet. 2015, 24, 4997–5014. [Google Scholar] [CrossRef] [PubMed]
- Hoff, S.; Halbritter, J.; Epting, D.; Frank, V.; Nguyen, T.M.; van Reeuwijk, J.; Boehlke, C.; Schell, C.; Yasunaga, T.; Helmstadter, M.; et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat. Genet. 2013, 45, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Sohara, E.; Luo, Y.; Zhang, J.; Manning, D.K.; Beier, D.R.; Zhou, J. Nek8 regulates the expression and localization of polycystin-1 and polycystin-2. J. Am. Soc. Nephrol. 2008, 19, 469–476. [Google Scholar] [CrossRef]
- Grampa, V.; Delous, M.; Zaidan, M.; Odye, G.; Thomas, S.; Elkhartoufi, N.; Filhol, E.; Niel, O.; Silbermann, F.; Lebreton, C.; et al. Novel NEK8 Mutations Cause Severe Syndromic Renal Cystic Dysplasia through YAP Dysregulation. PLoS Genet. 2016, 12, e1005894. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.V.; Haycraft, C.J.; Besschetnova, T.Y.; Turbe-Doan, A.; Stottmann, R.W.; Herron, B.J.; Chesebro, A.L.; Qiu, H.; Scherz, P.J.; Shah, J.V.; et al. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 2008, 40, 403–410. [Google Scholar] [CrossRef]
- Graser, S.; Stierhof, Y.D.; Lavoie, S.B.; Gassner, O.S.; Lamla, S.; Le Clech, M.; Nigg, E.A. Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 2007, 179, 321–330. [Google Scholar] [CrossRef]
- Chaki, M.; Airik, R.; Ghosh, A.K.; Giles, R.H.; Chen, R.; Slaats, G.G.; Wang, H.; Hurd, T.W.; Zhou, W.; Cluckey, A.; et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012, 150, 533–548. [Google Scholar] [CrossRef]
- Humbert, M.C.; Weihbrecht, K.; Searby, C.C.; Li, Y.; Pope, R.M.; Sheffield, V.C.; Seo, S. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc. Natl. Acad. Sci. USA 2012, 109, 19691–19696. [Google Scholar] [CrossRef] [PubMed]
- Joo, K.; Kim, C.G.; Lee, M.S.; Moon, H.Y.; Lee, S.H.; Kim, M.J.; Kweon, H.S.; Park, W.Y.; Kim, C.H.; Gleeson, J.G.; et al. CCDC41 is required for ciliary vesicle docking to the mother centriole. Proc. Natl. Acad. Sci. USA 2013, 110, 5987–5992. [Google Scholar] [CrossRef] [PubMed]
- Schueler, M.; Braun, D.A.; Chandrasekar, G.; Gee, H.Y.; Klasson, T.D.; Halbritter, J.; Bieder, A.; Porath, J.D.; Airik, R.; Zhou, W.; et al. DCDC2 mutations cause a renal-hepatic ciliopathy by disrupting Wnt signaling. Am. J. Hum. Genet. 2015, 96, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.; Bizet, A.A.; Lachaux, A.; Gonzales, E.; Filhol, E.; Collardeau-Frachon, S.; Jeanpierre, C.; Henry, C.; Fabre, M.; Viremouneix, L.; et al. DCDC2 Mutations Cause Neonatal Sclerosing Cholangitis. Hum. Mutat. 2016, 37, 1025–1029. [Google Scholar] [CrossRef]
- Parisi, M.A. The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity. Transl. Sci. Rare Dis. 2019, 4, 25–49. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.C.; Tong, Z.J.; Westfall, J.E.; Ault, J.G.; Page-McCaw, P.S.; Ferland, R.J. Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum. Mol. Genet. 2009, 18, 3926–3941. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, M.; Cox, J.J.; Gayral, S.; Hampshire, D.J.; Ayub, M.; Blockmans, M.; Pernot, E.; Kisseleva, M.V.; Compere, P.; Schiffmann, S.N.; et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 2009, 41, 1027–1031. [Google Scholar] [CrossRef]
- Cantagrel, V.; Silhavy, J.L.; Bielas, S.L.; Swistun, D.; Marsh, S.E.; Bertrand, J.Y.; Audollent, S.; Attie-Bitach, T.; Holden, K.R.; Dobyns, W.B.; et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet. 2008, 83, 170–179. [Google Scholar] [CrossRef]
- Duldulao, N.A.; Lee, S.; Sun, Z. Cilia localization is essential for in vivo functions of the Joubert syndrome protein Arl13b/Scorpion. Development 2009, 136, 4033–4042. [Google Scholar] [CrossRef]
- Tuz, K.; Bachmann-Gagescu, R.; O’Day, D.R.; Hua, K.; Isabella, C.R.; Phelps, I.G.; Stolarski, A.E.; O’Roak, B.J.; Dempsey, J.C.; Lourenco, C.; et al. Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet. 2014, 94, 62–72. [Google Scholar] [CrossRef]
- Alkanderi, S.; Molinari, E.; Shaheen, R.; Elmaghloob, Y.; Stephen, L.A.; Sammut, V.; Ramsbottom, S.A.; Srivastava, S.; Cairns, G.; Edwards, N.; et al. ARL3 Mutations Cause Joubert Syndrome by Disrupting Ciliary Protein Composition. Am. J. Hum. Genet. 2018, 103, 612–620. [Google Scholar] [CrossRef]
- Schrick, J.J.; Vogel, P.; Abuin, A.; Hampton, B.; Rice, D.S. ADP-ribosylation factor-like 3 is involved in kidney and photoreceptor development. Am. J. Pathol. 2006, 168, 1288–1298. [Google Scholar] [CrossRef]
- Lee, J.E.; Silhavy, J.L.; Zaki, M.S.; Schroth, J.; Bielas, S.L.; Marsh, S.E.; Olvera, J.; Brancati, F.; Iannicelli, M.; Ikegami, K.; et al. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat. Genet. 2012, 44, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Hartill, V.; Szymanska, K.; Sharif, S.M.; Wheway, G.; Johnson, C.A. Meckel-Gruber Syndrome: An Update on Diagnosis, Clinical Management, and Research Advances. Front. Pediatr. 2017, 5, 244. [Google Scholar] [CrossRef] [PubMed]
- Weatherbee, S.D.; Niswander, L.A.; Anderson, K.V. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum. Mol. Genet. 2009, 18, 4565–4575. [Google Scholar] [CrossRef] [PubMed]
- Dawe, H.R.; Smith, U.M.; Cullinane, A.R.; Gerrelli, D.; Cox, P.; Badano, J.L.; Blair-Reid, S.; Sriram, N.; Katsanis, N.; Attie-Bitach, T.; et al. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum. Mol. Genet. 2007, 16, 173–186. [Google Scholar] [CrossRef]
- Tammachote, R.; Hommerding, C.J.; Sinders, R.M.; Miller, C.A.; Czarnecki, P.G.; Leightner, A.C.; Salisbury, J.L.; Ward, C.J.; Torres, V.E.; Gattone, V.H.; et al. Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3. Hum. Mol. Genet. 2009, 18, 3311–3323. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Simms, R.J.; Abdelhamed, Z.; Dawe, H.R.; Szymanska, K.; Logan, C.V.; Wheway, G.; Pitt, E.; Gull, K.; Knowles, M.A.; et al. A meckelin-filamin a interaction mediates ciliogenesis. Hum. Mol. Genet. 2012, 21, 1272–1286. [Google Scholar] [CrossRef]
- Garcia-Gonzalo, F.R.; Corbit, K.C.; Sirerol-Piquer, M.S.; Ramaswami, G.; Otto, E.A.; Noriega, T.R.; Seol, A.D.; Robinson, J.F.; Bennett, C.L.; Josifova, D.J.; et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011, 43, 776–784. [Google Scholar] [CrossRef]
- Valente, E.M.; Logan, C.V.; Mougou-Zerelli, S.; Lee, J.H.; Silhavy, J.L.; Brancati, F.; Iannicelli, M.; Travaglini, L.; Romani, S.; Illi, B.; et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010, 42, 619–625. [Google Scholar] [CrossRef]
- Lee, J.H.; Silhavy, J.L.; Lee, J.E.; Al-Gazali, L.; Thomas, S.; Davis, E.E.; Bielas, S.L.; Hill, K.J.; Iannicelli, M.; Brancati, F.; et al. Evolutionarily assembled cis-regulatory module at a human ciliopathy locus. Science 2012, 335, 966–969. [Google Scholar] [CrossRef]
- Christopher, K.J.; Wang, B.; Kong, Y.; Weatherbee, S.D. Forward genetics uncovers Transmembrane protein 107 as a novel factor required for ciliogenesis and Sonic hedgehog signaling. Dev. Biol. 2012, 368, 382–392. [Google Scholar] [CrossRef]
- Chih, B.; Liu, P.; Chinn, Y.; Chalouni, C.; Komuves, L.G.; Hass, P.E.; Sandoval, W.; Peterson, A.S. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat. Cell Biol. 2011, 14, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Bardet-Biedl Syndrome. Atlas Inherit. Retin. Dis. 2018, 1085, 171–174. [Google Scholar] [CrossRef]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peranen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C.; et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.J.; May-Simera, H.; Eichers, E.R.; Kai, M.; Hill, J.; Jagger, D.J.; Leitch, C.C.; Chapple, J.P.; Munro, P.M.; Fisher, S.; et al. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates (vol 37, pg 1135, 2005). Nat. Genet. 2005, 37, 1381. [Google Scholar] [CrossRef]
- Scott, C.A.; Marsden, A.N.; Rebagliati, M.R.; Zhang, Q.; Chamling, X.; Searby, C.C.; Baye, L.M.; Sheffield, V.C.; Slusarski, D.C. Nuclear/cytoplasmic transport defects in BBS6 underlie congenital heart disease through perturbation of a chromatin remodeling protein. PLoS Genet. 2017, 13, e1006936. [Google Scholar] [CrossRef] [PubMed]
- Marion, V.; Stoetzel, C.; Schlicht, D.; Messaddeq, N.; Koch, M.; Flori, E.; Danse, J.M.; Mandel, J.L.; Dollfus, H. Transient ciliogenesis involving Bardet-Biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 1820–1825. [Google Scholar] [CrossRef]
- Jin, H.; White, S.R.; Shida, T.; Schulz, S.; Aguiar, M.; Gygi, S.P.; Bazan, J.F.; Nachury, M.V. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 2010, 141, 1208–1219. [Google Scholar] [CrossRef]
- Cardenas-Rodriguez, M.; Osborn, D.P.; Irigoin, F.; Grana, M.; Romero, H.; Beales, P.L.; Badano, J.L. Characterization of CCDC28B reveals its role in ciliogenesis and provides insight to understand its modifier effect on Bardet-Biedl syndrome. Hum. Genet. 2013, 132, 91–105. [Google Scholar] [CrossRef]
- Seo, S.; Zhang, Q.; Bugge, K.; Breslow, D.K.; Searby, C.C.; Nachury, M.V.; Sheffield, V.C. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PLoS Genet. 2011, 7, e1002358. [Google Scholar] [CrossRef]
- Loktev, A.V.; Zhang, Q.; Beck, J.S.; Searby, C.C.; Scheetz, T.E.; Bazan, J.F.; Slusarski, D.C.; Sheffield, V.C.; Jackson, P.K.; Nachury, M.V. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev. Cell 2008, 15, 854–865. [Google Scholar] [CrossRef]
- Scheidecker, S.; Etard, C.; Pierce, N.W.; Geoffroy, V.; Schaefer, E.; Muller, J.; Chennen, K.; Flori, E.; Pelletier, V.; Poch, O.; et al. Exome sequencing of Bardet-Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18). J. Med. Genet. 2014, 51, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Wang, Z.; Diener, D.; Rosenbaum, J. Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control. Curr. Biol. 2007, 17, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Bhogaraju, S.; Cajanek, L.; Fort, C.; Blisnick, T.; Weber, K.; Taschner, M.; Mizuno, N.; Lamla, S.; Bastin, P.; Nigg, E.A.; et al. Molecular Basis of Tubulin Transport Within the Cilium by IFT74 and IFT81. Science 2013, 341, 1009–1012. [Google Scholar] [CrossRef] [PubMed]
- Lindstrand, A.; Frangakis, S.; Carvalho, C.M.; Richardson, E.B.; McFadden, K.A.; Willer, J.R.; Pehlivan, D.; Liu, P.; Pediaditakis, I.L.; Sabo, A.; et al. Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. Am. J. Hum. Genet. 2016, 99, 318–336. [Google Scholar] [CrossRef] [PubMed]
- Otto, E.A.; Hurd, T.W.; Airik, R.; Chaki, M.; Zhou, W.; Stoetzel, C.; Patil, S.B.; Levy, S.; Ghosh, A.K.; Murga-Zamalloa, C.A.; et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 2010, 42, 840–850. [Google Scholar] [CrossRef]
- Bizet, A.A.; Becker-Heck, A.; Ryan, R.; Weber, K.; Filhol, E.; Krug, P.; Halbritter, J.; Delous, M.; Lasbennes, M.C.; Linghu, B.; et al. Mutations in TRAF3IP1/IFT54 reveal a new role for IFT proteins in microtubule stabilization. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef]
- Berbari, N.F.; Kin, N.W.; Sharma, N.; Michaud, E.J.; Kesterson, R.A.; Yoder, B.K. Mutations in Traf3ip1 reveal defects in ciliogenesis, embryonic development, and altered cell size regulation. Dev. Biol. 2011, 360, 66–76. [Google Scholar] [CrossRef]
- Collin, G.B.; Marshall, J.D.; Ikeda, A.; So, W.V.; Russell-Eggitt, I.; Maffei, P.; Beck, S.; Boerkoel, C.F.; Sicolo, N.; Martin, M.; et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat. Genet. 2002, 31, 74–78. [Google Scholar] [CrossRef]
- Hearn, T. ALMS1 and Alstrom syndrome: A recessive form of metabolic, neurosensory and cardiac deficits. J. Mol. Med. 2019, 97, 1–17. [Google Scholar] [CrossRef]
- Marshall, J.D.; Maffei, P.; Collin, G.B.; Naggert, J.K. Alstrom syndrome: Genetics and clinical overview. Curr. Genom. 2011, 12, 225–235. [Google Scholar] [CrossRef]
- Li, G.C.; Vega, R.; Nelms, K.; Gekakis, N.; Goodnow, C.; McNamara, P.; Wu, H.; Hong, N.A.; Glynne, R. A role for Alstrom syndrome protein, Alms1, in kidney ciliogenesis and cellular quiescence. PLoS Genet. 2007, 3, e8. [Google Scholar] [CrossRef] [PubMed]
- Collin, G.B.; Cyr, E.; Bronson, R.; Marshall, J.D.; Gifford, E.J.; Hicks, W.; Murray, S.A.; Zheng, Q.Y.; Smith, R.S.; Nishina, P.M.; et al. Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum. Mol. Genet. 2005, 14, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, M.I.; Giorgio, G.; Feather, S.A.; Bulfone, A.; Wright, V.; Ghiani, M.; Selicorni, A.; Gammaro, L.; Scolari, F.; Woolf, A.S.; et al. Identification of the gene for oral-facial-digital type I syndrome. Am. J. Hum. Genet. 2001, 68, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Bruel, A.L.; Franco, B.; Duffourd, Y.; Thevenon, J.; Jego, L.; Lopez, E.; Deleuze, J.F.; Doummar, D.; Giles, R.H.; Johnson, C.A.; et al. Fifteen years of research on oral-facial-digital syndromes: From 1 to 16 causal genes. J. Med. Genet. 2017, 54, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt-Dias, M.; Hildebrandt, F.; Pellman, D.; Woods, G.; Godinho, S.A. Centrosomes and cilia in human disease. Trends Genet. 2011, 27, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Zullo, A.; Iaconis, D.; Barra, A.; Cantone, A.; Messaddeq, N.; Capasso, G.; Dolle, P.; Igarashi, P.; Franco, B. Kidney-specific inactivation of Ofd1 leads to renal cystic disease associated with upregulation of the mTOR pathway. Hum. Mol. Genet. 2010, 19, 2792–2803. [Google Scholar] [CrossRef]
- Walczak-Sztulpa, J.; Eggenschwiler, J.; Osborn, D.; Brown, D.A.; Emma, F.; Klingenberg, C.; Hennekam, R.C.; Torre, G.; Garshasbi, M.; Tzschach, A.; et al. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet. 2010, 86, 949–956. [Google Scholar] [CrossRef]
- Takahara, M.; Katoh, Y.; Nakamura, K.; Hirano, T.; Sugawa, M.; Tsurumi, Y.; Nakayama, K. Ciliopathy-associated mutations of IFT122 impair ciliary protein trafficking but not ciliogenesis. Hum. Mol. Genet. 2018, 27, 516–528. [Google Scholar] [CrossRef]
- Lang, G.D.; Young, I.D. Cranioectodermal Dysplasia in Sibs. J. Med. Genet. 1991, 28, 424. [Google Scholar] [CrossRef][Green Version]
- Eke, T.; Woodruff, G.; Young, I.D. A new oculorenal syndrome: Retinal dystrophy and tubulointerstitial nephropathy in cranioectodermal dysplasia. Br. J. Ophthalmol. 1996, 80, 490–491. [Google Scholar] [CrossRef]
- Caparros-Martin, J.A.; De Luca, A.; Cartault, F.; Aglan, M.; Temtamy, S.; Otaify, G.A.; Mehrez, M.; Valencia, M.; Vazquez, L.; Alessandri, J.L.; et al. Specific variants in WDR35 cause a distinctive form of Ellis-van Creveld syndrome by disrupting the recruitment of the EvC complex and SMO into the cilium. Hum. Mol. Genet. 2015, 24, 4126–4137. [Google Scholar] [CrossRef] [PubMed]
- Mill, P.; Lockhart, P.J.; Fitzpatrick, E.; Mountford, H.S.; Hall, E.A.; Reijns, M.A.; Keighren, M.; Bahlo, M.; Bromhead, C.J.; Budd, P.; et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am. J. Hum. Genet. 2011, 88, 508–515. [Google Scholar] [CrossRef]
- Bredrup, C.; Saunier, S.; Oud, M.M.; Fiskerstrand, T.; Hoischen, A.; Brackman, D.; Leh, S.M.; Midtbo, M.; Filhol, E.; Bole-Feysot, C.; et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet. 2011, 89, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Watnick, T.; Germino, G. From cilia to cyst. Nat. Genet. 2003, 34, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Otto, E. Cilia and centrosomes: A unifying pathogenic concept for cystic kidney disease? Nat. Rev. Genet. 2005, 6, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Tian, X.; Igarashi, P.; Pazour, G.J.; Somlo, S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 2013, 45, 1004–1012. [Google Scholar] [CrossRef]
- Boletta, A. Emerging evidence of a link between the polycystins and the mTOR pathways. Pathogenetics 2009, 2, 6. [Google Scholar] [CrossRef]
- Wallace, D.P. Cyclic AMP-mediated cyst expansion. Biochim. Biophys. Acta 2011, 1812, 1291–1300. [Google Scholar] [CrossRef]
- Ma, M.; Gallagher, A.R.; Somlo, S. Ciliary Mechanisms of Cyst Formation in Polycystic Kidney Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a028209. [Google Scholar] [CrossRef]
- Boguslawska, J.; Kryst, P.; Poletajew, S.; Piekielko-Witkowska, A. TGF-beta and microRNA Interplay in Genitourinary Cancers. Cells 2019, 8, E1619. [Google Scholar] [CrossRef]
- Basten, S.G.; Willekers, S.; Vermaat, J.S.; Slaats, G.G.; Voest, E.E.; van Diest, P.J.; Giles, R.H. Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia 2013, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Schraml, P.; Frew, I.J.; Thoma, C.R.; Boysen, G.; Struckmann, K.; Krek, W.; Moch, H. Sporadic clear cell renal cell carcinoma but not the papillary type is characterized by severely reduced frequency of primary cilia. Mod. Pathol. 2009, 22, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Boveri, T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J. Cell Sci. 2008, 121, 1–84. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.; Margall-Ducos, G.; Guidotti, J.E.; Bregerie, O.; Celati, C.; Brechot, C.; Desdouets, C. The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. J. Cell Sci. 2007, 120, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Harlander, S.; Schonenberger, D.; Toussaint, N.C.; Prummer, M.; Catalano, A.; Brandt, L.; Moch, H.; Wild, P.J.; Frew, I.J. Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice. Nat. Med. 2017, 23, 869. [Google Scholar] [CrossRef]
- Mans, D.A.; Lolkema, M.P.; van Beest, M.; Daenen, L.G.; Voest, E.E.; Giles, R.H. Mobility of the von Hippel-Lindau tumour suppressor protein is regulated by kinesin-2. Exp. Cell Res. 2008, 314, 1229–1236. [Google Scholar] [CrossRef]
- Fabbri, L.; Bost, F.; Mazure, N.M. Primary Cilium in Cancer Hallmarks. Int. J. Mol. Sci. 2019, 20, 1336. [Google Scholar] [CrossRef]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef]
- Montani, M.; Heinimann, K.; von Teichman, A.; Rudolph, T.; Perren, A.; Moch, H. VHL-gene deletion in single renal tubular epithelial cells and renal tubular cysts: Further evidence for a cyst-dependent progression pathway of clear cell renal carcinoma in von Hippel-Lindau disease. Am. J. Surg. Pathol. 2010, 34, 806–815. [Google Scholar] [CrossRef]
- Mitchell, T.J.; Turajlic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.R.; O’Brien, T.; Martincorena, I.; Tarpey, P.; Angelopoulos, N.; Yates, L.R.; et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018, 173, 611–623. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Q. VHL and Hypoxia Signaling: Beyond HIF in Cancer. Biomedicines 2018, 6, E35. [Google Scholar] [CrossRef] [PubMed]
- Schermer, B.; Ghenoiu, C.; Bartram, M.; Muller, R.U.; Kotsis, F.; Hohne, M.; Kuhn, W.; Rapka, M.; Nitschke, R.; Zentgraf, H.; et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J. Cell Biol. 2006, 175, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Lolkema, M.P.; Mans, D.A.; Snijckers, C.M.; van Noort, M.; van Beest, M.; Voest, E.E.; Giles, R.H. The von Hippel-Lindau tumour suppressor interacts with microtubules through kinesin-2. FEBS Lett. 2007, 581, 4571–4576. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, E.W.; Walz, G.; Benzing, T. Von hippel-lindau: A tumor suppressor links microtubules to ciliogenesis and cancer development. Cancer Res. 2007, 67, 4537–4540. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, H.; Vicari, D.; Wild, P.J.; Frew, I.J. Combined Deletion of Vhl and Kif3a Accelerates Renal Cyst Formation. J. Am. Soc. Nephrol. 2015, 26, 2778–2788. [Google Scholar] [CrossRef] [PubMed]
- Noonan, H.R.; Metelo, A.M.; Kamei, C.N.; Peterson, R.T.; Drummond, I.A.; Iliopoulos, O. Loss of vhl in the zebrafish pronephros recapitulates early stages of human clear cell renal cell carcinoma. Dis. Model. Mech. 2016, 9, 873–884. [Google Scholar] [CrossRef]
- Thoma, C.R.; Frew, I.J.; Hoerner, C.R.; Montani, M.; Moch, H.; Krek, W. pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat. Cell Biol. 2007, 9, 588–595. [Google Scholar] [CrossRef]
- O’Toole, S.M.; Watson, D.S.; Novoselova, T.V.; Romano, L.E.L.; King, P.J.; Bradshaw, T.Y.; Thompson, C.L.; Knight, M.M.; Sharp, T.V.; Barnes, M.R.; et al. Oncometabolite induced primary cilia loss in pheochromocytoma. Endocr. Relat. Cancer 2019, 26, 165–180. [Google Scholar] [CrossRef]
- Dere, R.; Perkins, A.L.; Bawa-Khalfe, T.; Jonasch, D.; Walker, C.L. beta-Catenin Links von Hippel-Lindau to AuroraKinase A and Loss of Primary Cilia in Renal Cell Carcinoma. J. Am. Soc. Nephrol. 2015, 26, 553–564. [Google Scholar] [CrossRef]
- Natoli, T.A.; Gareski, T.C.; Dackowski, W.R.; Smith, L.; Bukanov, N.O.; Russo, R.J.; Husson, H.; Matthews, D.; Piepenhagen, P.; Ibraghimov-Beskrovnaya, O. Pkd1 and Nek8 mutations affect cell-cell adhesion and cilia in cysts formed in kidney organ cultures. Am. J. Physiol. Renal. Physiol. 2008, 294, F73–F83. [Google Scholar] [CrossRef]
- Bowers, A.J.; Boylan, J.F. Nek8, a NIMA family kinase member, is overexpressed in primary human breast tumors. Gene 2004, 328, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Trapp, M.L.; Galtseva, A.; Manning, D.K.; Beier, D.R.; Rosenblum, N.D.; Quarmby, L.M. Defects in ciliary localization of Nek8 is associated with cystogenesis. Pediatr. Nephrol. 2008, 23, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Mahjoub, M.R.; Trapp, M.L.; Quarmby, L.M. NIMA-related kinases defective in murine models of polycystic kidney diseases localize to primary cilia and centrosomes. J. Am. Soc. Nephrol. 2005, 16, 3485–3489. [Google Scholar] [CrossRef] [PubMed]
- Zalli, D.; Bayliss, R.; Fry, A.M. The Nek8 protein kinase, mutated in the human cystic kidney disease nephronophthisis, is both activated and degraded during ciliogenesis. Hum. Mol. Genet. 2012, 21, 1155–1171. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.F.; Zhou, J.; Hu, Q.Y.; Liu, S.C.; Chen, G. The tumor suppressor pVHL down-regulates never-in-mitosis A-related kinase 8 via hypoxia-inducible factors to maintain cilia in human renal cancer cells. J. Biol. Chem. 2015, 290, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Habbig, S.; Bartram, M.P.; Muller, R.U.; Schwarz, R.; Andriopoulos, N.; Chen, S.H.; Sogmuller, J.G.; Hoehne, M.; Burst, V.; Liebau, M.C.; et al. NPHP4, a cilia-associated protein, negatively regulates the Hippo pathway. J. Cell Biol. 2011, 193, 633–642. [Google Scholar] [CrossRef]
- Slater, A.A.; Alokail, M.; Gentle, D.; Yao, M.; Kovacs, G.; Maher, E.R.; Latif, F. DNA methylation profiling distinguishes histological subtypes of renal cell carcinoma. Epigenetics 2013, 8, 252–267. [Google Scholar] [CrossRef]
- Bender, C.; Ullrich, A. PRKX, TTBK2 and RSK4 expression causes Sunitinib resistance in kidney carcinoma- and melanoma-cell lines. Int. J. Cancer 2012, 131, E45–E55. [Google Scholar] [CrossRef]
- Goetz, S.C.; Liem, K.F.; Anderson, K.V. The Spinocerebellar Ataxia-Associated Gene Tau Tubulin Kinase 2 Controls the Initiation of Ciliogenesis. Cell 2012, 151, 847–858. [Google Scholar] [CrossRef]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Kim, S.; Lee, K.; Choi, J.H.; Ringstad, N.; Dynlacht, B.D. Nek2 activation of Kif24 ensures cilium disassembly during the cell cycle. Nat. Commun. 2015, 6, 8087. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, T.J.; Wheway, G.; Slaats, G.G.; SYSCILIA Study Group; Huynen, M.A.; Giles, R.H. The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia 2013, 2, 7. [Google Scholar] [CrossRef]
- Mao, H.M.; Tang, Z.M.; Li, H.E.; Sun, B.; Tan, M.J.; Fan, S.H.; Zhu, Y.; Sun, Y. Neddylation inhibitor MLN4924 suppresses cilia formation by modulating AKT1. Protein Cell 2019, 10, 726–744. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Si, Y.; Yu, H.F.; Zhang, L.Q.; Xie, P.; Jiang, W.G. MLN4924 (Pevonedistat), a protein neddylation inhibitor, suppresses proliferation and migration of human clear cell renal cell carcinoma. Sci. Rep. UK 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Powell, R.T.; Stephan, C.; Uray, I.P.; Talley, T.; Karki, M.; Tripathi, D.N.; Park, Y.S.; Mancini, M.A.; Davies, P.; et al. Bexarotene—A novel modulator of AURKA and the primary cilium in VHL-deficient cells. J. Cell Sci. 2018, 131, jcs219923. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Li, F.; Zhou, Y.; Zhang, Y.; Wang, Z.; Zhang, R.; Zhu, J.; Ren, Y.; Tan, Y.; et al. Therapeutic target database 2020: Enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2020, 48, D1031–D1041. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Mao, W.; Chen, Q.; Zhuang, Q.; Wang, L.; Dai, J.; Wang, H.; Huang, Z. Endostar, a modified recombinant human endostatin, suppresses angiogenesis through inhibition of Wnt/beta-catenin signaling pathway. PLoS ONE 2014, 9, e107463. [Google Scholar] [CrossRef]
- Burnett, J.C.; Rossi, J.J.; Tiemann, K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol. J. 2011, 6, 1130–1146. [Google Scholar] [CrossRef]
- Dominguez, J.M.; Fuertes, A.; Orozco, L.; del Monte-Millan, M.; Delgado, E.; Medina, M. Evidence for irreversible inhibition of glycogen synthase kinase-3beta by tideglusib. J. Biol. Chem. 2012, 287, 893–904. [Google Scholar] [CrossRef]
- Cheung, A.K.; Ip, J.C.; Lung, H.L.; Wu, J.Z.; Tsao, S.W.; Lung, M.L. Polo-like kinase inhibitor Ro5203280 has potent antitumor activity in nasopharyngeal carcinoma. Mol. Cancer Ther. 2013, 12, 1393–1401. [Google Scholar] [CrossRef]
- Schoffski, P. Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology. Oncologist 2009, 14, 559–570. [Google Scholar] [CrossRef]
- Santamaria, A.; Neef, R.; Eberspacher, U.; Eis, K.; Husemann, M.; Mumberg, D.; Prechtl, S.; Schulze, V.; Siemeister, G.; Wortmann, L.; et al. Use of the novel Plk1 inhibitor ZK-thiazolidinone to elucidate functions of Plk1 in early and late stages of mitosis. Mol. Biol. Cell 2007, 18, 4024–4036. [Google Scholar] [CrossRef]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lenart, P.; Petronczki, M.; Krssak, M.; Gurtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef]
- Ma, W.W.; Messersmith, W.A.; Dy, G.K.; Weekes, C.D.; Whitworth, A.; Ren, C.; Maniar, M.; Wilhelm, F.; Eckhardt, S.G.; Adjei, A.A.; et al. Phase I study of Rigosertib, an inhibitor of the phosphatidylinositol 3-kinase and Polo-like kinase 1 pathways, combined with gemcitabine in patients with solid tumors and pancreatic cancer. Clin. Cancer Res. 2012, 18, 2048–2055. [Google Scholar] [CrossRef]
- Hikichi, Y.; Honda, K.; Hikami, K.; Miyashita, H.; Kaieda, I.; Murai, S.; Uchiyama, N.; Hasegawa, M.; Kawamoto, T.; Sato, T.; et al. TAK-960, a novel, orally available, selective inhibitor of polo-like kinase 1, shows broad-spectrum preclinical antitumor activity in multiple dosing regimens. Mol. Cancer Ther. 2012, 11, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, A.; Yugandhar, D.; Srivastava, A.K. BET inhibitors in cancer therapeutics: A patent review. Expert Opin. Ther. Pat. 2016, 26, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Garland, L.L.; Taylor, C.; Pilkington, D.L.; Cohen, J.L.; Von Hoff, D.D. A phase I pharmacokinetic study of HMN-214, a novel oral stilbene derivative with polo-like kinase-1-interacting properties, in patients with advanced solid tumors. Clin. Cancer Res. 2006, 12, 5182–5189. [Google Scholar] [CrossRef]
- Mullard, A. 2012 FDA drug approvals. Nat. Rev. Drug Discov. 2013, 12, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. 2018 FDA drug approvals. Nat. Rev. Drug Discov. 2019, 18, 85–89. [Google Scholar] [CrossRef]
- Ge, X.; Yamamoto, S.; Tsutsumi, S.; Midorikawa, Y.; Ihara, S.; Wang, S.M.; Aburatani, H. Interpreting expression profiles of cancers by genome-wide survey of breadth of expression in normal tissues. Genomics 2005, 86, 127–141. [Google Scholar] [CrossRef]
- D’Amato, C.; Rosa, R.; Marciano, R.; D’Amato, V.; Formisano, L.; Nappi, L.; Raimondo, L.; Di Mauro, C.; Servetto, A.; Fulciniti, F.; et al. Inhibition of Hedgehog signalling by NVP-LDE225 (Erismodegib) interferes with growth and invasion of human renal cell carcinoma cells. Br. J. Cancer 2014, 111, 1168–1179. [Google Scholar] [CrossRef]
- Peukert, S.; He, F.; Dai, M.; Zhang, R.; Sun, Y.; Miller-Moslin, K.; McEwan, M.; Lagu, B.; Wang, K.; Yusuff, N.; et al. Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. ChemMedChem 2013, 8, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Xin, M. Hedgehog inhibitors: A patent review (2013–present). Expert Opin. Ther. Pat. 2015, 25, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, H.; Evron, T.; Vardy, E.; Han, G.W.; Huang, X.P.; Hufeisen, S.J.; Mangano, T.J.; Urban, D.J.; Katritch, V.; et al. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ibuki, N.; Ghaffari, M.; Pandey, M.; Iu, I.; Fazli, L.; Kashiwagi, M.; Tojo, H.; Nakanishi, O.; Gleave, M.E.; Cox, M.E. TAK-441, a novel investigational smoothened antagonist, delays castration-resistant progression in prostate cancer by disrupting paracrine hedgehog signaling. Int. J. Cancer 2013, 133, 1955–1966. [Google Scholar] [CrossRef]
- Manetti, F.; Petricci, E. Evaluation of WO2014207069 A1: Multitarget Hedgehog pathway inhibitors and uses thereof. Expert Opin. Ther. Pat. 2016, 26, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Miller-Moslin, K.; Peukert, S.; Jain, R.K.; McEwan, M.A.; Karki, R.; Llamas, L.; Yusuff, N.; He, F.; Li, Y.; Sun, Y.; et al. 1-amino-4-benzylphthalazines as orally bioavailable smoothened antagonists with antitumor activity. J. Med. Chem. 2009, 52, 3954–3968. [Google Scholar] [CrossRef] [PubMed]
- Peukert, S.; Jain, R.K.; Geisser, A.; Sun, Y.; Zhang, R.; Bourret, A.; Carlson, A.; Dasilva, J.; Ramamurthy, A.; Kelleher, J.F. Identification and structure-activity relationships of ortho-biphenyl carboxamides as potent Smoothened antagonists inhibiting the Hedgehog signaling pathway. Bioorg. Med. Chem. Lett. 2009, 19, 328–331. [Google Scholar] [CrossRef]
- Axten, J.M. Protein kinase R(PKR)-like endoplasmic reticulum kinase (PERK) inhibitors: A patent review (2010-2015). Expert Opin. Ther. Pat. 2017, 27, 37–48. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Estrada, A.A.; Chan, B.K.; Baker-Glenn, C.; Beresford, A.; Burdick, D.J.; Chambers, M.; Chen, H.; Dominguez, S.L.; Dotson, J.; Drummond, J.; et al. Discovery of highly potent, selective, and brain-penetrant aminopyrazole leucine-rich repeat kinase 2 (LRRK2) small molecule inhibitors. J. Med. Chem. 2014, 57, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.S.; Uto, Y.; Won, M.; Nakamura, H. Hypoxia-inducible factor (HIF) inhibitors: A patent survey (2011–2015). Expert Opin. Ther. Pat. 2016, 26, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Farshi, P.; Deshmukh, R.R.; Nwankwo, J.O.; Arkwright, R.T.; Cvek, B.; Liu, J.; Dou, Q.P. Deubiquitinases (DUBs) and DUB inhibitors: A patent review. Expert Opin. Ther. Pat. 2015, 25, 1191–1208. [Google Scholar] [CrossRef]
- Helal, I.; Reed, B.; Schrier, R.W. Emergent Early Markers of Renal Progression in Autosomal-Dominant Polycystic Kidney Disease Patients: Implications for Prevention and Treatment. Am. J. Nephrol. 2012, 36, 162–167. [Google Scholar] [CrossRef]
- Adiyanti, S.S.; Loho, T. Acute Kidney Injury (AKI) biomarker. Acta Med. Indones. 2012, 44, 246–255. [Google Scholar] [PubMed]
- Hersch, S.M.; Rosas, H.D. Biomarkers to Enable the Development of Neuroprotective Therapies for Huntington’s Disease. In Neurobiology of Huntington’s Disease: Applications to Drug Discovery; Lo, D.C., Hughes, R.E., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2011. [Google Scholar]
- El Shamieh, S.; Visvikis-Siest, S. Genetic biomarkers of hypertension and future challenges integrating epigenomics. Clin. Chim. Acta 2012, 414, 259–265. [Google Scholar] [CrossRef]
- Pich, E.M.; Vargas, G.; Domenici, E. Biomarkers for antipsychotic therapies. In Handbook of Experimental Pharmacology; Gross, G., Geyer, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Villanova, F.; Di Meglio, P.; Nestle, F.O. Biomarkers in psoriasis and psoriatic arthritis. Ann. Rheum. Dis. 2013, 72, 104–110. [Google Scholar] [CrossRef]
- Rodon, J.; Saura, C.; Dienstmann, R.; Vivancos, A.; Ramon Cajal, S.; Baselga, J.; Tabernero, J. Molecular prescreening to select patient population in early clinical trials. Nat. Rev. Clin. Oncol. 2012, 9, 359–366. [Google Scholar] [CrossRef]
- Vilmar, A.C.; Santoni-Rugiu, E.; Sorensen, J.B. Class III beta-tubulin in advanced NSCLC of adenocarcinoma subtype predicts superior outcome in a randomized trial. Clin. Cancer Res. 2011, 17, 5205–5214. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Diseases | Mutated Genes | Features |
|---|---|---|
| PKD | PKD1, PKD2 | Renal cyst formation |
| Meckel-Gruber syndrome | MKS1, TMEM67, TMEM216, TMEM107, TMEM231, CEP290/NPHP6, NPHP8/RPGRIP1L, CC2D2A, NPHP3, TCTN2, B9D1, B9D2 | Renal cyst formation |
| Nephronophthisis | NPHP1, NPHP3, NPHP2 (INVS), NPHP4, NPHP5/IQCB1, NPHP6/CEP290, NPHP7/GLIS2, NPHP8/RPGRIP1L, NPHP9/NEK8, TMEM67/MKS3, TTC21B/JBTS11, WDR19, ZNF423, CEP164, ANKS6, CEP83, DCDC2 MAPKBP1 | Renal fibrosis and cyst formation |
| Joubert syndrome | INPP5E, TMEM216, AHI1, NPHP1, NPHP6/CEP290, TMEM67/MKS3, RPGRIP1L/NPHP8, ARL13B, OFD1, TTC21B, TMEM237, CEP41, TMEM138, TCTN3, ZNF423, TMEM231, CSPP1, PDE6D, TCTN2, B9D1, MKS1, TMEM107, B9D2, ARL3 | Renal cyst formation |
| Oral-facial-digital syndrome type I | OFD1 | Cystic kidney disease |
| Cranioectodermal dysplasia | IFT122, WDR35, IFT43, WDR19/IFT144 | Renal failure |
| Bardet-Biedl | BBS1, BBS2, ARL6, BBS4, BBS5, BBS6/MKKS, BBS7, BBS8, BBS9, BBS10, TRIM32, BBS12, CCDC28B, CEP290/NPHP6, TMEM67/MKS3, MKS1, SDCCAG8, LZTFL1, BBIP1, IFT27, IFT74, | Chronic renal failure in children |
| Senior-Loken syndrome | NPHP1, NPHP4, NPHP5, NPHP6, SDCCAG8, WDR19/IFT144, TRAF3IP1 | Juvenile nephronophthisis, renal cyst |
| Alström syndrome | ALMS1 | Renal failure |
| RCC | VHL, NPHP4, NPHP9/NEK8, TTBK2 | Cyst and cancer |
| Von Hippel-Lindau | VHL | Cyst and cancer |
| Ciliary Genes | Type of Cancer | Name of Drug | Clinical Trial | References |
|---|---|---|---|---|
| CTNNB1 | Solid cancer | Recombinant human endostatin | approved | [179,180] |
| DRD5 | Solid cancer | DS-8273 | Phase1 | https://clinicaltrials.gov/ct2/show/NCT02076451 |
| GSK3B | Acute myeloid leukemia, osteosarcoma | LY2090314, Tideglusib | Phase2 | https://clinicaltrials.gov/ct2/show/NCT01214603 [181] |
| PLK1 | Solid cancer, Acute myeloid leukemia | Rigosertib, Volasertib | Phase3, Phase3 | [182,183,184,185,186,187,188,189] |
| SMO | Solid cancer, skin cancer | LDE225 LY2940680, BMS-833923, LEQ-506, TAK-441 | Approved (basal cell carcinoma) Phase1, Phase2 | [190,191,192,193,194,195,196,197,198,199,200] |
| TTK | Solid cancer | BAY1161909, BAY1161909 | Phase1 Phase1 | [201,202,203] |
| VDAC3 | Solid cancer | PRLX93936 | Phase1/2 | https://clinicaltrials.gov/ct2/show/NCT01695590 |
| VHL | Renal cell cancer | Pyrrolidine carboxamide derivative 1 | Patented-recorded Target | [204,205] |
| Ciliary Genes | Biomarker Gene Location | Disease | Biomarker Type | Molecular Type | Biomarker Measure | Interactors | Reference |
|---|---|---|---|---|---|---|---|
| PKD1 | 16p13.3 | Polycystic kidney disease | Prognostic | Gene | Mutation | - | [206] |
| CLUAP1 | 16p13.3 | Acute kidney injury, Huntington disease | Diagnostic, Prognostic | Protein | Elevated level | - | [207,208] |
| DRD1 | 5q35.1 | Hypertension | Associative | Gene | SNP | - | [209] |
| DRD2 | 11q23.2 | Hypertension, schizophrenia | Associative, Theragnostic | Gene | SNP | - | [209,210] |
| NUP62 | 19q13.33 | Psoriatic arthitis | Diagnostic | Gene | Expression | - | [211] |
| PTCH1 | 9q22.32- | Medulloblastoma | Theragnostic | Gene | Mutation | - | [212] |
| SMO | 7q32.1- | Medulloblastoma | Theragnostic | Gene | Mutation | - | [212] |
| TUBB3 | 16q24.3 | Non-small cell lung cancer | Prognostic | Protein | Absence | ERCC1 | [213] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamiok-Ostrowska, A.; Piekiełko-Witkowska, A. Ciliary Genes in Renal Cystic Diseases. Cells 2020, 9, 907. https://doi.org/10.3390/cells9040907
Adamiok-Ostrowska A, Piekiełko-Witkowska A. Ciliary Genes in Renal Cystic Diseases. Cells. 2020; 9(4):907. https://doi.org/10.3390/cells9040907
Chicago/Turabian StyleAdamiok-Ostrowska, Anna, and Agnieszka Piekiełko-Witkowska. 2020. "Ciliary Genes in Renal Cystic Diseases" Cells 9, no. 4: 907. https://doi.org/10.3390/cells9040907
APA StyleAdamiok-Ostrowska, A., & Piekiełko-Witkowska, A. (2020). Ciliary Genes in Renal Cystic Diseases. Cells, 9(4), 907. https://doi.org/10.3390/cells9040907