Sphingosine Kinase 1 Regulates the Survival of Breast Cancer Stem Cells and Non-stem Breast Cancer Cells by Suppression of STAT1

 , , ,
, , ,
Abstract
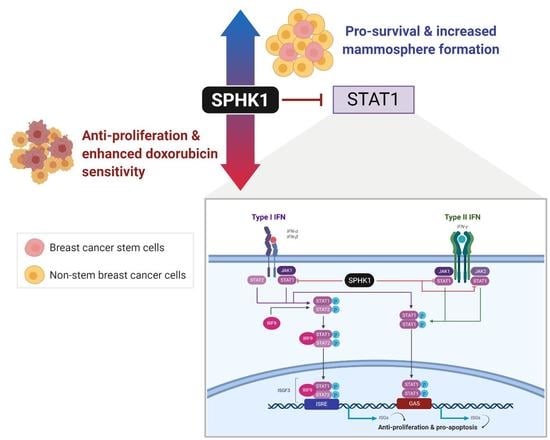
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Immunoblotting
2.3. S1P ELISA
2.4. Lentivirus Production and Transduction
2.5. Cell Proliferation Assay
2.6. Caspase Activation Assay
2.7. Proteomic Profiling
2.8. ISRE and GAS Luciferase Reporter Assay
2.9. Apoptosis Assay
3. Results
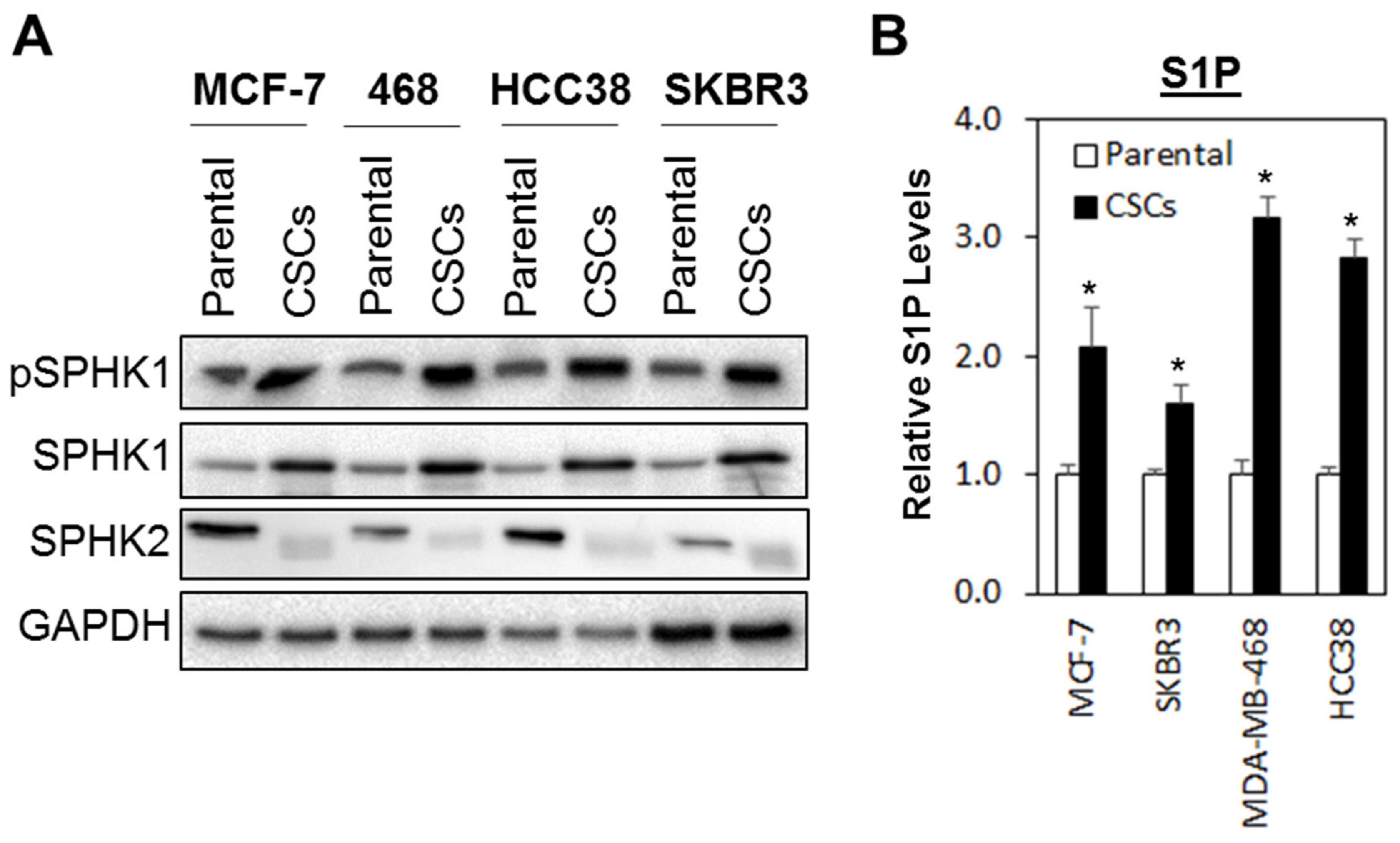
3.1. The SPHK1-S1P Axis Is Hyperactivated in Breast CSCs
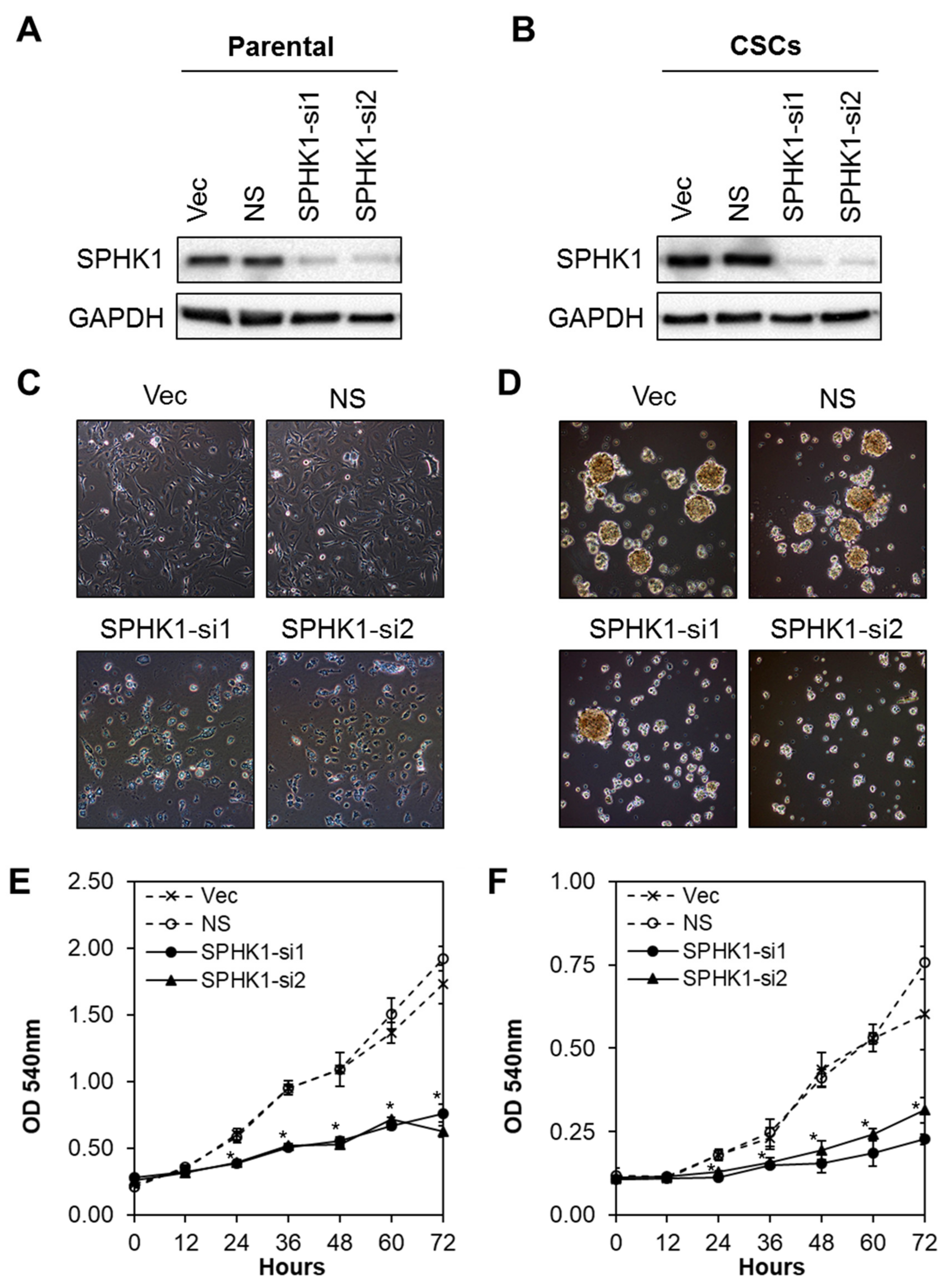
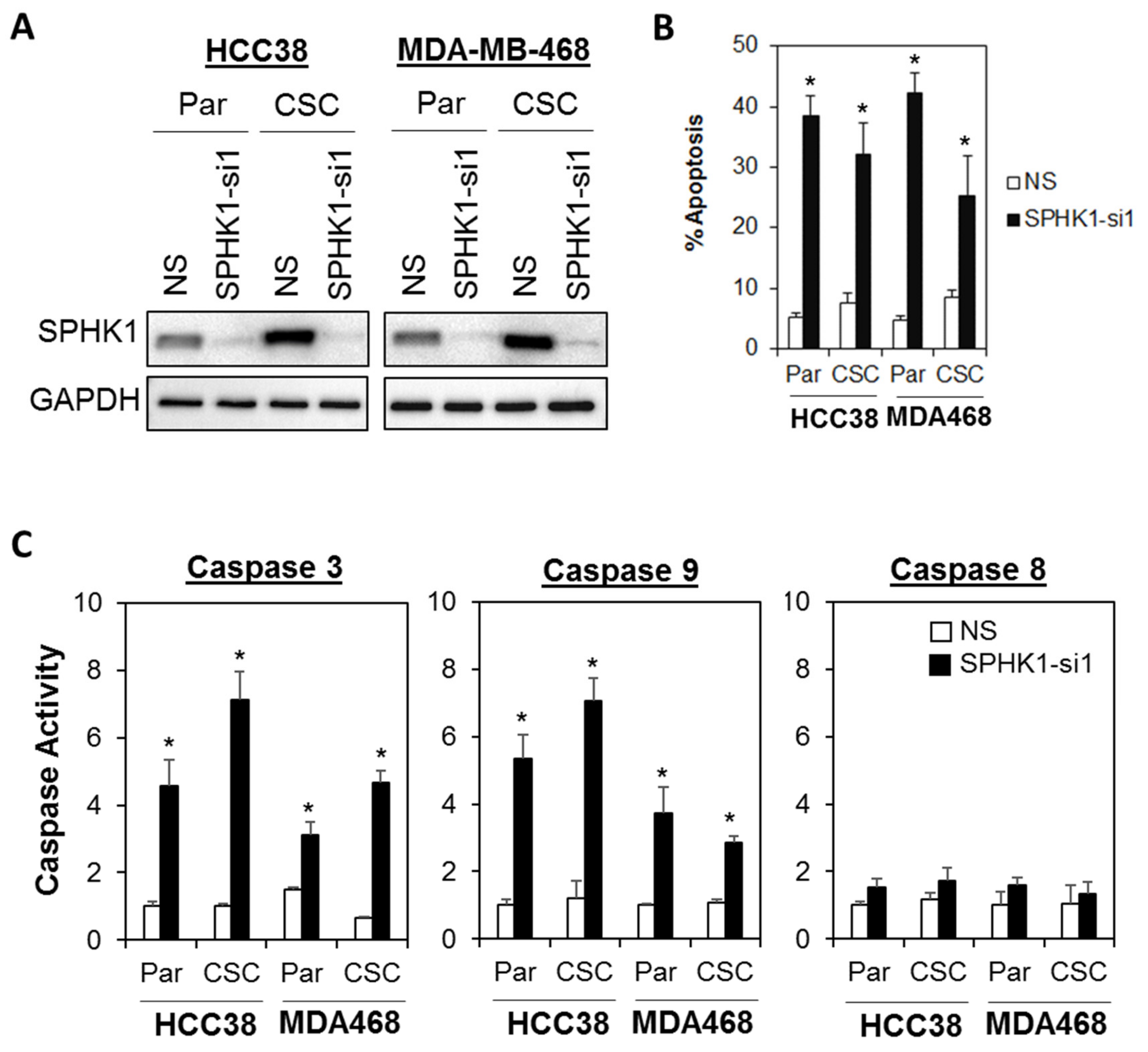
3.2. Depletion of SPHK1 Inhibits Breast CSCs’ and Non-CSCs’ Survival and Proliferation
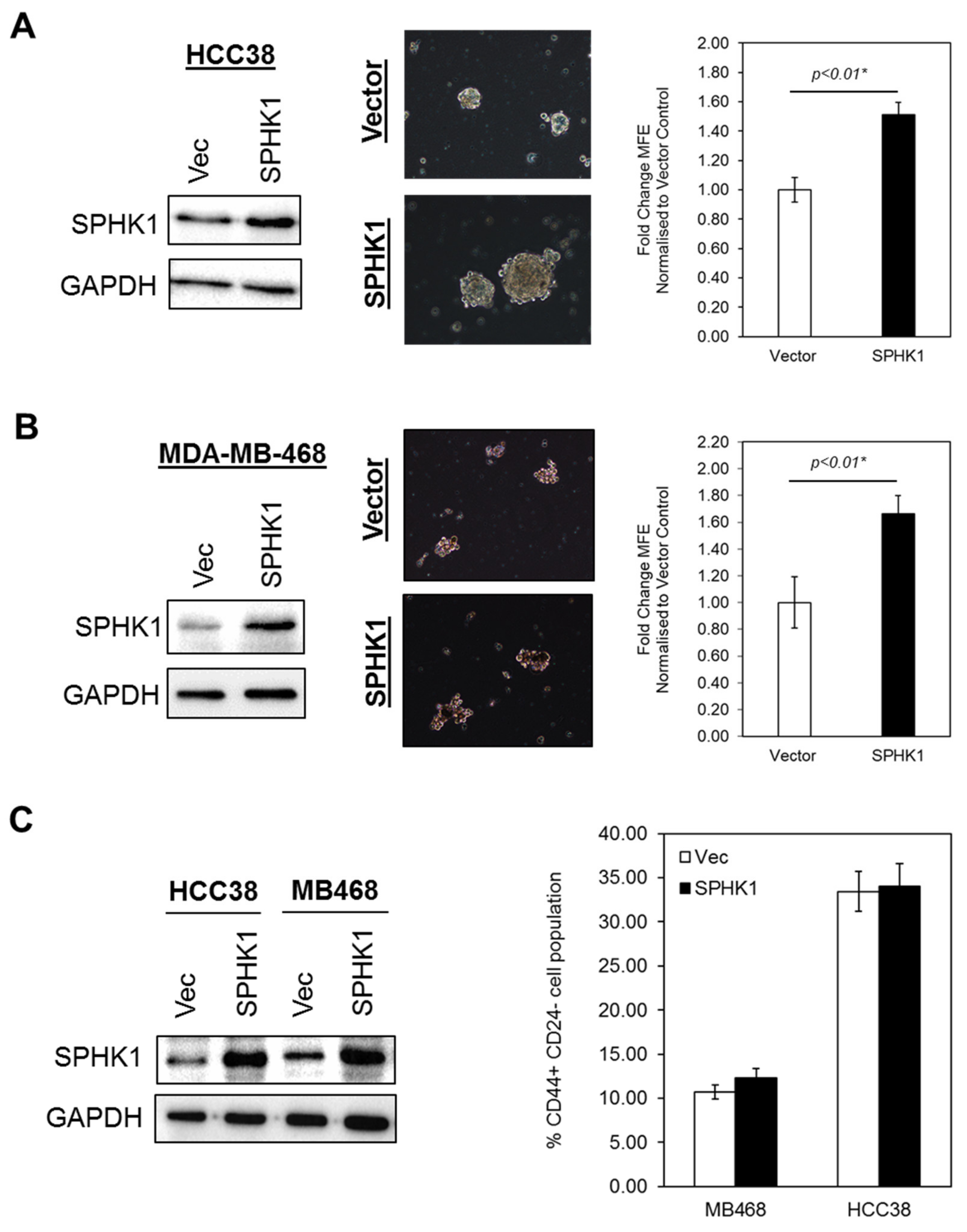
3.3. Ectopic Expression of SPHK1 Enhances Breast CSCs’ Survival and Mammosphere Forming Efficiency, but not Cell Fate Transition
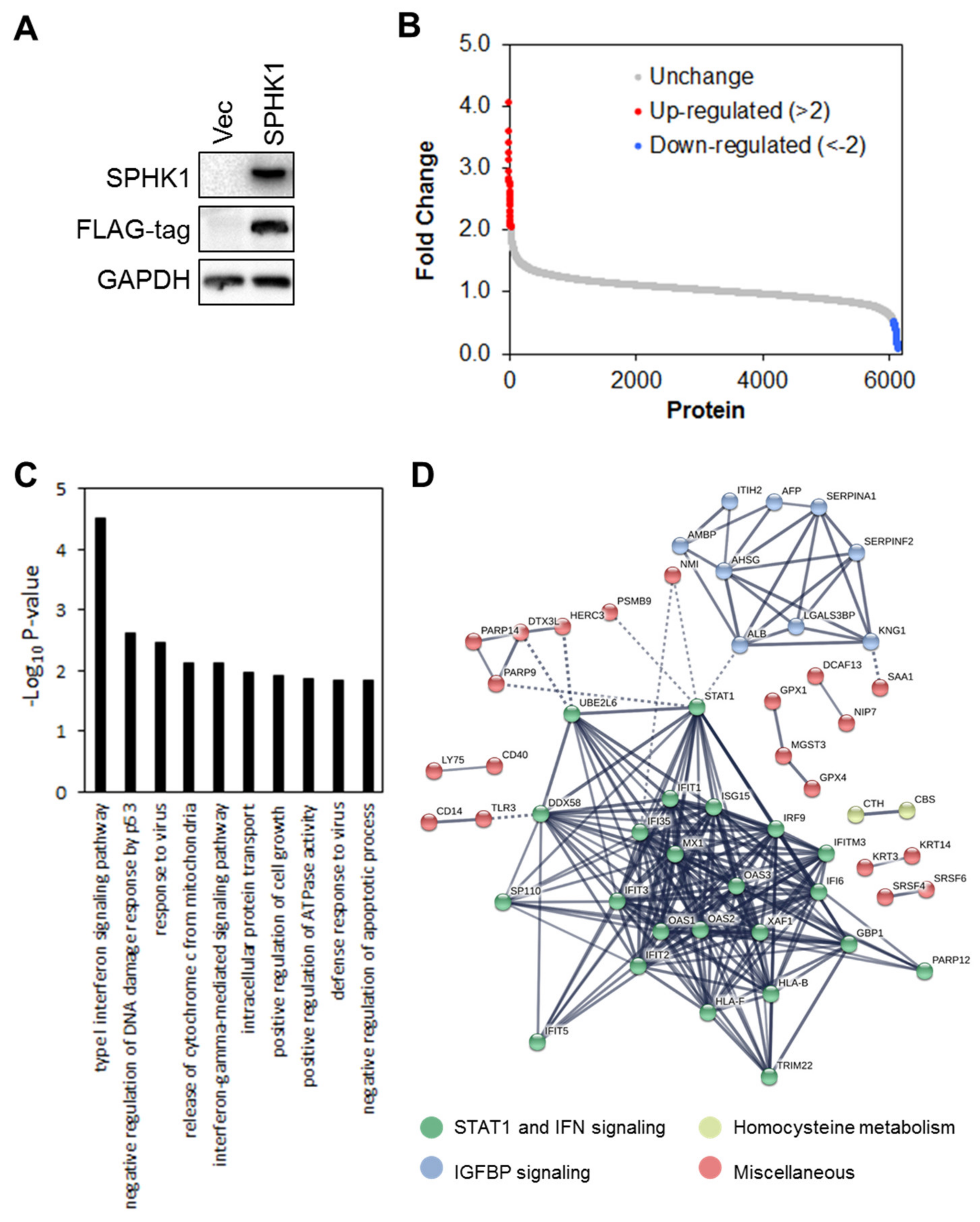
3.4. Proteomic Profiling Identifies STAT1 as the Major Target Regulated by SPHK1
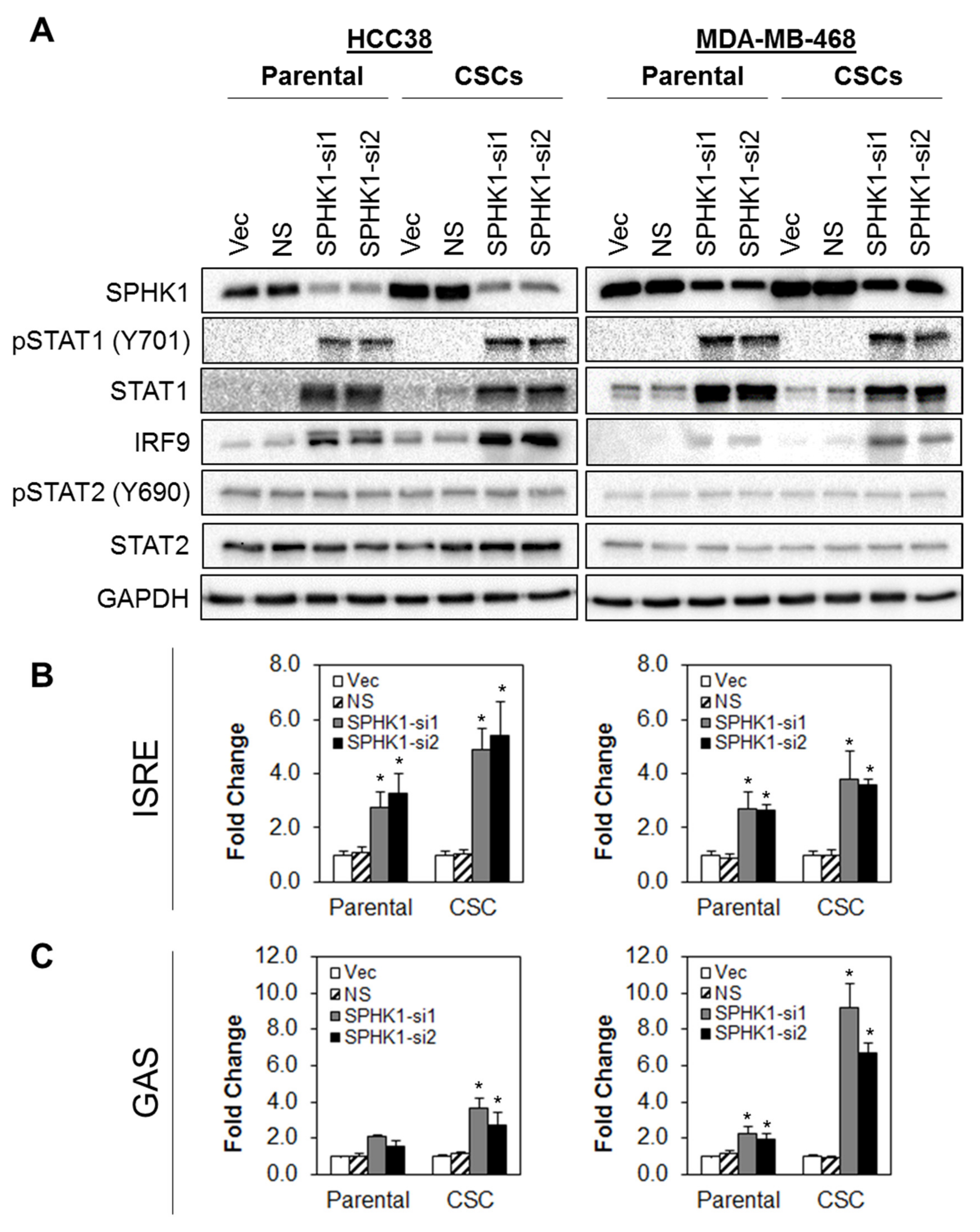
3.5. Depletion of SPHK1 Induces STAT1 and Activates Type I and II Interferon Signalling
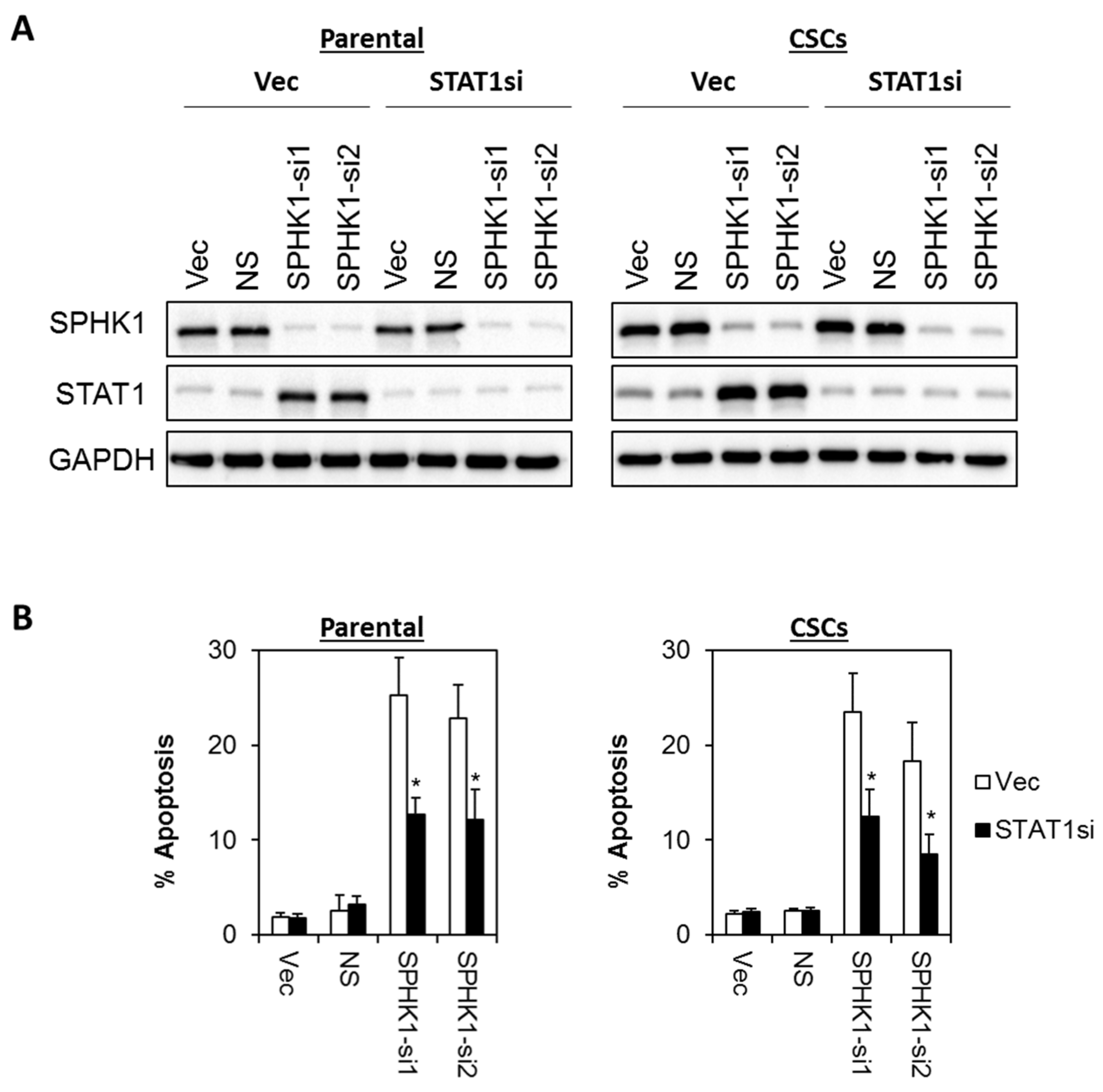
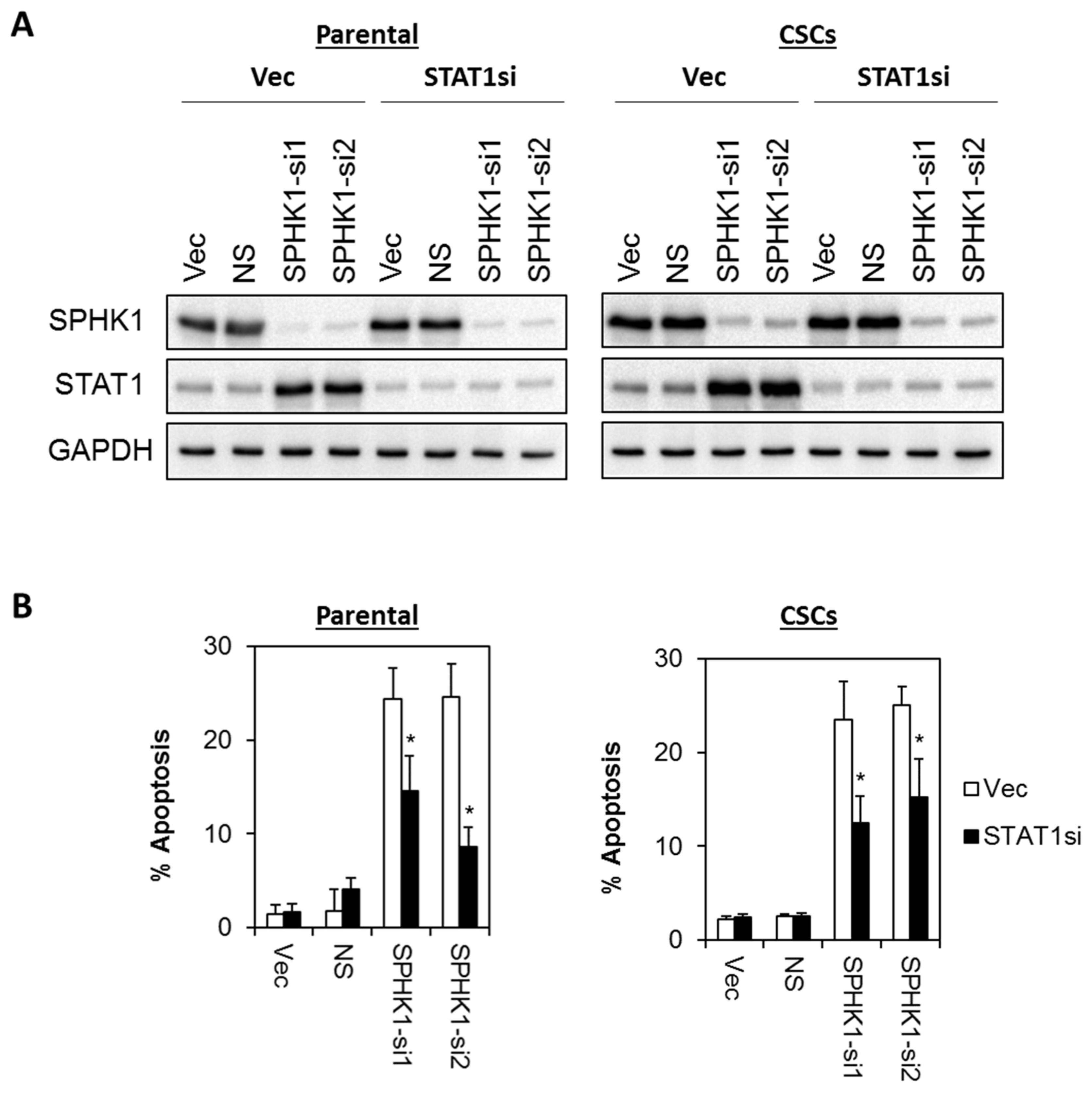
3.6. Apoptosis Induced by SPHK1 Depletion Is Mediated through STAT1
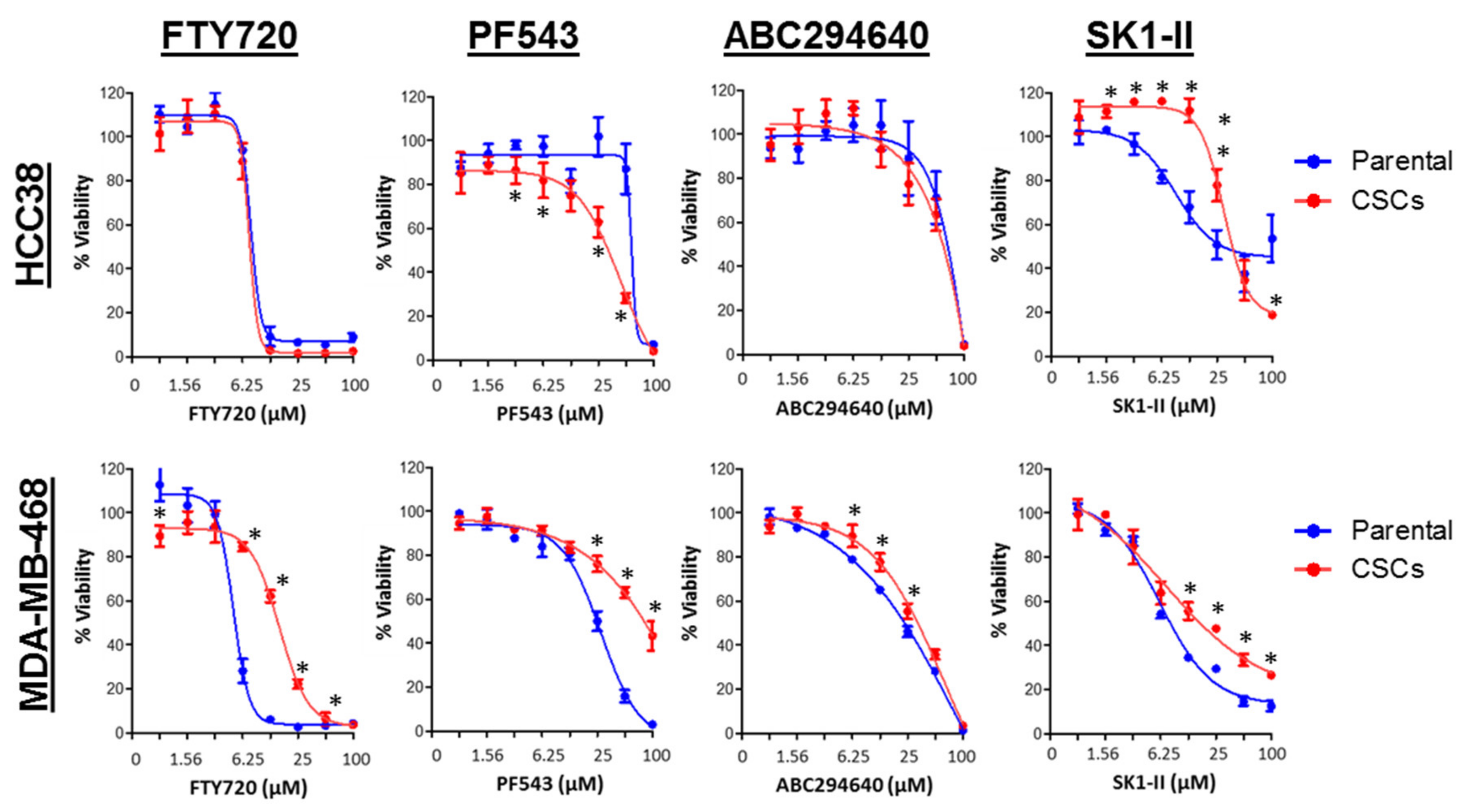
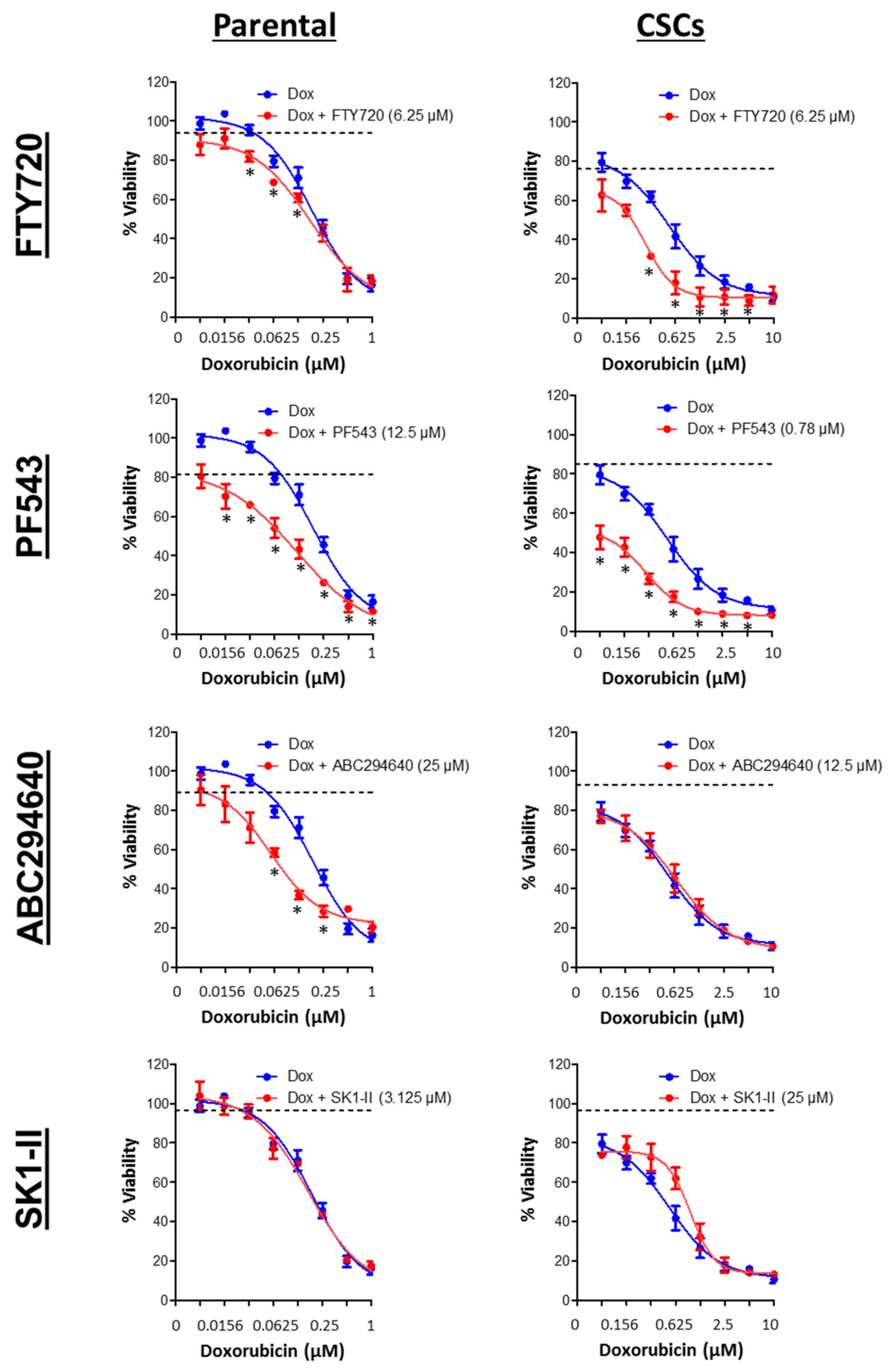
3.7. SPHK1 Inhibitors Enhance Doxorubicin Sensitivity in Basal-Like Breast CSCs and Non-CSCs.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Barbato, L.; Bocchetti, M.; Di Biase, A.; Regad, T. Cancer Stem Cells and Targeting Strategies. Cells 2019, 8. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell. Signal. 2013, 25, 961–969. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.M.F.; Vermeulen, L. Wnt Signaling in Cancer Stem Cell Biology. Cancers 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.B.; Hong, I.S.; Kim, R.J.; Lee, S.Y.; Park, S.J.; Lee, E.S.; Park, J.H.; Yun, C.H.; Chung, J.U.; Lee, K.J.; et al. Wnt/beta-Catenin Small-Molecule Inhibitor CWP232228 Preferentially Inhibits the Growth of Breast Cancer Stem-like Cells. Cancer Res. 2015, 75, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y.; Hong, I.S.; Nam, J.S. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef]
- Farnie, G.; Clarke, R.B. Mammary stem cells and breast cancer--role of Notch signalling. Stem Cell Rev. 2007, 3, 169–175. [Google Scholar] [CrossRef]
- D’Angelo, R.C.; Ouzounova, M.; Davis, A.; Choi, D.; Tchuenkam, S.M.; Kim, G.; Luther, T.; Quraishi, A.A.; Senbabaoglu, Y.; Conley, S.J.; et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol. Cancer Ther. 2015, 14, 779–787. [Google Scholar] [CrossRef]
- Zhao, D.; Mo, Y.; Li, M.T.; Zou, S.W.; Cheng, Z.L.; Sun, Y.P.; Xiong, Y.; Guan, K.L.; Lei, Q.Y. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J. Clin. Investig. 2014, 124, 5453–5465. [Google Scholar] [CrossRef]
- Suman, S.; Das, T.P.; Damodaran, C. Silencing NOTCH signaling causes growth arrest in both breast cancer stem cells and breast cancer cells. Br. J. Cancer 2013, 109, 2587–2596. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Reviews. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.H.; Lai, A.G. Aberrations in Notch-Hedgehog signalling reveal cancer stem cells harbouring conserved oncogenic properties associated with hypoxia and immunoevasion. Br. J. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef] [PubMed]
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells 2018, 7. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Reviews. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kim, J.H.; Kim, S.L.; Lee, D.S. Disruption of the NF-kappaB/IL-8 Signaling Axis by Sulconazole Inhibits Human Breast Cancer Stem Cell Formation. Cells 2019, 8. [Google Scholar] [CrossRef]
- Shostak, K.; Chariot, A. NF-kappaB, stem cells and breast cancer: The links get stronger. Breast Cancer Res 2011, 13, 214. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, H.; Gu, P.; Bai, J.; Margolick, J.B.; Zhang, Y. NF-kappaB pathway inhibitors preferentially inhibit breast cancer stem-like cells. Breast Cancer Res. Treat. 2008, 111, 419–427. [Google Scholar] [CrossRef]
- Liu, S.; Wicha, M.S. Targeting breast cancer stem cells. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 4006–4012. [Google Scholar] [CrossRef]
- Clarke, M.F. Clinical and Therapeutic Implications of Cancer Stem Cells. N. Engl. J. Med. 2019, 380, 2237–2245. [Google Scholar] [CrossRef] [PubMed]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Gentile, P.; Fabbri, G.; Cervelli, V.; Orlandi, A. The Role of Breast Cancer Stem Cells as a Prognostic Marker and a Target to Improve the Efficacy of Breast Cancer Therapy. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Choi, J.H.; Nam, J.S. Targeting Cancer Stem Cells in Triple-Negative Breast Cancer. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; El Buri, A.; Adams, D.R.; Pyne, S. Sphingosine 1-phosphate and cancer. Adv. Biol. Regul. 2018, 68, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Pyne, S.; Pyne, N.J. Translational aspects of sphingosine 1-phosphate biology. Trends Mol. Med. 2011, 17, 463–472. [Google Scholar] [CrossRef]
- Pyne, S.; Adams, D.R.; Pyne, N.J. Sphingosine 1-phosphate and sphingosine kinases in health and disease: Recent advances. Prog. Lipid Res. 2016, 62, 93–106. [Google Scholar] [CrossRef]
- Pyne, N.J.; McNaughton, M.; Boomkamp, S.; MacRitchie, N.; Evangelisti, C.; Martelli, A.M.; Jiang, H.R.; Ubhi, S.; Pyne, S. Role of sphingosine 1-phosphate receptors, sphingosine kinases and sphingosine in cancer and inflammation. Adv. Biol. Regul. 2016, 60, 151–159. [Google Scholar] [CrossRef]
- Fang, Z.; Pyne, S.; Pyne, N.J. Ceramide and sphingosine 1-phosphate in adipose dysfunction. Prog. Lipid Res. 2019, 74, 145–159. [Google Scholar] [CrossRef]
- Pyne, N.J.; Ohotski, J.; Bittman, R.; Pyne, S. The role of sphingosine 1-phosphate in inflammation and cancer. Adv. Biol. Regul. 2014, 54, 121–129. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate is a missing link between chronic inflammation and colon cancer. Cancer Cell 2013, 23, 5–7. [Google Scholar] [CrossRef]
- Pyne, S.; Chapman, J.; Steele, L.; Pyne, N.J. Sphingomyelin-derived lipids differentially regulate the extracellular signal-regulated kinase 2 (ERK-2) and c-Jun N-terminal kinase (JNK) signal cascades in airway smooth muscle. Eur. J. Biochem. 1996, 237, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Pyne, S.; Edwards, J.; Ohotski, J.; Pyne, N.J. Sphingosine 1-phosphate receptors and sphingosine kinase 1: Novel biomarkers for clinical prognosis in breast, prostate, and hematological cancers. Front. Oncol. 2012, 2, 168. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Tonelli, F.; Lim, K.G.; Long, J.S.; Edwards, J.; Pyne, S. Sphingosine 1-phosphate signalling in cancer. Biochem. Soc. Trans. 2012, 40, 94–100. [Google Scholar] [CrossRef]
- Pyne, N.J.; Tonelli, F.; Lim, K.G.; Long, J.; Edwards, J.; Pyne, S. Targeting sphingosine kinase 1 in cancer. Adv. Biol. Regul. 2012, 52, 31–38. [Google Scholar] [CrossRef]
- Pyne, S.; Bittman, R.; Pyne, N.J. Sphingosine kinase inhibitors and cancer: Seeking the golden sword of Hercules. Cancer Res. 2011, 71, 6576–6582. [Google Scholar] [CrossRef]
- Song, L.; Xiong, H.; Li, J.; Liao, W.; Wang, L.; Wu, J.; Li, M. Sphingosine kinase-1 enhances resistance to apoptosis through activation of PI3K/Akt/NF-kappaB pathway in human non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 1839–1849. [Google Scholar] [CrossRef]
- Acharya, S.; Yao, J.; Li, P.; Zhang, C.; Lowery, F.J.; Zhang, Q.; Guo, H.; Qu, J.; Yang, F.; Wistuba, I.I.; et al. Sphingosine Kinase 1 Signaling Promotes Metastasis of Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 4211–4226. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Reviews. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef]
- Long, J.S.; Edwards, J.; Watson, C.; Tovey, S.; Mair, K.M.; Schiff, R.; Natarajan, V.; Pyne, N.J.; Pyne, S. Sphingosine kinase 1 induces tolerance to human epidermal growth factor receptor 2 and prevents formation of a migratory phenotype in response to sphingosine 1-phosphate in estrogen receptor-positive breast cancer cells. Mol. Cell. Biol. 2010, 30, 3827–3841. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Loo, S.Y.; Huang, B.; Wong, L.; Tan, S.S.; Tan, T.Z.; Lee, S.C.; Thiery, J.P.; Lim, Y.C.; Yong, W.P.; et al. SPHK1 regulates proliferation and survival responses in triple-negative breast cancer. Oncotarget 2014, 5, 5920–5933. [Google Scholar] [CrossRef] [PubMed]
- Ruckhaberle, E.; Rody, A.; Engels, K.; Gaetje, R.; von Minckwitz, G.; Schiffmann, S.; Grosch, S.; Geisslinger, G.; Holtrich, U.; Karn, T.; et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res. Treat. 2008, 112, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; You, H.; Tan, J.X.; Li, F.; Qiu, Z.; Li, H.Z.; Huang, H.Y.; Zheng, K.; Ren, G.S. Overexpression of sphingosine kinase 1 is predictive of poor prognosis in human breast cancer. Oncol. Lett. 2017, 14, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Takabe, K.; Hait, N.C. Metastatic triple-negative breast cancer is dependent on SphKs/S1P signaling for growth and survival. Cell. Signal. 2017, 32, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.; Long, J.S.; Orange, C.; Tannahill, C.L.; Mallon, E.; McGlynn, L.M.; Pyne, S.; Pyne, N.J.; Edwards, J. High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am. J. Pathol. 2010, 177, 2205–2215. [Google Scholar] [CrossRef]
- Ohotski, J.; Edwards, J.; Elsberger, B.; Watson, C.; Orange, C.; Mallon, E.; Pyne, S.; Pyne, N.J. Identification of novel functional and spatial associations between sphingosine kinase 1, sphingosine 1-phosphate receptors and other signaling proteins that affect prognostic outcome in estrogen receptor-positive breast cancer. Int. J. Cancer 2013, 132, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Ohotski, J.; Long, J.S.; Orange, C.; Elsberger, B.; Mallon, E.; Doughty, J.; Pyne, S.; Pyne, N.J.; Edwards, J. Expression of sphingosine 1-phosphate receptor 4 and sphingosine kinase 1 is associated with outcome in oestrogen receptor-negative breast cancer. Br. J. Cancer 2012, 106, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Hirata, N.; Yamada, S.; Shoda, T.; Kurihara, M.; Sekino, Y.; Kanda, Y. Sphingosine-1-phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand-independent Notch activation. Nat. Commun. 2014, 5, 4806. [Google Scholar] [CrossRef]
- Wang, Y.C.; Tsai, C.F.; Chuang, H.L.; Chang, Y.C.; Chen, H.S.; Lee, J.N.; Tsai, E.M. Benzyl butyl phthalate promotes breast cancer stem cell expansion via SPHK1/S1P/S1PR3 signaling. Oncotarget 2016, 7, 29563–29576. [Google Scholar] [CrossRef]
- Tiong, K.H.; Tan, B.S.; Choo, H.L.; Chung, F.F.; Hii, L.W.; Tan, S.H.; Khor, N.T.; Wong, S.F.; See, S.J.; Tan, Y.F.; et al. Fibroblast growth factor receptor 4 (FGFR4) and fibroblast growth factor 19 (FGF19) autocrine enhance breast cancer cells survival. Oncotarget 2016, 7, 57633–57650. [Google Scholar] [CrossRef] [PubMed]
- Soo, J.S.; Ng, C.H.; Tan, S.H.; Malik, R.A.; Teh, Y.C.; Tan, B.S.; Ho, G.F.; See, M.H.; Taib, N.A.; Yip, C.H.; et al. Metformin synergizes 5-fluorouracil, epirubicin, and cyclophosphamide (FEC) combination therapy through impairing intracellular ATP production and DNA repair in breast cancer stem cells. Apoptosis Int. J. Program. Cell Death 2015, 20, 1371–1387. [Google Scholar] [CrossRef]
- Fillmore, C.M.; Kuperwasser, C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Hii, L.W.; Chung, F.F.; Soo, J.S.; Tan, B.S.; Mai, C.W.; Leong, C.O. Histone deacetylase (HDAC) inhibitors and doxorubicin combinations target both breast cancer stem cells and non-stem breast cancer cells simultaneously. Breast Cancer Res. Treat. 2019. [Google Scholar] [CrossRef]
- Soo, H.C.; Chung, F.F.; Lim, K.H.; Yap, V.A.; Bradshaw, T.D.; Hii, L.W.; Tan, S.H.; See, S.J.; Tan, Y.F.; Leong, C.O.; et al. Cudraflavone C Induces Tumor-Specific Apoptosis in Colorectal Cancer Cells through Inhibition of the Phosphoinositide 3-Kinase (PI3K)-AKT Pathway. PLoS ONE 2017, 12, e0170551. [Google Scholar] [CrossRef]
- Er, J.L.; Goh, P.N.; Lee, C.Y.; Tan, Y.J.; Hii, L.W.; Mai, C.W.; Chung, F.F.; Leong, C.O. Identification of inhibitors synergizing gemcitabine sensitivity in the squamous subtype of pancreatic ductal adenocarcinoma (PDAC). Apoptosis Int. J. Program. Cell Death 2018, 23, 343–355. [Google Scholar] [CrossRef]
- Chung, F.F.; Tan, P.F.; Raja, V.J.; Tan, B.S.; Lim, K.H.; Kam, T.S.; Hii, L.W.; Tan, S.H.; See, S.J.; Tan, Y.F.; et al. Jerantinine A induces tumor-specific cell death through modulation of splicing factor 3b subunit 1 (SF3B1). Sci. Rep. 2017, 7, 42504. [Google Scholar] [CrossRef]
- Stone, E.L.; Citossi, F.; Singh, R.; Kaur, B.; Gaskell, M.; Farmer, P.B.; Monks, A.; Hose, C.; Stevens, M.F.; Leong, C.O.; et al. Antitumour benzothiazoles. Part 32: DNA adducts and double strand breaks correlate with activity; synthesis of 5F203 hydrogels for local delivery. Bioorganic Med. Chem. 2015, 23, 6891–6899. [Google Scholar] [CrossRef]
- Huang, F.K.; Zhang, G.; Lawlor, K.; Nazarian, A.; Philip, J.; Tempst, P.; Dephoure, N.; Neubert, T.A. Deep Coverage of Global Protein Expression and Phosphorylation in Breast Tumor Cell Lines Using TMT 10-plex Isobaric Labeling. J. Proteome Res. 2017, 16, 1121–1132. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Functions of the multifaceted family of sphingosine kinases and some close relatives. J. Biol. Chem. 2007, 282, 2125–2129. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Ding, G.; Sonoda, H.; Kajimoto, T.; Haga, Y.; Khosrowbeygi, A.; Gao, S.; Miwa, N.; Jahangeer, S.; Nakamura, S. Involvement of N-terminal-extended form of sphingosine kinase 2 in serum-dependent regulation of cell proliferation and apoptosis. J. Biol. Chem. 2005, 280, 36318–36325. [Google Scholar] [CrossRef] [PubMed]
- Blick, T.; Hugo, H.; Widodo, E.; Waltham, M.; Pinto, C.; Mani, S.A.; Weinberg, R.A.; Neve, R.M.; Lenburg, M.E.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 235–252. [Google Scholar] [CrossRef]
- Meissl, K.; Macho-Maschler, S.; Muller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.R.; Vermi, W.; Luo, J.; Lucini, L.; Rickert, C.; Fowler, A.M.; Lonardi, S.; Arthur, C.; Young, L.J.; Levy, D.E.; et al. STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast Cancer Res. 2012, 14, R16. [Google Scholar] [CrossRef]
- Klover, P.J.; Muller, W.J.; Robinson, G.W.; Pfeiffer, R.M.; Yamaji, D.; Hennighausen, L. Loss of STAT1 from mouse mammary epithelium results in an increased Neu-induced tumor burden. Neoplasia 2010, 12, 899–905. [Google Scholar] [CrossRef]
- Raven, J.F.; Williams, V.; Wang, S.; Tremblay, M.L.; Muller, W.J.; Durbin, J.E.; Koromilas, A.E. Stat1 is a suppressor of ErbB2/Neu-mediated cellular transformation and mouse mammary gland tumor formation. Cell Cycle 2011, 10, 794–804. [Google Scholar] [CrossRef]
- Mogensen, T.H. IRF and STAT Transcription Factors From Basic Biology to Roles in Infection, Protective Immunity, and Primary Immunodeficiencies. Front. Immunol. 2018, 9, 3047. [Google Scholar] [CrossRef]
- Lewis, A.C.; Wallington-Beddoe, C.T.; Powell, J.A.; Pitson, S.M. Targeting sphingolipid metabolism as an approach for combination therapies in haematological malignancies. Cell Death Discov. 2018, 4, 4. [Google Scholar] [CrossRef]
- Tonelli, F.; Lim, K.G.; Loveridge, C.; Long, J.; Pitson, S.M.; Tigyi, G.; Bittman, R.; Pyne, S.; Pyne, N.J. FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen-independent prostate cancer cells. Cell. Signal. 2010, 22, 1536–1542. [Google Scholar] [CrossRef]
- Hla, T.; Brinkmann, V. Sphingosine 1-phosphate (S1P): Physiology and the effects of S1P receptor modulation. Neurology 2011, 76, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef] [PubMed]
- French, K.J.; Zhuang, Y.; Maines, L.W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J.J.; Green, C.L.; Keller, S.N.; Smith, C.D. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J. Pharmacol. Exp. Ther. 2010, 333, 129–139. [Google Scholar] [CrossRef] [PubMed]
- McNaughton, M.; Pitman, M.; Pitson, S.M.; Pyne, N.J.; Pyne, S. Proteasomal degradation of sphingosine kinase 1 and inhibition of dihydroceramide desaturase by the sphingosine kinase inhibitors, SKi or ABC294640, induces growth arrest in androgen-independent LNCaP-AI prostate cancer cells. Oncotarget 2016, 7, 16663–16675. [Google Scholar] [CrossRef] [PubMed]
- Alsanafi, M.; Kelly, S.L.; Jubair, K.; McNaughton, M.; Tate, R.J.; Merrill, A.H., Jr.; Pyne, S.; Pyne, N.J. Native and Polyubiquitinated Forms of Dihydroceramide Desaturase Are Differentially Linked to Human Embryonic Kidney Cell Survival. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Studer, S.; Leaf, N.; Kiosses, W.B.; Nguyen, N.; Matsuki, K.; Negishi, H.; Taniguchi, T.; Oldstone, M.B.; Rosen, H. S1PR1-mediated IFNAR1 degradation modulates plasmacytoid dendritic cell interferon-alpha autoamplification. Proc. Natl. Acad. Sci. USA 2016, 113, 1351–1356. [Google Scholar] [CrossRef]
- Yester, J.W.; Bryan, L.; Waters, M.R.; Mierzenski, B.; Biswas, D.D.; Gupta, A.S.; Bhardwaj, R.; Surace, M.J.; Eltit, J.M.; Milstien, S.; et al. Sphingosine-1-phosphate inhibits IL-1-induced expression of C-C motif ligand 5 via c-Fos-dependent suppression of IFN-beta amplification loop. Faseb J. 2015, 29, 4853–4865. [Google Scholar] [CrossRef]
- Murakami, M.; Ito, H.; Hagiwara, K.; Kobayashi, M.; Hoshikawa, A.; Takagi, A.; Kojima, T.; Tamiya-Koizumi, K.; Sobue, S.; Ichihara, M.; et al. Sphingosine kinase 1/S1P pathway involvement in the GDNF-induced GAP43 transcription. J. Cell. Biochem. 2011, 112, 3449–3458. [Google Scholar] [CrossRef]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef]
- Haricharan, S.; Li, Y. STAT signaling in mammary gland differentiation, cell survival and tumorigenesis. Mol. Cell. Endocrinol. 2014, 382, 560–569. [Google Scholar] [CrossRef]
- Bailey, S.G.; Cragg, M.S.; Townsend, P.A. Role of STAT1 in the breast. Jak-Stat 2012, 1, 197–199. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Koromilas, A.E.; Sexl, V. The tumor suppressor function of STAT1 in breast cancer. Jak-Stat 2013, 2, e23353. [Google Scholar] [CrossRef] [PubMed]
- Baran-Marszak, F.; Feuillard, J.; Najjar, I.; Le Clorennec, C.; Bechet, J.M.; Dusanter-Fourt, I.; Bornkamm, G.W.; Raphael, M.; Fagard, R. Differential roles of STAT1alpha and STAT1beta in fludarabine-induced cell cycle arrest and apoptosis in human B cells. Blood 2004, 104, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Townsend, P.A.; Scarabelli, T.M.; Davidson, S.M.; Knight, R.A.; Latchman, D.S.; Stephanou, A. STAT-1 interacts with p53 to enhance DNA damage-induced apoptosis. J. Biol. Chem. 2004, 279, 5811–5820. [Google Scholar] [CrossRef]
- Powell, J.A.; Pitman, M.R.; Zebol, J.R.; Moretti, P.A.B.; Neubauer, H.A.; Davies, L.T.; Lewis, A.C.; Dagley, L.F.; Webb, A.I.; Costabile, M.; et al. Kelch-like protein 5-mediated ubiquitination of lysine 183 promotes proteasomal degradation of sphingosine kinase 1. Biochem. J. 2019, 476, 3211–3226. [Google Scholar] [CrossRef]
- Taha, T.A.; Osta, W.; Kozhaya, L.; Bielawski, J.; Johnson, K.R.; Gillanders, W.E.; Dbaibo, G.S.; Hannun, Y.A.; Obeid, L.M. Down-regulation of sphingosine kinase-1 by DNA damage: Dependence on proteases and p53. J. Biol. Chem. 2004, 279, 20546–20554. [Google Scholar] [CrossRef]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef]
- Gao, P.; Smith, C.D. Ablation of sphingosine kinase-2 inhibits tumor cell proliferation and migration. Mol. Cancer Res. 2011, 9, 1509–1519. [Google Scholar] [CrossRef]
- Schrecengost, R.S.; Keller, S.N.; Schiewer, M.J.; Knudsen, K.E.; Smith, C.D. Downregulation of Critical Oncogenes by the Selective SK2 Inhibitor ABC294640 Hinders Prostate Cancer Progression. Mol. Cancer Res. 2015, 13, 1591–1601. [Google Scholar] [CrossRef]
- Wallington-Beddoe, C.T.; Bennett, M.K.; Vandyke, K.; Davies, L.; Zebol, J.R.; Moretti, P.A.B.; Pitman, M.R.; Hewett, D.R.; Zannettino, A.C.W.; Pitson, S.M. Sphingosine kinase 2 inhibition synergises with bortezomib to target myeloma by enhancing endoplasmic reticulum stress. Oncotarget 2017, 8, 43602–43616. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
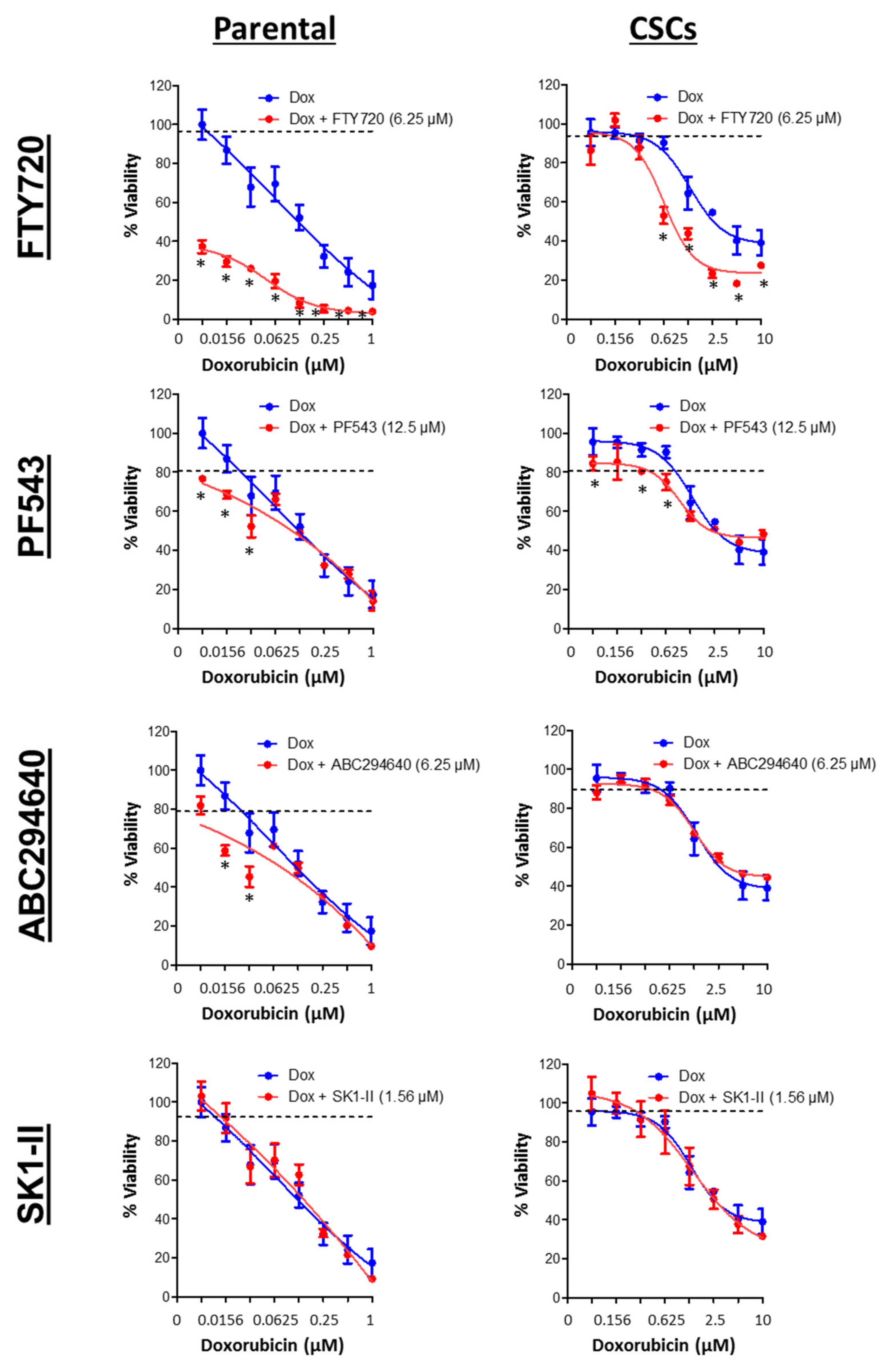{kind=link}
| Drugs | HCC38 | MDA-MB-468 | ||
|---|---|---|---|---|
| Parental | CSCs | Parental | CSCs | |
| Doxorubicin (µM) | 0.20 ± 0.02 | 0.59 ± 0.14 | 0.15 ± 0.04 | 2.22 ± 1.03 |
| FTY720 (µM) | 9.50 ± 0.13 | 9.04 ± 0.33 | 5.30 ± 0.14 | 16.29 ± 0.81 |
| PF543 (µM) | 72.86 ± 4.68 | 33.89 ± 3.67 | 21.55 ± 7.67 | 85.98 ± 15.70 |
| ABC294640 (µM) | 65.20 ± 6.42 | 60.85 ± 5.10 | 22.57 ± 1.02 | 31.56 ± 3.01 |
| SK1-II (µM) | 29.70 ± 11.56 | 41.36 ± 4.73 | 7.58 ± 0.52 | 15.72 ± 5.21 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hii, L.-W.; Chung, F.F.-L.; Mai, C.W.; Yee, Z.Y.; Chan, H.H.; Raja, V.J.; Dephoure, N.E.; Pyne, N.J.; Pyne, S.; Leong, C.-O. Sphingosine Kinase 1 Regulates the Survival of Breast Cancer Stem Cells and Non-stem Breast Cancer Cells by Suppression of STAT1. Cells 2020, 9, 886. https://doi.org/10.3390/cells9040886
Hii L-W, Chung FF-L, Mai CW, Yee ZY, Chan HH, Raja VJ, Dephoure NE, Pyne NJ, Pyne S, Leong C-O. Sphingosine Kinase 1 Regulates the Survival of Breast Cancer Stem Cells and Non-stem Breast Cancer Cells by Suppression of STAT1. Cells. 2020; 9(4):886. https://doi.org/10.3390/cells9040886
Chicago/Turabian StyleHii, Ling-Wei, Felicia Fei-Lei Chung, Chun Wai Mai, Zong Yang Yee, Hong Hao Chan, Vijay Joseph Raja, Noah Elias Dephoure, Nigel J. Pyne, Susan Pyne, and Chee-Onn Leong. 2020. "Sphingosine Kinase 1 Regulates the Survival of Breast Cancer Stem Cells and Non-stem Breast Cancer Cells by Suppression of STAT1" Cells 9, no. 4: 886. https://doi.org/10.3390/cells9040886
APA StyleHii, L.-W., Chung, F. F.-L., Mai, C. W., Yee, Z. Y., Chan, H. H., Raja, V. J., Dephoure, N. E., Pyne, N. J., Pyne, S., & Leong, C.-O. (2020). Sphingosine Kinase 1 Regulates the Survival of Breast Cancer Stem Cells and Non-stem Breast Cancer Cells by Suppression of STAT1. Cells, 9(4), 886. https://doi.org/10.3390/cells9040886