1. Introduction
Connexins (Cx) are a family of proteins that directly connect the cytoplasm of adjacent cells through the formation of gap junction (GJ) channels. In humans, twenty members form the connexin family, while at least seventeen members have been reported in rats. Based on sequence homology, connexin genes can be grouped into four classes, α, β, γ and δ [
1]. Each connexin consists of four transmembrane regions, two extracellular loops and one intracellular loop, with both the amino and carboxyl terminals facing the cytosol. After synthesis, connexin proteins oligomerize into hexameric structures termed hemichannels and are transported to the plasma membrane, where they may dock to hemichannels from adjacent cells to form a functional GJ channel. The carboxyl terminal is where most differences between connexins are found, and a large part of the protein interactions and post-translational modifications of connexins are thought to occur in this region [
2]. Although traditionally associated with establishing direct communication between adjacent cells, the localization of connexins at the plasma membrane as undocked connexin hemichannels as well as its presence in mitochondria, nucleus and extracellular vesicles, suggest that connexins play other non-canonical biological roles [
3]. Given all of the above, fine-tuned regulatory mechanisms are required to modulate connexin levels, function and localization.
Although GJ intercellular communication can be modulated through the gating of the channel pore, mechanisms that regulate the number of connexin-containing channels at the plasma membrane have also been implicated in the regulation of intercellular communication. Given the short half-life of Cx43, mechanisms of protein degradation play an important role in the regulation of Cx43 levels and intercellular communication. We and others have shown that ubiquitination of Cx43, modulated by E3 ligases, including neural precursor cell expressed developmentally down-regulated protein 4 (Nedd4), and deubiquitinating enzymes, such as associated molecule with the SH3 domain of STAM (AMSH), dictates the final fate of the protein [
4,
5,
6,
7]. At the plasma membrane, Cx43 is modified by Lysine 63-linked polyubiquitin chains, which are recognized by the endocytic adaptor epidermal growth factor receptor substrate 15 (Eps15) to trigger the internalization of the protein [
7]. Furthermore, Cx43 sorting from early endosomes to the lysosome is also reliant on ubiquitination to mediate the interaction with the endosomal sorting complex required for transport (ESCRT) components hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs) and tumour susceptibility gene 101 (Tsg101) [
8,
9]. The role of Cx43 ubiquitination in signalling lysosomal GJ degradation is important not only in the endocytic pathway but also in the autophagy process. Indeed, GJ degradation by macroautophagy is also dependent, at least partially, on prior ubiquitination of Cx43 [
10,
11], a process that requires not only the ubiquitin-binding domain containing autophagy receptor p62, but also, and surprisingly, the endocytic adaptor Eps15 [
11]. However, preventing the ubiquitination of Cx43 did not fully abrogate its autophagic degradation, suggesting that alternative mechanisms for targeting Cx43 to autophagosomes exist.
Autophagy is one of the major catabolic pathways in the cell, in which substrates are delivered to the lysosome for degradation. In macroautophagy, a double membrane structure termed the phagophore, grows around the target substrate, eventually closing to form an autophagosome, which subsequently fuses with the lysosome. During phagophore formation, the ubiquitin-like protein microtubule-associated protein 1 light chain 3 (MAPLC3) is conjugated to phosphatydilethanolamine (PE) present on the nascent phagophore, through a mechanism closely resembling ubiquitin conjugation. The E1-like protein autophagy-related protein 7 (ATG7) activates LC3, to allow its transfer to the E2-like protein ATG3. In the final step of the conjugation process, the E3-like complex ATG12-ATG5-ATG16L1 facilitates the transfer of LC3 to PE [
12]. The LC3/GABARAP (γ-aminobutyric acid receptor–associated protein) family is comprised of 6 members in humans: LC3A, LC3B, LC3C, GABARAP, GABARAPL1 and GABARAPL2. Although macroautophagy was initially thought of as an unselective process for the bulk degradation of cytosolic content, it is now understood that this process can be highly selective. Substrates to be degraded are labelled with ubiquitin chains, which, in turn, are recognized by ubiquitin-binding domains on autophagy receptors such as p62, neighbour of BRCA1 gene 1 (NBR1), NDP52 and Optineurin. These receptors bridge the substrate to the nascent phagophore by binding to LC3/GABARAP family proteins present in the inner membrane through LC3-interacting region (LIR) motifs. The core consensus sequence of the LIR motif consists of [W/F/Y]-X1-X2-[L/I/V] [
12,
13]. More recently, a GABARAP interacting motif (GIM) has been described, consisting of the consensus sequence [W/F]-[V/I]-X2-V [
14]. Although the ubiquitin-dependent binding of Cx43 to LC3 was previously shown to be mediated by p62 [
11], the existence of a putative LIR motif in the amino terminal as well as the fact that hampering Cx43 ubiquitination and p62 silencing does not completely abolish the interaction of Cx43 with LC3, prompted us to investigate whether Cx43 can directly bind to LC3 through a LIR motif, without the need of ubiquitin.
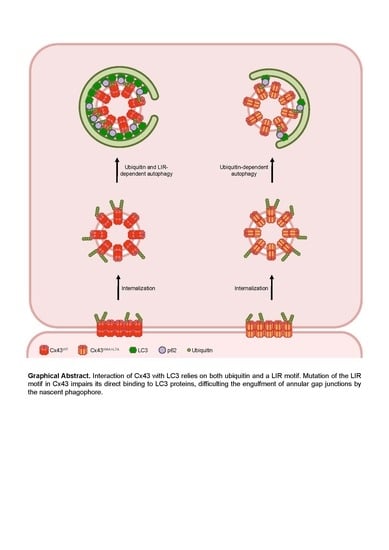
In this study we identify a functional LIR domain in several members of the connexin family, which mediates binding to both LC3B and GABARAP proteins. We also show that connexin ubiquitination alone is insufficient to promote binding to LC3 proteins. Mutation of the LIR motif of Cx43 also rendered the protein resistant to nutrient deprivation-induced degradation. Altogether, the results presented herein support a model in which connexin proteins are targeted for autophagosomal degradation through direct binding to LC3/GABARAP family proteins present on the nascent phagophore membrane.
2. Materials and Methods
2.1. Antibodies and Reagents
Goat polyclonal antibodies against Cx43 (AB1600), GST (AB9919-200), V5 (AB0096-500) and calnexin (AB0041-500) were obtained from SICGEN (Cantanhede, Portugal). Rabbit polyclonal antibodies against Cx43 (H-150) and mouse monoclonal antibodies against p62 (sc-28359) were obtained from Santa Cruz Biotechnology (Heidelberg, Germany). Rabbit polyclonal antibodies against GABARAP (ab109364) and Nedd4 (ab14592) were obtained from Abcam (Cambridge, UK). Rabbit polyclonal antibodies against LC3B (PA1-16930) and mouse monoclonal antibodies against V5 (46-0705) were obtained from Invitrogen (Paisley, UK). Mouse monoclonal antibodies against ubiquitin (P4D1) were obtained from Covance (San Diego, CA, USA). Mouse monoclonal antibodies against EEA1 (610457) were obtained from BD Transduction Laboratories (San Jose, CA, USA). Rabbit polyclonal antibodies against ATG7 (A2856), Phorbol 12-myristate 13-acetate (PMA), 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) and cycloheximide (CHX) were obtained from Sigma (Saint Louis, MO, USA). Bafilomycin A1 was obtained from Bioaustralis (Smithfield, Australia).
2.2. Cell Culture and Transfections
HEK293A cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics (100 U/mL penicillin, 100 g/mL streptomycin and 8 μg/mL blasticidin) and maintained at 37 °C under 5% CO2. Transient transfection of cells was performed with Lipofectamine 2000 (Grand Island, NY, Invitrogen), according to the manufacturer’s recommendations.
2.3. Plasmid Constructions
Rat Cx43 cDNA was cloned into a modified pENTR GFP C2 vector [
15]. Site-directed mutagenesis was performed to generate the Cx43
W4A+L7A, Cx43
Y265A, Cx43
Y286A and Cx43
Y265,286A mutants from Cx43 cDNA. Plasmids expressing V5-Cx43
WT and V5-Cx43
W4A+L7A were generated by cloning the appropriate cDNA into a pENTR V5 vector. Rat Cx37 and Cx40 cDNA were amplified from rat aorta tissue using RT-PCR and cloned into a pENTR V5 vector. Rat Cx46 and Cx50 cDNA were amplified from rat lens using RT-PCR and cloned into a pENTR V5 vector. Site-directed mutagenesis was performed to generate the W4A+L7A mutants from Cx37, Cx40, Cx46 and Cx50. Plasmids expressing GST-Cx43
WT_NT and GST-Cx43
W4A+L7A_NT were generated by subcloning the first 22 amino acids of Cx43
WT and Cx43
W4A+L7A into a pGEX4T1 vector. All constructs were verified by DNA sequencing. GFP-LC3B and GFP-GABARAP were obtained by insertion of human LC3B and GABARAP cDNAs into pEGFP-C1. Fusion proteins were subsequently cloned into pETDuet-1 for bacterial expression. The last five amino acids of the LC3B coding sequence and the last amino acid of GABARAP were deleted to mimic ATG4 cleavage. A 6xHis-tag was added C-terminally to purify the proteins [
16]. Plasmids expressing GFP-LC3B were kindly provided by Dr. Tamotsu Yoshimori (National Institute for Basic Biology, Okazaki, Japan).
2.4. Viral shRNA Infection
HEK293A cells were incubated with the lentiviral vectors and 8 µg/mL of polybrene for 20 min at room temperature. After 30 min centrifugation at 800× g and 32 °C, cells were plated and monitored for the expression of GFP. Lentiviral vectors containing shRNA targeting ATG7 and the control empty vector were kindly provided by Dr. A.M. Cuervo (Albert Einstein College of Medicine, Yeshiva University, New York, USA).
2.5. siRNA-Mediated Knockdown
siRNA targeting p62 (s16960 or s16961) and a non-targeting control sequence were obtained from Ambion. Cells were transfected with 20 nM siRNA using Lipofectamine 2000 (Grand Island, NY, Invitrogen) according to manufacturer’s recommendations. p62 knockdown was achieved after 24 h of transfection.
2.6. Immunoprecipitation and Western Blotting
Cells were rinsed with phosphate buffered saline (PBS) at 4 °C, resuspended in lysis buffer (190 mM NaCl, 50 mM Tris-HCl, 6 mM EDTA, 1% Triton X-100, pH 8.3) supplemented with protease inhibitor cocktail (Roche), 2 mM PMSF, 10 mM iodoacetamide, and incubated on ice for 10 min. The samples were then centrifuged at 10,000× g for 10 min and the supernatants used for immunoprecipitation. Briefly, protein G was incubated with goat polyclonal antibodies directed against Cx43 or V5. Incubations proceeded for 1 h at 4 °C, followed by incubation with supernatants for 3 h at 4 °C. The samples were then centrifuged and the protein G-sepharose sediments washed 3 times in an appropriate washing buffer (500 mM NaCl, 50 mM Tris-HCl, 6 mM EDTA, 1% Triton X-100, pH 8.3), resuspended in Laemmli buffer and denatured at 70 °C for 10 min.
For Western blot analysis of the immunoprecipitated proteins, samples were separated using SDS-PAGE, transferred to a nitrocellulose membrane and probed with appropriate antibodies. Inputs represent about 10% of the total amount of protein in the lysates before immunoprecipitation. Immunoprecipitation controls were performed by pooling the lysates of two samples transfected and/or treated in the same conditions, separating them in two fractions and then proceeding with the immunoprecipitation without adding antibody to one of the samples (No Ab). The corresponding pooled lysate appears in the Western blot panels to the right of the No Ab samples.
2.7. Bacterial Protein Expression and Purification
GST-Cx43
WT_NT and GST-Cx43
W4A+L7A_NT proteins were expressed in Escherichia coli BL21-CodonPlus (DE3)-RILP Cells (Agilent Technologies, Santa Clara, CA, USA). Bacteria were grown in Luria broth (LB) medium until OD600 ≈ 0.8–1, induced with 0.1 mM isopropylthiogalactoside (IPTG) and grown at 37 °C for 4 h. GST constructs were isolated from harvested cells using Glutathione Sepharose 4B (GE Healthcare, Buckinghamshire, UK) according to manufacturer’s recommendations. GFP-LC3B and GFP-GABARAP proteins were expressed in Escherichia coli Rosetta (DE3) pLysS cells. Cells were induced at an OD600 of 0.5 for 16 h at 18 °C with 0.1 mM IPTG. Harvested cells were resuspended in lysis buffer 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) at pH 7.5, 500 mM NaCl, 10 mM imidazole, 2 mM MgCl
2, 2 mM β-mercaptoethanol, complete protease inhibitor (Roche, Basel, Switzerland) and DNase I and lysed using a freeze–thaw cycle followed by brief 30 s sonication. Lysates were cleared using ultracentrifugation at 140,000 g for 30 min at 4 °C (Beckman, Brea, CA, USA, Ti45 rotor). Supernatants were applied to Ni-NTA columns (GE Healthcare, Buckinghamshire, UK), and constructs were eluted via a stepwise imidazole gradient (50, 75, 100, 150, 200, and 300 mM) [
16].
2.8. GFP-Trap Pull-Down Assay
Five microlitres of GFP-Trap beads slurry (ChromoTek, Munich, Germany) were mixed with a 5 μM solution of GFP-fused bait proteins (GFP-LC3B or GFP-GABARAP) and incubated on a rotating wheel at 4 °C for 1 h. Subsequently the beads were washed twice with 150 mM NaCl, 50 mM Tris at pH 7.4, and incubated with 20 μg of prey solution (GST-Cx43WT_NT or GST-Cx43W4A+L7A_NT). Precipitates were analysed using Western blot using goat polyclonal antibodies against GST.
2.9. Microscopy-Based Protein-Protein Interaction Assay
Five microlitres of glutathione Sepharose 4B beads slurry (GE Healthcare, Buckinghamshire, UK) were mixed with 20 µg of GST-fused bait proteins (GST-Cx43
WT_NT or GST-Cx43
W4A+L7A_NT) and incubated on a rotating wheel at 4 °C for at least 30 min. Subsequently the beads were washed twice with 150 mM NaCl, 50 mM Tris at pH 7.4 and resuspended in 6 µL of the same buffer. Of a 5 µM prey solution (GFP-LC3B or GFP-GABARAP), 10 µL was plated into the well of a 384-well glass-bottom microplate (Greiner Bio-One, Frickenhausen, Germany), after which 10% of the previous beads solution was added. Samples were then imaged on a confocal microscope. To quantify the protein recruitment to beads the maximum brightness along a straight line drawn through a single bead was taken (maximal fluorescence). Next, the average brightness of an empty portion of each picture was measured (background fluorescence) and subtracted from the maximal fluorescence for each bead [
16].
2.10. Triton X-100 Fractionation Assay
The detergent solubility assay with 1% Triton X-100 was performed essentially as described previously by others [
17]. Cells were resuspended in lysis buffer (190 mM NaCl, 50 mM Tris-HCl, 6 mM EDTA, 1% Triton X-100, pH 8.3) supplemented with protease inhibitor cocktail (Roche, Basel, Switzerland), 2 mM PMSF and 10 mM iodoacetamide. Samples were then ultracentrifuged at 100,000 g for 50 min and the supernatant recovered (Triton X-100 soluble fraction). The detergent-insoluble pellets were resuspended in lysis buffer supplemented with 0.1% SDS (Triton X-100 insoluble fraction) and sonicated. Laemmli buffer was added to 10% of each Triton X-100 soluble and insoluble fractions and denatured at 70 °C for 10 min before SDS-PAGE analysis. The remainder of each sample was used for immunoprecipitation.
2.11. Immunofluorescence
HEK293A cells grown on glass coverslips were fixed with 4% paraformaldehyde in PBS. The samples were then washed with PBS, permeabilised with 0.2% v/v Triton X-100 in PBS and blocked with 2% w/v BSA in PBS for 20 min prior to incubation with primary antibodies overnight at 4 °C. The samples were then washed three times with PBS before incubation with the secondary antibody for 1 h at room temperature. The specimens were rinsed in PBS and mounted with MOWIOL 4-88 Reagent (Calbiochem, San Diego, CA, USA). Nuclei were stained with DAPI. For controls primary antibodies were omitted. Imaging was performed on a laser-scanning confocal (Zeiss LSM 710, Carl Zeiss, Oberkochen, Germany) with Plan-Apochromat 63X/1.4 oil objective (Carl Zeiss, Oberkochen, Germany). Colocalization of Cx43 and GFP-LC3B or EEA1 was quantitated using Pearson’s correlation coefficient (PCC) using the Coloc 2 plugin of ImageJ software (
http://imagej.net/Coloc_2). Cells were selected as regions of interest (ROIs) using the freehand selection tool before running the plugin. Pearson’s R values (no threshold) reported were plotted in a graph.
2.12. Statistical Analysis
Data were displayed with means on a scatter dot plot. For data with non-Gaussian distribution, statistical significance was determined using a non-parametric Kruskall-Wallis test followed by a Dunn’s multiple comparison test, non-parametric Mann-Whitney test or multiple Student’s t-test with Holm-Sidak’s correction. Data was analysed and graphs were assembled with GraphPad Prism 6 for Windows, version 6.01 (GraphPad Software, Inc., San Diego, CA, USA). Results were considered significantly different for p < 0.05. The tests used in each experiment are indicated in the figure legends. Graphical data depict individual experiments except when noted in the figure legend.
4. Discussion
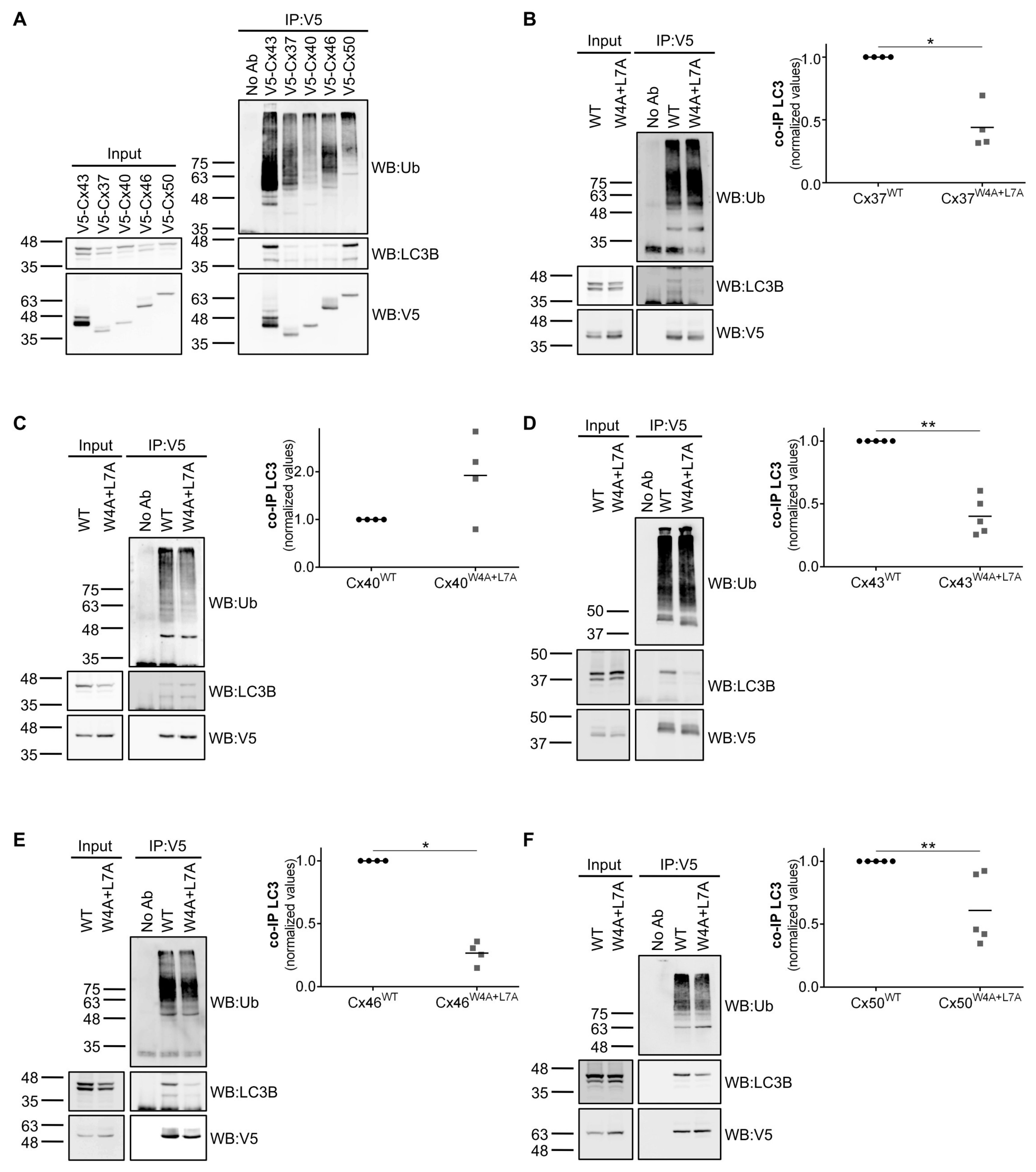
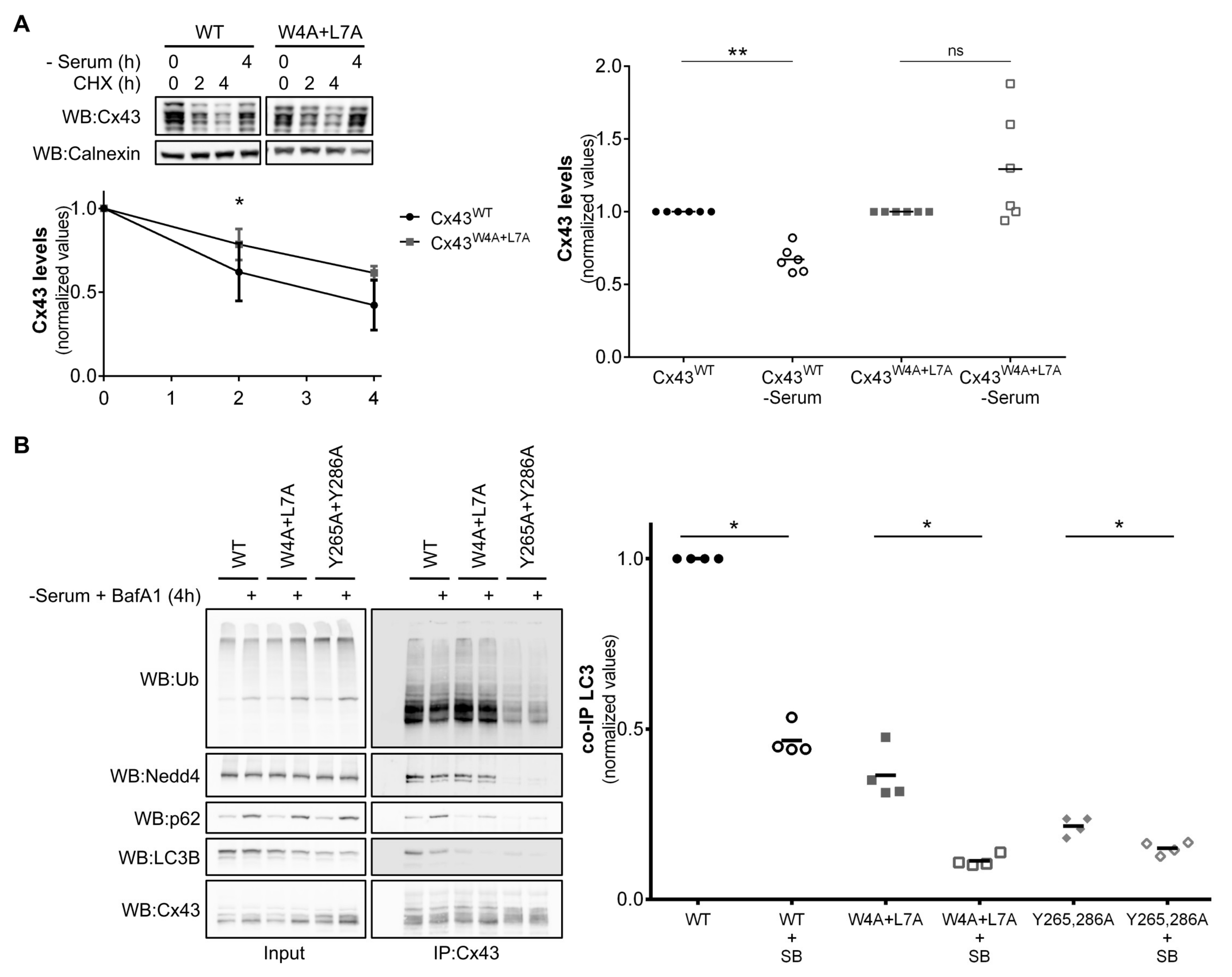
Previous studies demonstrated that connexin proteins are degraded by autophagy. We and others have reported that degradation of Cx43 by macroautophagy requires prior ubiquitination of Cx43 and its further recognition by p62 [
10,
11]. However, preventing Cx43 ubiquitination by siRNA depletion of the E3 ubiquitin ligase Nedd4, or mutation of Tyrosine 286, a core residue in the PPxY motif used for Nedd4 binding to Cx43, failed to completely abrogate degradation by autophagy, suggesting that a parallel pathway, independent of Cx43 ubiquitination, may exist to deliver the protein to the autophagosome. In this manuscript we describe a LIR motif, present in the amino terminal, that is conserved in almost all human and rat connexin family members. Mutation of the core consensus residues tryptophan 4 and leucine 7 to alanine in Cx37, Cx43, Cx46 and Cx50 lead to a substantial decrease in binding to both LC3B and GABARAP. Moreover, in the case of Cx43, interaction with LC3B occurs mostly in internalized vesicles, as evidenced by the Triton X-100 fractionation experiments and immunofluorescence imaging. Lastly, mutation of the LIR motif of Cx43 also renders the protein resistant to nutrient deprivation induced autophagy. Data from each mutant is summarized in
Table S1.
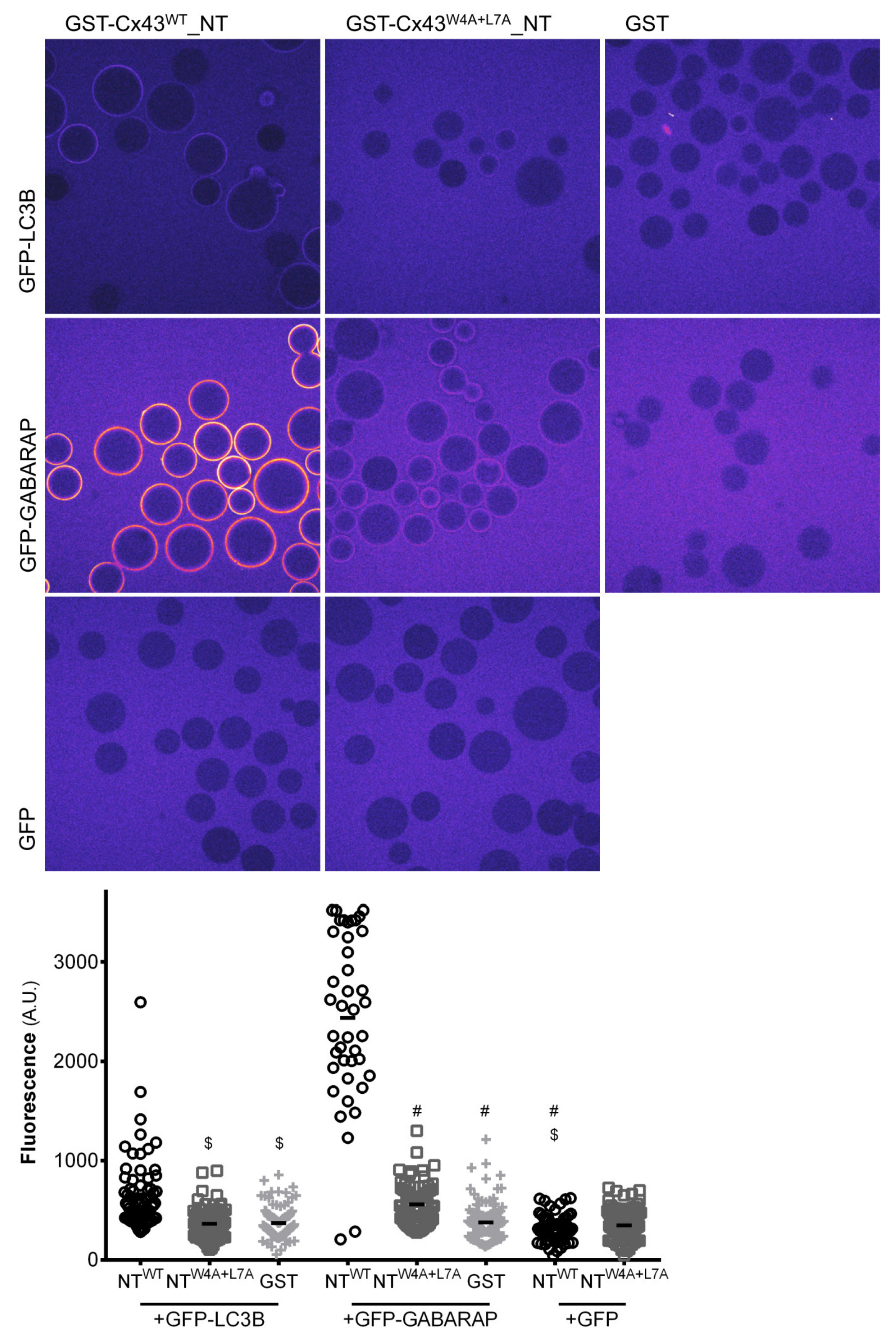
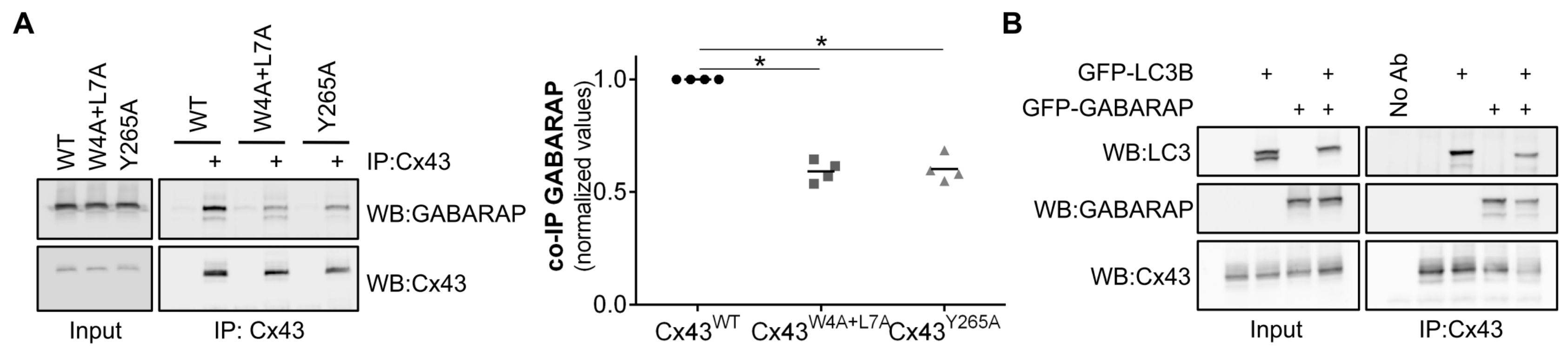
The results of in vitro protein-protein interaction assays suggest that the LIR motif present on the amino terminal of Cx43 binds preferentially to GABARAP as opposed to LC3B. A subset of LIR motifs that bind preferentially to GABARAP (GIM motif), characterized by the consensus sequence [W/F]-[V/I]-X2-V, was recently described [
14]. Curiously, none of the LIR motifs present in the human and rat connexin family members can be considered as GIMs. More intriguing is the observation that this preference for binding to GABARAP present in the in vitro assays performed using only the amino terminal of Cx43 is not carried over to the interaction experiments using full-length Cx43 expressed in mammalian cells. Indeed, when GFP-LC3B is co-expressed with GFP-GABARAP, Cx43 does not display any binding preference. It is plausible that post-translational modifications, which are not available during the production of the GST-Cx43
WT_NT chimera in bacteria, are necessary to facilitate the binding of Cx43 to LC3B. For instance, although the LIR consensus sequence is commonly defined as [W/F/Y]-X1-X2-[L/I/V], the presence of negative residues surrounding the core residues of the LIR motif has been described to modulate the interaction with LC3B [
13]. Accordingly, it is conceivable that in vivo phosphorylation of amino acid residues surrounding the LIR motif of Cx43 promote interaction with LC3. Furthermore, changes in the topology and conformation adopted by Cxs when expressed in cells may justify the discrepancy of in vitro and in vivo data. Along the synthetic pathway, Cx monomers oligomerize to form a hexameric structure, the connexon, before reaching the plasma membrane. Thus, a fully mature Cx channel exists as a structure that would contain 6 LIR motifs in close proximity. Binding to LC3B in vivo may be facilitated by the presence of several LIR motifs clustered in a limited region and in a specific orientation, which is not easily replicated on the surface of the beads used in the in vitro experiments. Other regions of the protein may also facilitate the binding of LC3 to Cx43, as will be discussed further below.
Although all the five alpha subfamily connexins (Cx37, Cx40, Cx43, Cx46 and Cx50) tested can bind to LC3 proteins, only mutation of the LIR motif of Cx40 did not impair its interaction with either LC3B or GABARAP. It is possible that in the case of Cx40, the interaction we observed with LC3B/GABARAP occurred through other regions of the protein or was mediated by ubiquitination. Moreover, differences in the amino terminal of Cx40 may also contribute to render the LIR motif nonfunctional. It will be interesting to investigate this issue in the future considering that Cx40 can co-oligomerize and is found co-expressed with Cx37 and Cx43 in several tissues.
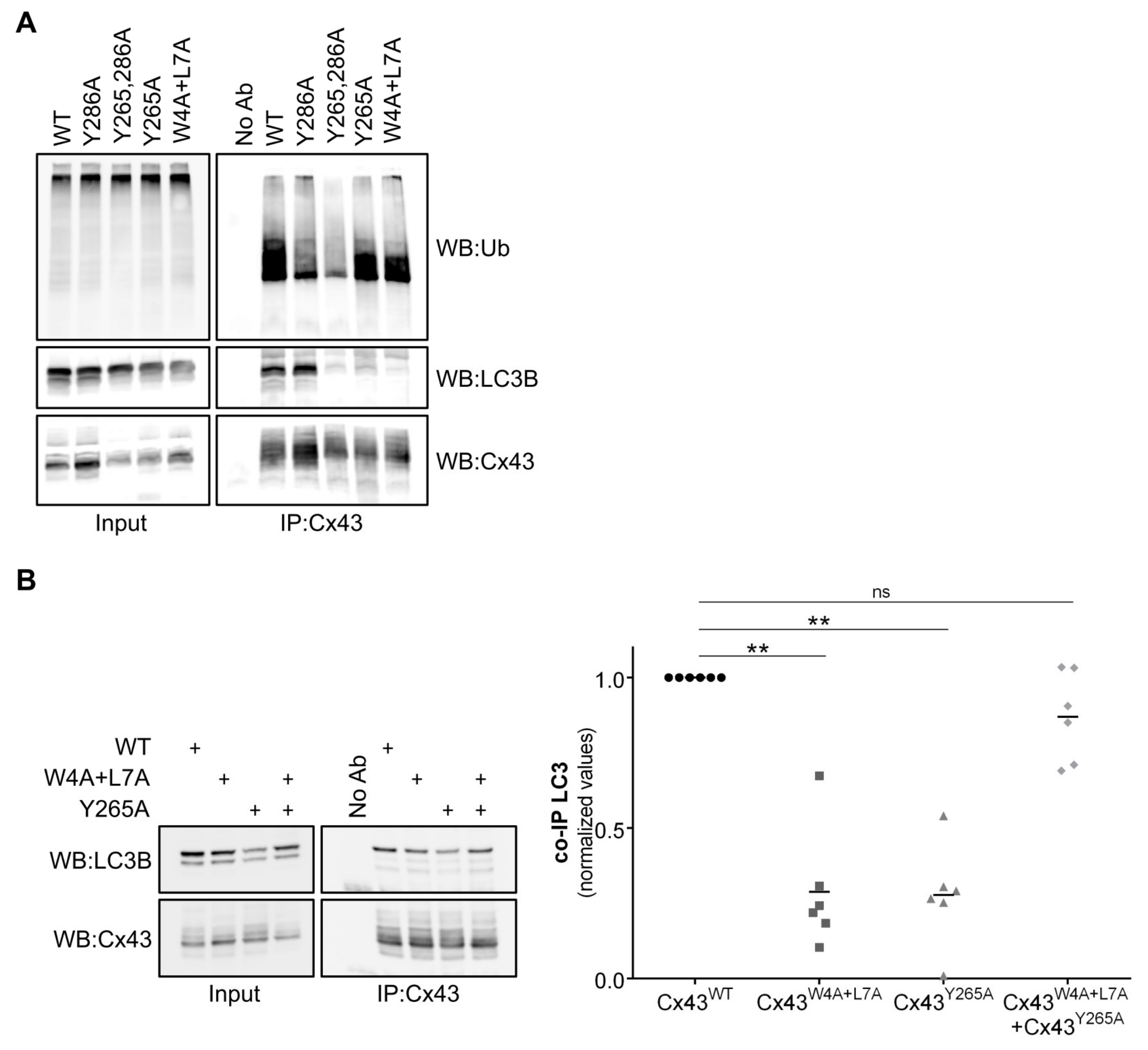
Surprisingly, we demonstrated that mutation of Tyrosine 265 on the carboxyl terminal of Cx43 also prevented binding to LC3B and GABARAP, although this residue was not part of any discernible LIR motif. Tyrosine 265 and 286 are part of two different endocytic tyrosine-sorting signals that were previously shown to play a role in the endocytosis of Cx43 [
6,
20], whereas Tyrosine 286 is additionally part of a PPxY motif required for binding to Nedd4 and subsequent Cx43 ubiquitination [
5,
21]. At a first glance, the impaired interaction of Cx43
Y265A with LC3B may suggest that the interaction could only occur after Cx43 internalization. Nevertheless, Cx43
Y286A, which also accumulates at the plasma membrane, still retains a relatively high binding level to LC3B, although to a lesser extent when compared to Cx43
WT, likely due to ubiquitination defects that hamper p62-mediated binding. Thus, some other mechanism, beyond the mere subcellular localization of Cx43
Y265A must be in play to explain its diminished binding to LC3B. Curiously, when Cx43
W4A+L7A was co-expressed with Cx43
Y265A, binding to LC3B was restored to normal levels. Presumably, when the two mutants oligomerize into the same hemichannel, they are able to compensate for each other’s mutation. This suggests a model in which direct LC3B binding to Cx43 depends not only on the LIR motif in the amino terminal, but also on the carboxyl terminal of another Cx present in the hemichannel. Furthermore, it is plausible that more than a strict dependence on Y265, mutation of this residue simply induces a conformational change in the carboxyl terminal that physically prevents access of LC3B to the LIR motif. It has been reported that Y265 phosphorylation by Src kinase impacts on different aspects of the Cx43 life cycle [
23], raising the possibility that Y265 phosphorylation may also modulate binding to LC3B. Another intriguing possibility is that the interaction of LC3B with Cx43 requires two binding sites, the LIR motif on the amino terminal, which would bind to the LIR docking site on LC3B, and an additional region in the carboxyl terminal, either including or modulated by Y265. Notably, LC3B does not bind to other proteins exclusively through its LIR docking site [
12]. Indeed, it has recently been reported that LC3B can interact with ubiquitin interacting motifs (UIM) through an UIM-docking site (UDS) [
24], and also with cardiolipin [
25] and Lamin B1 [
26].
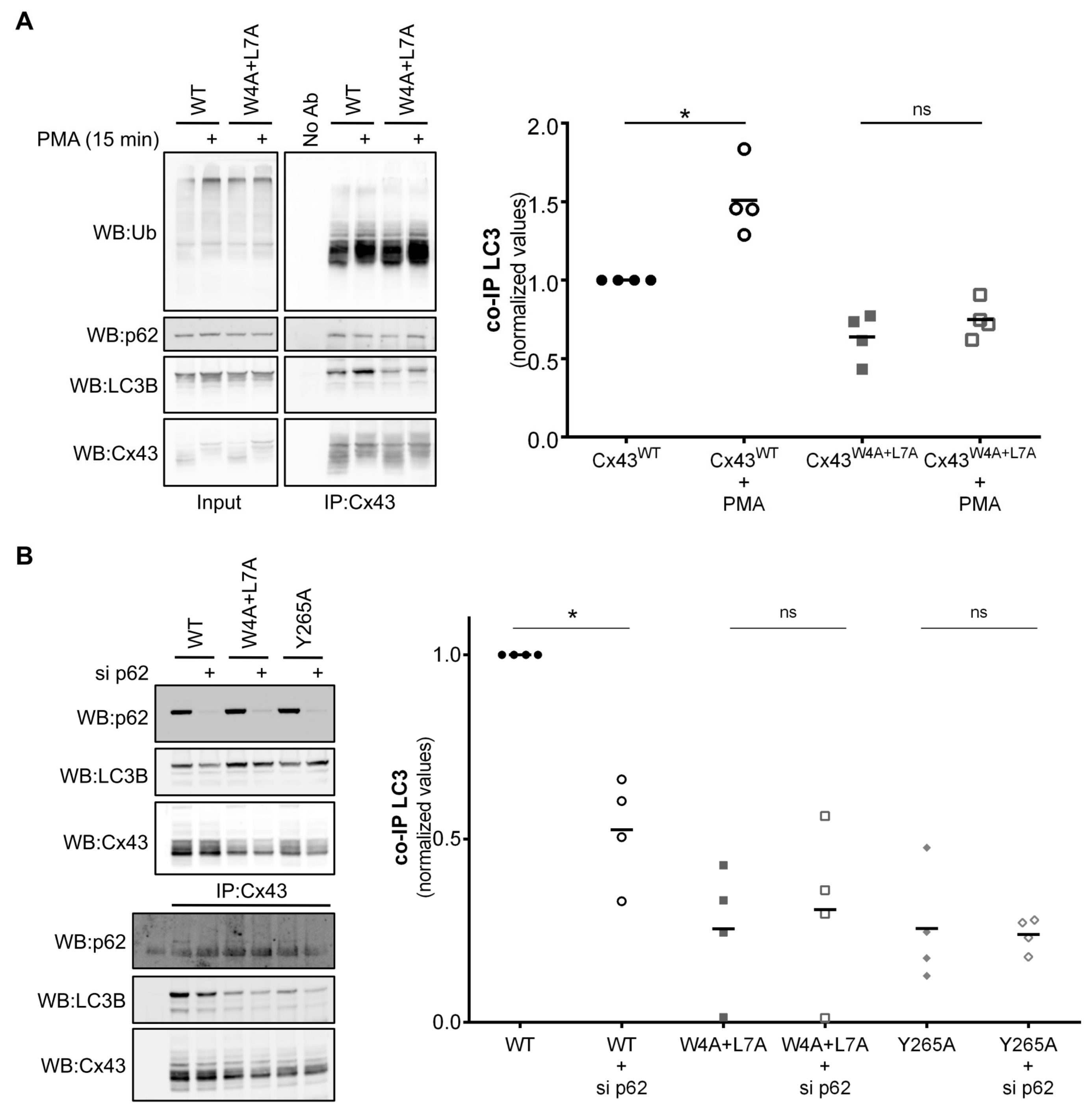
Triton X-100 fractionation experiments showed that the interaction occurred mainly in the Triton X-100 resistant fraction, which contains Cx43 present in GJ plaques and also internalized GJs. In addition, immunofluorescence imaging showed that the two proteins colocalized mainly in intracellular vesicles, thus supporting a model in which Cx43/LC3B interaction occurs mainly in internalized compartments. Treatment with PMA, which induces Cx43 phosphorylation, ubiquitination and internalization, lead to an increase in LC3B binding to Cx43
WT, while having no effect on binding with Cx43
W4A+L7A, suggesting that inducing the phosphorylation and ubiquitination of Cx43 cannot overcome the lack of a functional LIR motif. However, and in accordance with previous reports [
11], Cx43 ubiquitination still plays an important role in LC3B binding, as shown by the decrease in interaction when p62 is depleted. Our ATG7 silencing experiments also suggest that Cx43 binds preferentially to the non-conjugated form of LC3B, as while ATG7 depletion decreased the levels of LC3B-II, binding of LC3B to Cx43 was not impaired. This data raises an important question as to the function of this direct interaction of Cx43 with LC3B, within the context of the existence of a parallel pathway involving ubiquitination and p62 binding. An important fact to consider is that an autophagy substrate containing multiple points of LC3 interaction is not without precedent. For example, during mitophagy, ubiquitinated proteins on the surface of the mitochondria recruit autophagy receptors to assist in targeting the mitochondria to autophagosomes [
12]. However, in addition to this mechanism, LIR motif containing proteins on the surface of the mitochondria, such as Nix and FUNDC1, can also bind directly to LC3 to assist in mitophagy [
27]. In addition, the phospholipid cardiolipin, which is transported to the outer mitochondrial membrane following damage, can also bind to LC3 in a non-LIR motif dependent manner [
25]. Although plasma membrane proteins committed for degradation usually reach the lysosome through the endolysosomal pathway, GJs can be delivered to lysosomes through autophagy. This particularity of Cx proteins may stem from their unique internalization mechanism. Plasma membrane proteins are normally internalized through single membrane structures, whereas GJ plaques are internalized into one of the adjacent cells through a double membrane structure commonly referred to as an annular gap junction (AGJ). Perhaps the structure of AGJs is unsuited for easy processing through the endolysosomal pathway, requiring their degradation through autophagy. In such a case, as with mitophagy, the presence of multiple binding spots for LC3 may allow for more efficient engulfment of the AGJ by the phagophore. Another intriguing possibility is that this interaction is used by Cx43 to promote the recruitment of non-conjugated LC3B to AGJs to assist with its degradation by macroautophagy. Notably, Cx43 is also known to interact with several autophagy-related proteins, including ATG16, Vps35, Beclin-1 and Vps15 [
28], raising the possibility that Cx43 may function as a scaffold for autophagy machinery to further facilitate phagophore formation around AGJs. Although LC3/GABARAP family members offer some redundancy in function, specific functions for particular family members have been described, with the LC3 subfamily participating in the elongation of the phagophore membrane, while the GABARAP subfamily is involved in later stages of autophagosome formation [
12]. The ability of Cxs to interact with both subfamilies envisions a model in which the sequential binding of Cx43 to different LC3/GABARAP subfamily members is important to ensure AGJ engulfment and processing during autophagosome maturation.
Altogether, the results presented in this work lead us to propose a model in which Cxs bind directly to LC3/GABARAP proteins to facilitate their degradation by autophagy. Given the importance of GJIC in multiple tissues and organs, and its involvement in specific pathologies such as heart failure, understanding the mechanisms that regulate connexin degradation may shed new light upon the role of the connexin family in these diseases.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






