Proton Irradiation Increases the Necessity for Homologous Recombination Repair Along with the Indispensability of Non-Homologous End Joining

 , , , ,
, , , ,  ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Cell lines and Cell Culture
2.3. Irradiation
2.3.1. X-ray Photon Irradiation
2.3.2. Proton Irradiation
2.4. Colony Formation Assay
2.5. Immunofluorescence Staining
2.6. Superresolution STED Microscopy
2.7. Alkaline Single Cell Gel Electrophoresis (Comet) Assay
2.8. Statistical Analysis and Reproducibility
3. Results
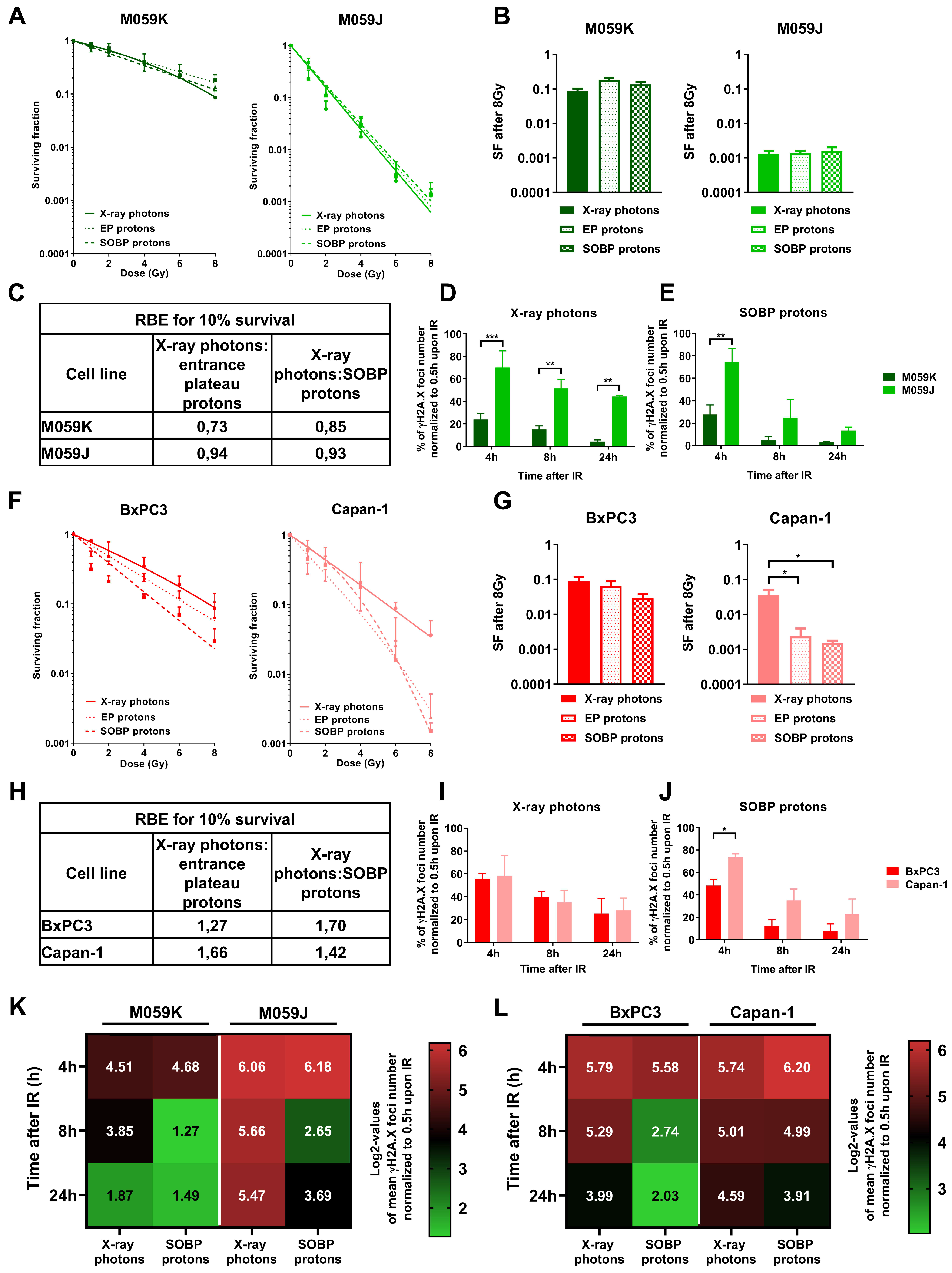
3.1. Proton Irradiation Induces Larger DNA repair γH2A.X Foci when Compared to Irradiation with X-ray Photons
3.2. Deficiency in Lig IV Strongly Sensitizes MEFs to Irradiation with Proton Beams and X-ray Photons
3.3. Lig IV is Essential for the Repair of DNA Damage Induced in MEFs by Irradiation with X-ray Photons and SOBP Protons
3.4. Impairment of NHEJ or HRR shows a Different Impact on Sensitivity of Cancer Cells to Irradiation with X-ray Photons or Protons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.M.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Prim. 2019, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.G.; Alexander, B.; Baumann, M.; Bratman, S.V.; Brown, J.M.; Camphausen, K.; Choyke, P.; Citrin, D.; Contessa, J.N.; Dicker, A.; et al. Combining precision radiotherapy with molecular targeting and immunomodulatory agents: A guideline by the American Society for Radiation Oncology. Lancet Oncol. 2018, 19, e240–e251. [Google Scholar] [CrossRef]
- Orth, M.; Lauber, K.; Niyazi, M.; Friedl, A.A.; Li, M.; Maihöfer, C.; Schüttrumpf, L.; Ernst, A.; Niemöller, O.M.; Belka, C. Current concepts in clinical radiation oncology. Radiat. Environ. Biophys. 2014, 53, 1–29. [Google Scholar] [CrossRef]
- Baumann, B.C.; Mitra, N.; Harton, J.; Xiao, Y.; Wojcieszynski, A.; Gabriel, P.E.; Zhong, H.; Geng, H.; Doucette, A.; Wei, J.J.; et al. Comparative effectiveness of proton therapy versus photon therapy as part of concurrent chemoradiotherapy for locally advanced cancer. J. Clin. Oncol. 2019, 37, 6521. [Google Scholar] [CrossRef]
- Durante, M.; Loeffler, J.S. Charged particles in radiation oncology. Nat. Rev. Clin. Oncol. 2010, 7, 37–43. [Google Scholar] [CrossRef]
- McDonald, M.W.; Zolali-Meybodi, O.; Lehnert, S.J.; Estabrook, N.C.; Liu, Y.; Cohen-Gadol, A.A.; Moore, M.G. Reirradiation of Recurrent and Second Primary Head and Neck Cancer With Proton Therapy. Int. J. Radiat. Oncol. 2016, 96, 808–819. [Google Scholar] [CrossRef]
- Rombi, B.; Vennarini, S.; Vinante, L.; Ravanelli, D.; Amichetti, M. Proton radiotherapy for pediatric tumors: Review of first clinical results. Ital. J. Pediatr. 2014, 40, 74. [Google Scholar] [CrossRef]
- Amichetti, M.; Cianchetti, M.; Amelio, D.; Enrici, R.M.; Minniti, G. Proton therapy in chordoma of the base of the skull: A systematic review. Neurosurg. Rev. 2009, 32, 403–416. [Google Scholar] [CrossRef]
- Tallen, G.; Resch, A.; Calaminus, G.; Wiener, A.; Leiss, U.; Pletschko, T.; Friedrich, C.; Langer, T.; Grabow, D.; Driever, P.H.; et al. Strategies to improve the quality of survival for childhood brain tumour survivors. Eur. J. Paediatr. Neurol. 2015, 19, 619–639. [Google Scholar] [CrossRef]
- Timmermann, B. Proton Beam Therapy for Childhood Malignancies: Status Report. Klin. Pädiatrie 2010, 222, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Tommasino, F.; Durante, M. Proton radiobiology. Cancers 2015, 7, 353–381. [Google Scholar] [CrossRef] [PubMed]
- Jones, B. Proton radiobiology and its clinical implications. Ecancermedicalscience 2017, 11, 777. [Google Scholar] [CrossRef] [PubMed]
- Oeck, S.; Szymonowicz, K.; Wiel, G.; Krysztofiak, A.; Lambert, J.; Koska, B.; Iliakis, G.; Timmermann, B.; Jendrossek, V. Relating Linear Energy Transfer to the Formation and Resolution of DNA Repair Foci After Irradiation with Equal Doses of X-ray Photons, Plateau, or Bragg-Peak Protons. Int. J. Mol. Sci. 2018, 19, 3779. [Google Scholar] [CrossRef]
- Jermann, M. Particle Therapy Statistics in 2014. Int. J. Part. Ther. 2015, 2, 50–54. [Google Scholar] [CrossRef]
- Montay-Gruel, P.; Meziani, L.; Yakkala, C.; Vozenin, M.C. Expanding the therapeutic index of radiation therapy by normal tissue protection. Br. J. Radiol. 2019, 92. [Google Scholar] [CrossRef]
- Nickoloff, J.A. Photon, light ion, and heavy ion cancer radiotherapy: Paths from physics and biology to clinical practice. Ann. Transl. Med. 2015, 3, 336. [Google Scholar]
- Paganetti, H. Relative biological effectiveness (RBE) values for proton beam therapy. Variations as a function of biological endpoint, dose, and linear energy transfer. Phys. Med. Biol. 2014, 59, R419–R472. [Google Scholar] [CrossRef]
- Paganetti, H.; Niemierko, A.; Ancukiewicz, M.; Gerweck, L.E.; Goitein, M.; Loeffler, J.S.; Suit, H.D. Relative biological effectiveness (RBE) values for proton beam therapy. Int. J. Radiat. Oncol. 2002, 53, 407–421. [Google Scholar] [CrossRef]
- Paganetti, H.; van Luijk, P. Biological Considerations When Comparing Proton Therapy With Photon Therapy. Semin. Radiat. Oncol. 2013, 23, 77–87. [Google Scholar] [CrossRef]
- Karger, C.P.; Peschke, P. RBE and related modeling in carbon-ion therapy. Phys. Med. Biol. 2018, 63. [Google Scholar] [CrossRef]
- Willers, H.; Allen, A.; Grosshans, D.; McMahon, S.J.; von Neubeck, C.; Wiese, C.; Vikram, B. Toward A variable RBE for proton beam therapy. Radiother. Oncol. 2018, 128, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Paganetti, H.; Olko, P.; Kobus, H.; Becker, R.; Schmitz, T.; Waligorski, M.P.R.; Filges, D.; Müller-Gärtner, H.-W. Calculation of relative biological effectiveness for proton beams using biological weighting functions. Int. J. Radiat. Oncol. 1997, 37, 719–729. [Google Scholar] [CrossRef]
- Scalliet, P.; Gueulette, J. Radiobiological Characterization of Clinical Proton and Carbon-Ion Beams. arXiv 2018, arXiv:1804.08500. [Google Scholar]
- Held, K.D.; Kawamura, H.; Kaminuma, T.; Paz, A.E.S.; Yoshida, Y.; Liu, Q.; Willers, H.; Takahashi, A. Effects of Charged Particles on Human Tumor Cells. Front. Oncol. 2016, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Lühr, A.; von Neubeck, C.; Krause, M.; Troost, E.G.C. Relative biological effectiveness in proton beam therapy – Current knowledge and future challenges. Clin. Transl. Radiat. Oncol. 2018, 9, 35–41. [Google Scholar] [CrossRef]
- Suzuki, M.; Kase, Y.; Yamaguchi, H.; Kanai, T.; Ando, K. Relative biological effectiveness for cell-killing effect on various human cell lines irradiated with heavy-ion medical accelerator in Chiba (HIMAC) carbon-ion beams. Int. J. Radiat. Oncol. 2000, 48, 241–250. [Google Scholar] [CrossRef]
- Grosse, N.; Fontana, A.O.; Hug, E.B.; Lomax, A.; Coray, A.; Augsburger, M.; Paganetti, H.; Sartori, A.A.; Pruschy, M. Deficiency in Homologous Recombination Renders Mammalian Cells More Sensitive to Proton Versus Photon Irradiation. Int. J. Radiat. Oncol. 2014, 88, 175–181. [Google Scholar] [CrossRef]
- Fontana, A.O.; Augsburger, M.A.; Grosse, N.; Guckenberger, M.; Lomax, A.J.; Sartori, A.A.; Pruschy, M.N. Differential DNA repair pathway choice in cancer cells after proton- and photon-irradiation. Radiother. Oncol. 2015, 116, 374–380. [Google Scholar] [CrossRef]
- Liu, Q.; Ghosh, P.; Magpayo, N.; Testa, M.; Tang, S.; Gheorghiu, L.; Biggs, P.; Paganetti, H.; Efstathiou, J.A.; Lu, H.-M.; et al. Lung Cancer Cell Line Screen Links Fanconi Anemia/BRCA Pathway Defects to Increased Relative Biological Effectiveness of Proton Radiation. Int. J. Radiat. Oncol. 2015, 91, 1081–1089. [Google Scholar] [CrossRef]
- Peeler, C.R.; Mirkovic, D.; Titt, U.; Blanchard, P.; Gunther, J.R.; Mahajan, A.; Mohan, R.; Grosshans, D.R. Clinical evidence of variable proton biological effectiveness in pediatric patients treated for ependymoma. Radiother. Oncol. 2016, 121, 395–401. [Google Scholar] [CrossRef]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA Double-Strand Break Repair as Determinant of Cellular Radiosensitivity to Killing and Target in Radiation Therapy. Front. Oncol. 2013, 3, 113. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2015, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Siemann, M.; Pantelias, G.E.; Iliakis, G. Marked contribution of alternative end-joining to chromosome-translocation-formation by stochastically induced DNA double-strand-breaks in G2-phase human cells. Mutat. Res. Toxicol. Environ. Mutagen. 2015, 793, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Critchlow, S.E.; Bowater, R.P.; Jackson, S.P. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol. 1997, 7, 588–598. [Google Scholar] [CrossRef]
- Andres, S.N.; Modesti, M.; Tsai, C.J.; Chu, G.; Junop, M.S. Crystal structure of human XLF: A twist in nonhomologous DNA end-joining. Mol. Cell 2007, 28, 1093–1101. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, B.P.C.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair (Amst). 2014, 17, 21–29. [Google Scholar] [CrossRef]
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V.; et al. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 2015, 347, 185–188. [Google Scholar] [CrossRef]
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of Homologous Recombination in Eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A.; Salmena, L. BRCA1 and BRCA2 mutations and breast cancer. Discov. Med. 2011, 12, 445–453. [Google Scholar] [PubMed]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Bothmer, A.; Robbiani, D.F.; Di Virgilio, M.; Bunting, S.F.; Klein, I.A.; Feldhahn, N.; Barlow, J.; Chen, H.-T.; Bosque, D.; Callen, E.; et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol. Cell 2011, 42, 319–329. [Google Scholar] [CrossRef]
- Gerelchuluun, A.; Manabe, E.; Ishikawa, T.; Sun, L.; Itoh, K.; Sakae, T.; Suzuki, K.; Hirayama, R.; Asaithamby, A.; Chen, D.J.; et al. The Major DNA Repair Pathway after Both Proton and Carbon-Ion Radiation is NHEJ, but the HR Pathway is More Relevant in Carbon Ions. Radiat. Res. 2015, 183, 345–356. [Google Scholar] [CrossRef]
- Yang, S.-H.; Kuo, T.-C.; Wu, H.; Guo, J.-C.; Hsu, C.; Hsu, C.-H.; Tien, Y.-W.; Yeh, K.-H.; Cheng, A.-L.; Kuo, S.-H. Perspectives on the combination of radiotherapy and targeted therapy with DNA repair inhibitors in the treatment of pancreatic cancer. World J. Gastroenterol. 2016, 22, 7275. [Google Scholar] [CrossRef]
- Biau, J.; Chautard, E.; Verrelle, P.; Dutreix, M. Altering DNA Repair to Improve Radiation Therapy: Specific and Multiple Pathway Targeting. Front. Oncol. 2019, 9, 1009. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Asaithamby, A. Repurposing DNA repair factors to eradicate tumor cells upon radiotherapy. Transl. Cancer Res. 2017, 6, S822–S839. [Google Scholar] [CrossRef]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Hinshaw, H.D.; Jalal, S.I.; Sears, C.R.; Pawelczak, K.S.; Turchi, J.J. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol. Ther. 2016, 160, 65–83. [Google Scholar] [CrossRef]
- DiBiase, S.J.; Zeng, Z.C.; Chen, R.; Hyslop, T.; Curran, W.J.; Iliakis, G. DNA-dependent protein kinase stimulates an independently active, nonhomologous, end-joining apparatus. Cancer Res. 2000, 60, 1245–1253. [Google Scholar]
- Abbott, D.W.; Holt, J.T.; Freeman, M.L. Double-Strand Break Repair Deficiency and Radiation Sensitivity in BRCA2 Mutant Cancer Cells. JNCI J. Natl. Cancer Inst. 1998, 90, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Riballo, E.; Woodbine, L.; Stiff, T.; Walker, S.A.; Goodarzi, A.A.; Jeggo, P.A. XLF-Cernunnos promotes DNA ligase IV–XRCC4 re-adenylation following ligation. Nucleic Acids Res. 2009, 37, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.J. The linear quadratic model: Usage, interpretation and challenges. Phys. Med. Biol. 2018, 64, 01TR01. [Google Scholar] [CrossRef] [PubMed]
- Staab, A.; Zukowski, D.; Walenta, S.; Scholz, M.; Mueller-Klieser, W. Response of Chinese Hamster V79 Multicellular Spheroids Exposed to High-Energy Carbon Ions. Radiat. Res. 2004, 161, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Oeck, S.; Malewicz, N.M.; Hurst, S.; Rudner, J.; Jendrossek, V. The Focinator—A new open-source tool for high-throughput foci evaluation of DNA damage. Radiat. Oncol. 2015, 10, 163. [Google Scholar] [CrossRef]
- Oeck, S.; Malewicz, N.M.; Hurst, S.; Al-Refae, K.; Krysztofiak, A.; Jendrossek, V. The Focinator v2-0—Graphical Interface, Four Channels, Colocalization Analysis and Cell Phase Identification. Radiat. Res. 2017, 000, RR14746.1. [Google Scholar] [CrossRef]
- Staudt, T.; Lang, M.C.; Medda, R.; Engelhardt, J.; Hell, S.W. 2,2′-Thiodiethanol: A new water soluble mounting medium for high resolution optical microscopy. Microsc. Res. Tech. 2007, 70, 1–9. [Google Scholar] [CrossRef]
- Baddeley, A.; Rubak, E.; Turner, R. Spatial Point Patterns: Methodology and Applications with R; Chapman & Hall/CRC: London, UK, 2015; ISBN 978-1-4822-1020-0. [Google Scholar]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef]
- Gyori, B.M.; Venkatachalam, G.; Thiagarajan, P.S.; Hsu, D.; Clement, M.-V. OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2014, 2, 457–465. [Google Scholar] [CrossRef]
- Krämer, M.; Weyrather, W.K.; Scholz, M. The Increased Biological Effectiveness of Heavy Charged Particles: From Radiobiology to Treatment Planning. Technol. Cancer Res. Treat. 2003, 2, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Hojo, H.; Dohmae, T.; Hotta, K.; Kohno, R.; Motegi, A.; Yagishita, A.; Makinoshima, H.; Tsuchihara, K.; Akimoto, T. Difference in the relative biological effectiveness and DNA damage repair processes in response to proton beam therapy according to the positions of the spread out Bragg peak. Radiat. Oncol. 2017, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Reindl, J.; Girst, S.; Walsh, D.W.M.; Greubel, C.; Schwarz, B.; Siebenwirth, C.; Drexler, G.A.; Friedl, A.A.; Dollinger, G. Chromatin organization revealed by nanostructure of irradiation induced γH2AX, 53BP1 and Rad51 foci. Sci. Rep. 2017, 7, 40616. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.; Marshall, T.I.; Currell, F.J.; Kacperek, A.; Schettino, G.; Prise, K.M. Variations in the Processing of DNA Double-Strand Breaks Along 60-MeV Therapeutic Proton Beams. Int. J. Radiat. Oncol. 2016, 95, 86–94. [Google Scholar] [CrossRef]
- Henthorn, N.T.; Warmenhoven, J.W.; Sotiropoulos, M.; Aitkenhead, A.H.; Smith, E.A.K.; Ingram, S.P.; Kirkby, N.F.; Chadwick, A.L.; Burnet, N.G.; Mackay, R.I.; et al. Clinically relevant nanodosimetric simulation of DNA damage complexity from photons and protons. RSC Adv. 2019, 9, 6845–6858. [Google Scholar] [CrossRef]
- Ray, S.; Cekanaviciute, E.; Lima, I.P.; Sørensen, B.S.; Costes, S.V. Comparing Photon and Charged Particle Therapy Using DNA Damage Biomarkers. Int. J. Part. Ther. 2018, 5, 15–24. [Google Scholar] [CrossRef]
- Maeda, K.; Yasui, H.; Yamamori, T.; Matsuura, T.; Takao, S.; Suzuki, M.; Matsuda, A.; Inanami, O.; Shirato, H. A Nucleoside Anticancer Drug, 1-(3-C-Ethynyl-β-D-Ribo-Pentofuranosyl)Cytosine, Induces Depth-Dependent Enhancement of Tumor Cell Death in Spread-Out Bragg Peak (SOBP) of Proton Beam. PLoS ONE 2016, 11, e0166848. [Google Scholar] [CrossRef]
- Timm, S.; Lorat, Y.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered DNA damage concentrated in particle trajectories causes persistent large-scale rearrangements in chromatin architecture. Radiother. Oncol. 2018. [Google Scholar] [CrossRef]
- Szabó, E.R.; Brand, M.; Hans, S.; Hideghéty, K.; Karsch, L.; Lessmann, E.; Pawelke, J.; Schürer, M.; Beyreuther, E. Radiobiological effects and proton RBE determined by wildtype zebrafish embryos. PLoS ONE 2018, 13, e0206879. [Google Scholar] [CrossRef]
- Wu, W.; Wang, M.; Wu, W.; Singh, S.K.; Mussfeldt, T.; Iliakis, G. Repair of radiation induced DNA double strand breaks by backup NHEJ is enhanced in G2. DNA Repair (Amst). 2008, 7, 329–338. [Google Scholar] [CrossRef]
- Iliakis, G.; Wang, H.; Perrault, A.R.; Boecker, W.; Rosidi, B.; Windhofer, F.; Wu, W.; Guan, J.; Terzoudi, G.; Pantelias, G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet. Genome Res. 2004, 104, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair (Amst). 2015, 32, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Mladenova, V.; Sharif, M.; Chaudhary, S.; Mavragani, I.V.; Soni, A.; Saha, J.; Schipler, A.; Mladenov, E. Defined Biological Models of High-Let Radiation Lesions. Radiat. Prot. Dosimetry 2019, 183, 60–68. [Google Scholar] [CrossRef]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA Damage Response Factors from Diverse Pathways, Including DNA Crosslink Repair, Mediate Alternative End Joining. PLoS Genet. 2015, 11, 1–25. [Google Scholar] [CrossRef]
- Bright, S.J.; Flint, D.B.; Chakraborty, S.; McFadden, C.H.; Yoon, D.S.; Bronk, L.; Titt, U.; Mohan, R.; Grosshans, D.R.; Sumazin, P.; et al. Nonhomologous End Joining Is More Important Than Proton Linear Energy Transfer in Dictating Cell Death. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 1119–1125. [Google Scholar] [CrossRef]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef]
- Paganetti, H.; Giantsoudi, D. Relative Biological Effectiveness Uncertainties and Implications for Beam Arrangements and Dose Constraints in Proton Therapy. Semin. Radiat. Oncol. 2018, 28, 256–263. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymonowicz, K.; Krysztofiak, A.; van der Linden, J.; Kern, A.; Deycmar, S.; Oeck, S.; Squire, A.; Koska, B.; Hlouschek, J.; Vüllings, M.; et al. Proton Irradiation Increases the Necessity for Homologous Recombination Repair Along with the Indispensability of Non-Homologous End Joining. Cells 2020, 9, 889. https://doi.org/10.3390/cells9040889
Szymonowicz K, Krysztofiak A, van der Linden J, Kern A, Deycmar S, Oeck S, Squire A, Koska B, Hlouschek J, Vüllings M, et al. Proton Irradiation Increases the Necessity for Homologous Recombination Repair Along with the Indispensability of Non-Homologous End Joining. Cells. 2020; 9(4):889. https://doi.org/10.3390/cells9040889
Chicago/Turabian StyleSzymonowicz, Klaudia, Adam Krysztofiak, Jansje van der Linden, Ajvar Kern, Simon Deycmar, Sebastian Oeck, Anthony Squire, Benjamin Koska, Julian Hlouschek, Melanie Vüllings, and et al. 2020. "Proton Irradiation Increases the Necessity for Homologous Recombination Repair Along with the Indispensability of Non-Homologous End Joining" Cells 9, no. 4: 889. https://doi.org/10.3390/cells9040889
APA StyleSzymonowicz, K., Krysztofiak, A., van der Linden, J., Kern, A., Deycmar, S., Oeck, S., Squire, A., Koska, B., Hlouschek, J., Vüllings, M., Neander, C., Siveke, J. T., Matschke, J., Pruschy, M., Timmermann, B., & Jendrossek, V. (2020). Proton Irradiation Increases the Necessity for Homologous Recombination Repair Along with the Indispensability of Non-Homologous End Joining. Cells, 9(4), 889. https://doi.org/10.3390/cells9040889