Lamin A/C Assembly Defects in LMNA-Congenital Muscular Dystrophy Is Responsible for the Increased Severity of the Disease Compared with Emery–Dreifuss Muscular Dystrophy

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Material and Methods
2.1. The UMD-LMNA Locus Specific Database
2.2. Cells
2.2.1. Human Primary Fibroblasts
2.2.2. Mouse Primary Myoblasts
2.2.3. Lentiviral Vector Production and Transduction
2.3. Drug Treatments
2.4. Immunofluorescence
2.5. Western Blotting
2.6. Quantitative-RT-PCR
2.7. Statistical Analyses
3. Results
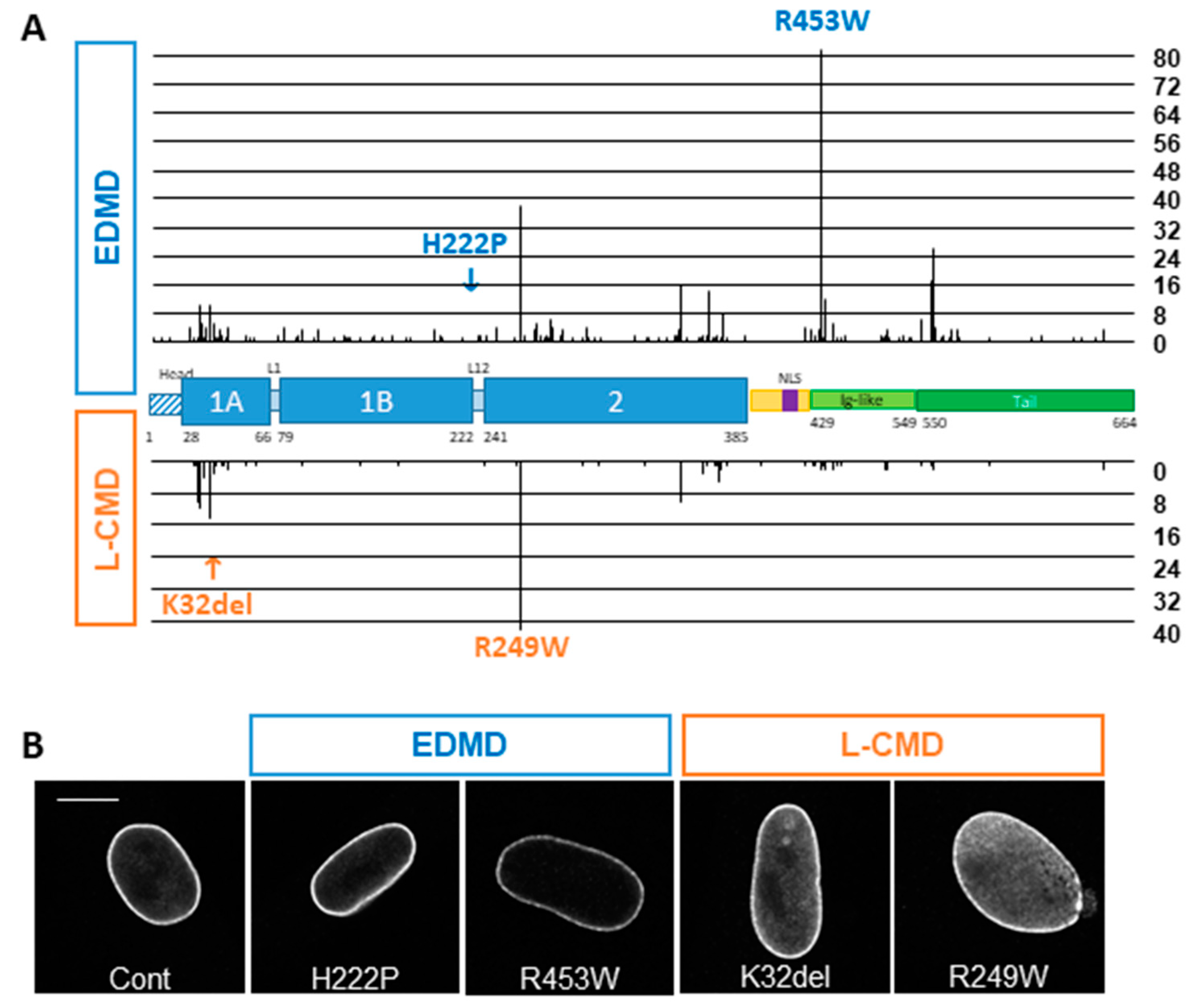
3.1. In Silico Analysis of the UMD-LMNA Database
3.2. Phenotype of EDMD and L-CMD Cells in Culture
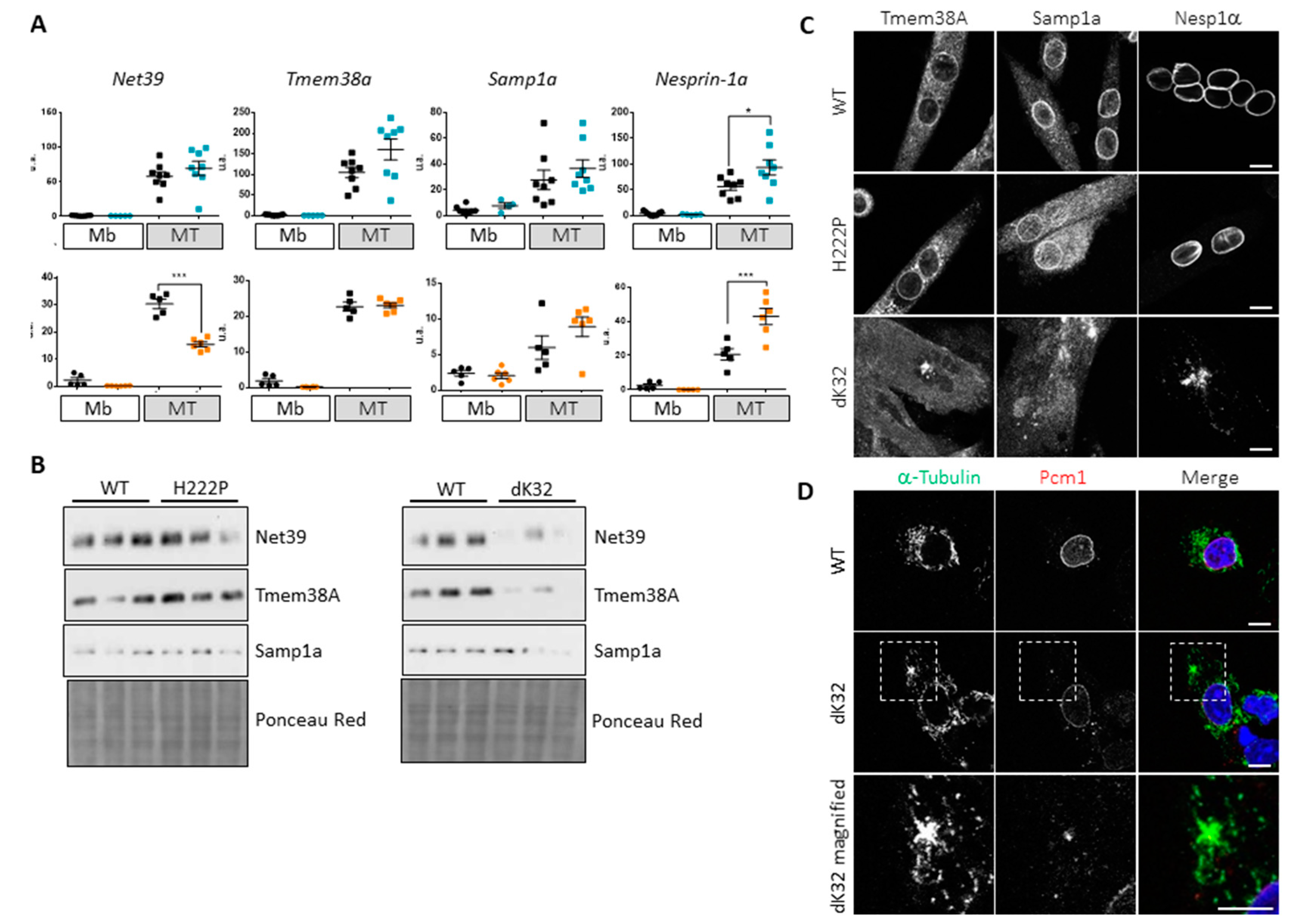
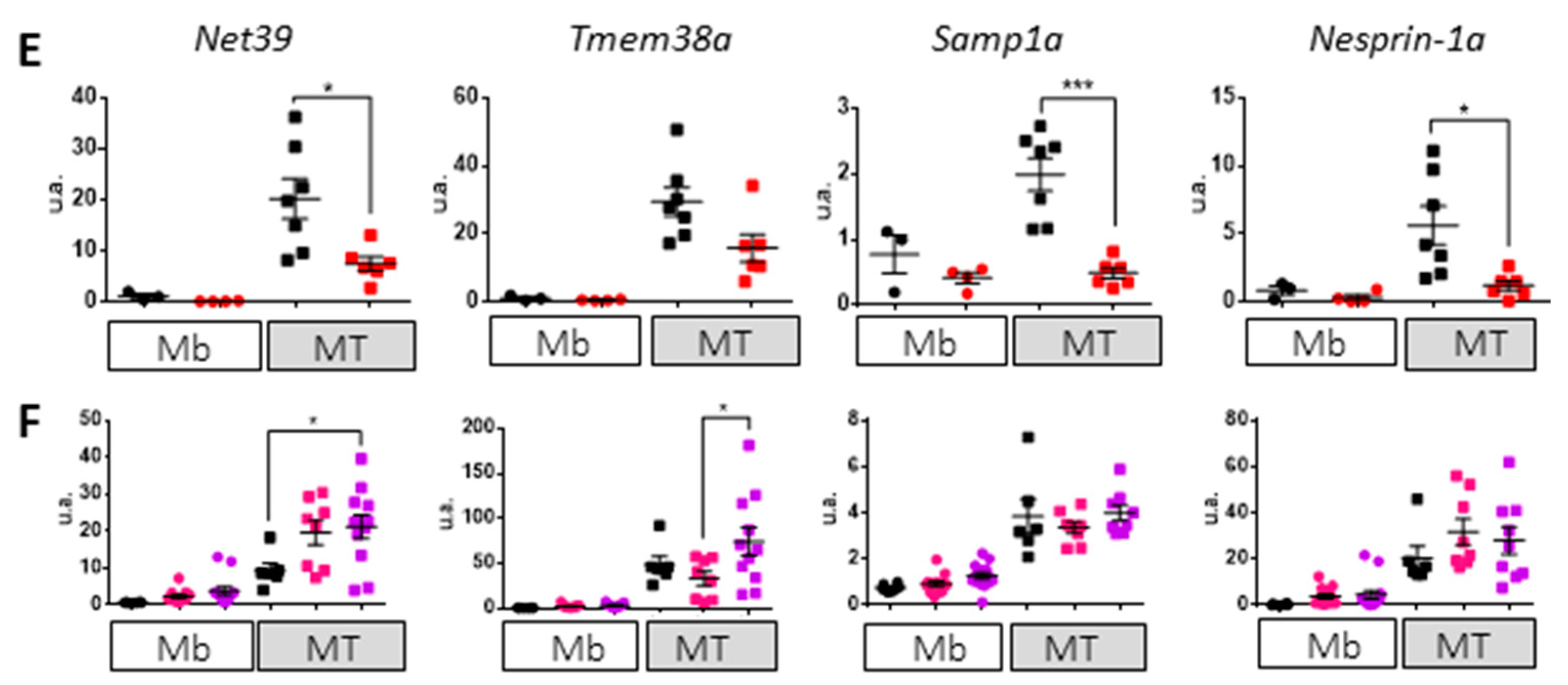
3.3. Impact of Lamin A/C Mislocalization in Myotubes
4. Discussion
4.1. L-CMD Mutations Strongly Affect Lamin A/C Properties
4.2. The Importance of Polymerized Lamin A/C for Chromatin Organization during Myoblast Differentiation
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schirmer, E.C.; Florens, L.; Guan, T.; Yates, J.R., 3rd; Gerace, L. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science 2003, 301, 1380–1382. [Google Scholar] [CrossRef]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Structure and Assembly. Cold Spring Harb Perspect Biol. 2016, 8. [Google Scholar] [CrossRef]
- Makarov, A.A.; Zou, J.; Houston, D.R.; Spanos, C.; Solovyova, A.S.; Cardenal-Peralta, C.; Rappsilber, J.; Schirmer, E.C. Lamin A molecular compression and sliding as mechanisms behind nucleoskeleton elasticity. Nat. Commun. 2019, 10, 3056. [Google Scholar] [CrossRef]
- Navarro, C.L.; Cau, P.; Levy, N. Molecular bases of progeroid syndromes. Hum. Mol. Genet. 2006, 15, R151–R161. [Google Scholar] [CrossRef]
- Torvaldson, E.; Kochin, V.; Eriksson, J.E. Phosphorylation of lamins determine their structural properties and signaling functions. Nucleus 2015, 6, 166–171. [Google Scholar] [CrossRef]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type Lamin Expression Compromises Nuclear Envelope Integrity Leading to Muscular Dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef]
- Thanisch, K.; Song, C.; Engelkamp, D.; Koch, J.; Wang, A.; Hallberg, E.; Foisner, R.; Leonhardt, H.; Stewart, C.L.; Joffe, B.; et al. Nuclear envelope localization of LEMD2 is developmentally dynamic and lamin A/C dependent yet insufficient for heterochromatin tethering. Differentiation 2017, 94, 58–70. [Google Scholar] [CrossRef]
- Malik, P.; Korfali, N.; Srsen, V.; Lazou, V.; Batrakou, D.G.; Zuleger, N.; Kavanagh, D.M.; Wilkie, G.S.; Goldberg, M.W.; Schirmer, E.C. Cell-specific and lamin-dependent targeting of novel transmembrane proteins in the nuclear envelope. Cell Mol. Life Sci. 2010, 67, 1353–1369. [Google Scholar] [CrossRef]
- Libotte, T.; Zaim, H.; Abraham, S.; Padmakumar, V.C.; Schneider, M.; Lu, W.; Munck, M.; Hutchison, C.; Wehnert, M.; Fahrenkrog, B.; et al. Lamin A/C-dependent localization of Nesprin-2, a giant scaffolder at the nuclear envelope. Mol. Biol. Cell 2005, 16, 3411–3424. [Google Scholar] [CrossRef]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef]
- Ungricht, R.; Kutay, U. Establishment of NE asymmetry-targeting of membrane proteins to the inner nuclear membrane. Curr. Opin. Cell Biol. 2015, 34, 135–141. [Google Scholar] [CrossRef]
- Andres, V.; Gonzalez, J.M. Role of A-type lamins in signaling, transcription, and chromatin organization. J. Cell Biol. 2009, 187, 945–957. [Google Scholar] [CrossRef]
- Parnaik, V.K. Role of nuclear lamins in nuclear organization, cellular signaling, and inherited diseases. Int. Rev. Cell Mol. Biol. 2008, 266, 157–206. [Google Scholar]
- Broers, J.; Ramaekers, F.; Bonne, G.; Ben Yaou, R.; Hutchison, C. The nuclear lamins: Laminopathies and their role in premature ageing. Physiol. Rev. 2006, 86, 967–1008. [Google Scholar] [CrossRef]
- Bertrand, A.T.; Chikhaoui, K.; Yaou, R.B.; Bonne, G. Clinical and genetic heterogeneity in laminopathies. Biochem. Soc. Trans. 2011, 39, 1687–1692. [Google Scholar] [CrossRef]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtiziberea, A.; Becane, H.M.; Reca, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Brull, A.; Morales Rodriguez, B.; Bonne, G.; Muchir, A.; Bertrand, A.T. The Pathogenesis and Therapies of Striated Muscle Laminopathies. Front. Physiol. 2018, 9, 1533. [Google Scholar] [CrossRef]
- Arimura, T.; Helbling-Leclerc, A.; Massart, C.; Varnous, S.; Niel, F.; Lacene, E.; Fromes, Y.; Toussaint, M.; Mura, A.M.; Keller, D.I.; et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 2005, 14, 155–169. [Google Scholar] [CrossRef]
- Bertrand, A.T.; Renou, L.; Papadopoulos, A.; Beuvin, M.; Lacene, E.; Massart, C.; Ottolenghi, C.; Decostre, V.; Maron, S.; Schlossarek, S.; et al. DelK32-lamin A/C has abnormal location and induces incomplete tissue maturation and severe metabolic defects leading to premature death. Hum. Mol. Genet. 2012, 21, 1037–1048. [Google Scholar] [CrossRef]
- Beroud, C.; Hamroun, D.; Collod-Beroud, G.; Boileau, C.; Soussi, T.; Claustres, M. UMD (Universal Mutation Database): 2005 update. Hum. Mutat. 2005, 26, 184–191. [Google Scholar] [CrossRef]
- Quijano-Roy, S.; Mbieleu, B.; Bonnemann, C.G.; Jeannet, P.Y.; Colomer, J.; Clarke, N.F.; Cuisset, J.M.; Roper, H.; De Meirleir, L.; D’Amico, A.; et al. De novo lmna mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 2008, 64, 177–186. [Google Scholar] [CrossRef]
- Zwerger, M.; Roschitzki-Voser, H.; Zbinden, R.; Denais, C.; Herrmann, H.; Lammerding, J.; Grutter, M.G.; Medalia, O. Altering lamina assembly reveals lamina-dependent and -independent functions for A-type lamins. J. Cell Sci. 2015, 128, 3607–3620. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Azzouz, M.; Deglon, N.; Aebischer, P.; Zurn, A.D. Complete and long-term rescue of lesioned adult motoneurons by lentiviral-mediated expression of glial cell line-derived neurotrophic factor in the facial nucleus. J. Neurosci 2000, 20, 5587–5593. [Google Scholar] [CrossRef]
- Robson, M.I.; de Las Heras, J.I.; Czapiewski, R.; Le Thanh, P.; Booth, D.G.; Kelly, D.A.; Webb, S.; Kerr, A.R.; Schirmer, E.C. Tissue-Specific Gene Repositioning by Muscle Nuclear Membrane Proteins Enhances Repression of Critical Developmental Genes during Myogenesis. Mol. Cell 2016, 62, 834–847. [Google Scholar] [CrossRef]
- Borrego-Pinto, J.; Jegou, T.; Osorio, D.S.; Aurade, F.; Gorjanacz, M.; Koch, B.; Mattaj, I.W.; Gomes, E.R. Samp1 is a component of TAN lines and is required for nuclear movement. J. Cell Sci. 2012, 125, 1099–1105. [Google Scholar] [CrossRef]
- Stroud, M.J.; Feng, W.; Zhang, J.; Veevers, J.; Fang, X.; Gerace, L.; Chen, J. Nesprin 1alpha2 is essential for mouse postnatal viability and nuclear positioning in skeletal muscle. J. Cell Biol. 2017, 216, 1915–1924. [Google Scholar] [CrossRef]
- Davidson, P.M.; Lammerding, J. Broken nuclei--lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014, 24, 247–256. [Google Scholar] [CrossRef]
- Moir, R.D.; Yoon, M.; Khuon, S.; Goldman, R.D. Nuclear lamins A and B1: Different pathways of assembly during nuclear envelope formation in living cells. J. Cell Biol. 2000, 151, 1155–1168. [Google Scholar] [CrossRef]
- Gruenbaum, Y.; Medalia, O. Lamins: The structure and protein complexes. Curr. Opin. Cell Biol. 2015, 32, 7–12. [Google Scholar] [CrossRef]
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Laine, J.; et al. Cellular micro-environments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 2014, 127, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Naetar, N.; Korbei, B.; Kozlov, S.; Kerenyi, M.A.; Dorner, D.; Kral, R.; Gotic, I.; Fuchs, P.; Cohen, T.V.; Bittner, R.; et al. Loss of nucleoplasmic LAP2alpha-lamin A complexes causes erythroid and epidermal progenitor hyperproliferation. Nat. Cell Biol. 2008, 10, 1341–1348. [Google Scholar] [CrossRef]
- Barateau, A.; Vadrot, N.; Vicart, P.; Ferreiro, A.; Mayer, M.; Heron, D.; Vigouroux, C.; Buendia, B. A Novel Lamin A Mutant Responsible for Congenital Muscular Dystrophy Causes Distinct Abnormalities of the Cell Nucleus. PLoS ONE 2017, 12, e0169189. [Google Scholar] [CrossRef]
- Azibani, F.; Brull, A.; Arandel, L.; Beuvin, M.; Nelson, I.; Jollet, A.; Ziat, E.; Prudhon, B.; Benkhelifa-Ziyyat, S.; Bitoun, M.; et al. Gene Therapy via Trans-Splicing for LMNA-Related Congenital Muscular Dystrophy. Mol. Ther. Nucleic Acids 2018, 10, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Pilat, U.; Dechat, T.; Bertrand, A.T.; Woisetschlager, N.; Gotic, I.; Spilka, R.; Biadasiewicz, K.; Bonne, G.; Foisner, R. The muscle dystrophy-causing DeltaK32 lamin A/C mutant does not impair the functions of the nucleoplasmic lamin-A/C-LAP2alpha complex in mice. J. Cell Sci. 2013, 126, 1753–1762. [Google Scholar] [CrossRef] [PubMed]
- Larsson, V.J.; Jafferali, M.H.; Vijayaraghavan, B.; Figueroa, R.A.; Hallberg, E. Mitotic spindle assembly and gamma-tubulin localisation depend on the integral nuclear membrane protein Samp1. J. Cell Sci. 2018, 131, jcs211664. [Google Scholar] [CrossRef]
- Jafferali, M.H.; Figueroa, R.A.; Hasan, M.; Hallberg, E. Spindle associated membrane protein 1 (Samp1) is required for the differentiation of muscle cells. Sci. Rep. 2017, 7, 16655. [Google Scholar] [CrossRef]
- Holt, I.; Duong, N.T.; Zhang, Q.; Lam le, T.; Sewry, C.A.; Mamchaoui, K.; Shanahan, C.M.; Morris, G.E. Specific localization of nesprin-1-alpha2, the short isoform of nesprin-1 with a KASH domain, in developing, fetal and regenerating muscle, using a new monoclonal antibody. BMC Cell Biol. 2016, 17, 26. [Google Scholar] [CrossRef]
- Tassin, A.M.; Maro, B.; Bornens, M. Fate of microtubule-organizing centers during myogenesis in vitro. J. Cell Biol. 1985, 100, 35–46. [Google Scholar] [CrossRef]
- Gimpel, P.; Lee, Y.L.; Sobota, R.M.; Calvi, A.; Koullourou, V.; Patel, R.; Mamchaoui, K.; Nedelec, F.; Shackleton, S.; Schmoranzer, J.; et al. Nesprin-1alpha-Dependent Microtubule Nucleation from the Nuclear Envelope via Akap450 Is Necessary for Nuclear Positioning in Muscle Cells. Curr. Biol. 2017, 27, 2999–3009 e9. [Google Scholar] [CrossRef]
- Azibani, F.; Muchir, A.; Vignier, N.; Bonne, G.; Bertrand, A.T. Striated muscle laminopathies. Semin. Cell Dev. Biol. 2014, 29C, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Lilina, A.V.; Chernyatina, A.A.; Guzenko, D.; Strelkov, S.V. Lateral A11 type tetramerization in lamins. J. Struct. Biol. 2020, 209, 107404. [Google Scholar] [CrossRef] [PubMed]
- Strelkov, S.V.; Schumacher, J.; Burkhard, P.; Aebi, U.; Herrmann, H. Crystal structure of the human lamin A coil 2B dimer: Implications for the head-to-tail association of nuclear lamins. J. Mol. Biol. 2004, 343, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Jo, I.; Kang, S.M.; Hong, S.; Kim, S.; Jeong, S.; Kim, Y.H.; Park, B.J.; Ha, N.C. Structural basis for lamin assembly at the molecular level. Nat. Commun. 2019, 10, 3757. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, F.; Degano, M. Disease-associated mutations in the coil 2B domain of human lamin A/C affect structural properties that mediate dimerization and intermediate filament formation. J. Struct. Biol. 2013, 181, 17–28. [Google Scholar] [CrossRef]
- Bank, E.M.; Ben-Harush, K.; Wiesel-Motiuk, N.; Barkan, R.; Feinstein, N.; Lotan, O.; Medalia, O.; Gruenbaum, Y. A laminopathic mutation disrupting lamin filament assembly causes disease-like phenotypes in C. elegans. Mol. Biol. Cell 2011, 22, 2716–2728. [Google Scholar] [CrossRef]
- Zwerger, M.; Jaalouk, D.E.; Lombardi, M.L.; Isermann, P.; Mauermann, M.; Dialynas, G.; Herrmann, H.; Wallrath, L.L.; Lammerding, J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum. Mol. Genet. 2013, 22, 2335–2349. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Pennypacker, J.K. RB and lamins in cell cycle regulation and aging. Adv. Exp. Med. Biol. 2014, 773, 127–142. [Google Scholar]
- Meinke, P.; Mattioli, E.; Haque, F.; Antoku, S.; Columbaro, M.; Straatman, K.R.; Worman, H.J.; Gundersen, G.G.; Lattanzi, G.; Wehnert, M.; et al. Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLoS Genet. 2014, 10, e1004605. [Google Scholar] [CrossRef]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef]
- Harr, J.C.; Luperchio, T.R.; Wong, X.; Cohen, E.; Wheelan, S.J.; Reddy, K.L. Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. J. Cell Biol. 2015, 208, 33–52. [Google Scholar] [CrossRef]
- Gesson, K.; Rescheneder, P.; Skoruppa, M.P.; von Haeseler, A.; Dechat, T.; Foisner, R. A-type lamins bind both hetero- and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 2016, 26, 462–473. [Google Scholar] [CrossRef]
- Demmerle, J.; Koch, A.J.; Holaska, J.M. The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J. Biol. Chem. 2012, 287, 22080–22088. [Google Scholar] [CrossRef]
- Somech, R.; Shaklai, S.; Geller, O.; Amariglio, N.; Simon, A.J.; Rechavi, G.; Gal-Yam, E.N. The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. J. Cell Sci. 2005, 118, 4017–4025. [Google Scholar] [CrossRef]
- De Las Heras, J.I.; Zuleger, N.; Batrakou, D.G.; Czapiewski, R.; Kerr, A.R.; Schirmer, E.C. Tissue-specific NETs alter genome organization and regulation even in a heterologous system. Nucleus 2017, 8, 81–97. [Google Scholar] [CrossRef]
- Bergqvist, C.; Niss, F.; Figueroa, R.A.; Beckman, M.; Maksel, D.; Jafferali, M.H.; Kulyte, A.; Strom, A.L.; Hallberg, E. Monitoring of chromatin organization in live cells by FRIC. Effects of the inner nuclear membrane protein Samp1. Nucleic Acids Res. 2019, 47, e49. [Google Scholar] [CrossRef]
- Mattioli, E.; Columbaro, M.; Jafferali, M.H.; Schena, E.; Hallberg, E.; Lattanzi, G. Samp1 Mislocalization in Emery-Dreifuss Muscular Dystrophy. Cells 2018, 7, 170. [Google Scholar] [CrossRef]
- Paulsen, J.; Sekelja, M.; Oldenburg, A.R.; Barateau, A.; Briand, N.; Delbarre, E.; Shah, A.; Sorensen, A.L.; Vigouroux, C.; Buendia, B.; et al. Chrom3D: Three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biol. 2017, 18, 21. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | Source (Reference Catalog) | WB | IF |
|---|---|---|---|
| Rabbit anti-Lamin A/C | Santa Cruz Biotechnologies (sc-20681) | 1:1000 | - |
| Goat anti-Lamin A/C | Santa Cruz Biotechnologies (sc-6215) | - | 1:50 |
| Mouse anti-Lamin A/C | Santa Cruz Biotechnologies (sc-376248) | - | 1:500 |
| Rabbit anti-Lamin B1 | Abcam (ab16048) | 1:1000 | - |
| Rabbit anti-Lamin B1 | Santa Cruz Biotechnologies (sc-6216) | - | 1:100 |
| Mouse anti-Emerin | Novocastra (NCL-emerin) | 1:500 | 1:50 |
| Mouse anti-Nesprin 1α | Glenn Morris (MANNES1E) | - | 1:50 |
| Rabbit anti-Tmem38A | Merck Millipore (#06-1005) | 1:200 | 1:50 |
| Rabbit anti-Net39 | Proteintech (20635-1-AP) | 1:200 | - |
| Rabbit anti-Pcm1 | Merck Millipore (HPA023370) | - | 1:500 |
| Rabbit anti-Samp1a | Merck Millipore (#06-1013) | 1:200 | 1:20 |
| Rabbit anti-Desmin | Abcam (ab15200) | - | 1:200 |
| Gene Name | Forward Primer | Reverse Primer | Ref |
|---|---|---|---|
| Net39 | 5′-CCCTGGCCCACTAGATAC-3′ | 5′-AGAGAAGGCTCCTATGGTCA-3′ | [25] |
| Tmem38A | 5′-CAGCTACTTCATCGTCTCCATC-3′ | 5′-CTCCCAAAACAGTGCAACATG-3′ | [25] |
| Samp1a | 5′-AGATTGAGGTGTACCGCCAC-3′ | 5′-TCACTGCTGCTTCTCTGACCT-3′ | [26] |
| Nesprin 1α | 5′-GGACTGAGCCTTTCGCTCTG-3′ | 5′-GCCACAGTCGCCACGTCTCT-3′ | [27] |
| Rplp0 | 5′-CTCCAAGCAGATGCAGCAGA-3′ | 5′-ATAGCCTTGCGCATCATGGT-3′ | [20] |
| Type | EDMD (n = 452) | L-CMD (n = 146) | p-Value | ||
|---|---|---|---|---|---|
| MS | 394 | (87.17%) | 119 | (81.51%) | 0.1017 |
| NS | 3 | (0.66%) | 0 | (0.00%) | > 0.99 |
| FS | 9 | (1.99%) | 2 | (1.37%) | > 0.99 |
| INF | 27 | (5.97%) | 21 | (14.38%) | 0.0024 |
| SPL | 19 | (4.20%) | 4 | (2.74%) | 0.6205 |
| Domain (AA Position) | EDMD (n = 452) | L-CMD (n = 146) | p-Value | ||
|---|---|---|---|---|---|
| Head (1–27) | 8 | (1.77%) | 0 | (0.00%) | 0.2093 |
| Coil 1a (28–66) | 60 | (13.27%) | 53 | (36.30%) | <0.0001 |
| L1 (67–78) | 1 | (0.22%) | 1 | (0.68%) | 0.429 |
| Coil 1b (79–222) | 35 | (7.74%) | 3 | (2.05%) | 0.0111 |
| L12 (223–240) | 4 | (0.88%) | 0 | (0.00%) | 0.5768 |
| Coil 2 (241–385) | 132 | (29.20%) | 66 | (45.21%) | 0.0006 |
| Tail N-term part (386–428) | 12 | (2.65%) | 1 | (0.68%) | 0.2042 |
| Ig-fold (429–549) | 191 | (42.26%) | 21 | (14.38%) | <0.0001 |
| Tail C-term part (550–664) | 9 | (1.99%) | 1 | (0.68%) | 0.4642 |
| Heptad Position | EDMD (n = 121) | L-CMD (n = 57) | p-Value | ||
|---|---|---|---|---|---|
| a/d | 28 | (23.14%) | 22 | (38.60%) | 0.0014 |
| e/g | 64 | (52.89%) | 31 | (54.39%) | 0.0355 |
| b/c | 20 | (16.53%) | 4 | (7.02%) | 0.4736 |
| f | 9 | (7.44%) | 0 | (0.00%) | 0.1236 |
| Residues Involved in Interaction between Dimers | EDMD (n = 452) | LCMD (n = 146) | p-Value | ||
| 67 | (14.82%) | 59 | (40.41%) | <0.0001 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertrand, A.T.; Brull, A.; Azibani, F.; Benarroch, L.; Chikhaoui, K.; Stewart, C.L.; Medalia, O.; Ben Yaou, R.; Bonne, G. Lamin A/C Assembly Defects in LMNA-Congenital Muscular Dystrophy Is Responsible for the Increased Severity of the Disease Compared with Emery–Dreifuss Muscular Dystrophy. Cells 2020, 9, 844. https://doi.org/10.3390/cells9040844
Bertrand AT, Brull A, Azibani F, Benarroch L, Chikhaoui K, Stewart CL, Medalia O, Ben Yaou R, Bonne G. Lamin A/C Assembly Defects in LMNA-Congenital Muscular Dystrophy Is Responsible for the Increased Severity of the Disease Compared with Emery–Dreifuss Muscular Dystrophy. Cells. 2020; 9(4):844. https://doi.org/10.3390/cells9040844
Chicago/Turabian StyleBertrand, Anne T., Astrid Brull, Feriel Azibani, Louise Benarroch, Khadija Chikhaoui, Colin L. Stewart, Ohad Medalia, Rabah Ben Yaou, and Gisèle Bonne. 2020. "Lamin A/C Assembly Defects in LMNA-Congenital Muscular Dystrophy Is Responsible for the Increased Severity of the Disease Compared with Emery–Dreifuss Muscular Dystrophy" Cells 9, no. 4: 844. https://doi.org/10.3390/cells9040844
APA StyleBertrand, A. T., Brull, A., Azibani, F., Benarroch, L., Chikhaoui, K., Stewart, C. L., Medalia, O., Ben Yaou, R., & Bonne, G. (2020). Lamin A/C Assembly Defects in LMNA-Congenital Muscular Dystrophy Is Responsible for the Increased Severity of the Disease Compared with Emery–Dreifuss Muscular Dystrophy. Cells, 9(4), 844. https://doi.org/10.3390/cells9040844