Role and Mechanisms of Mitophagy in Liver Diseases


Abstract
1. Introduction
2. Selective Autophagy Receptors
3. Mitophagy Signaling Pathways
3.1. PINK1-Parkin-Dependent Mitophagy
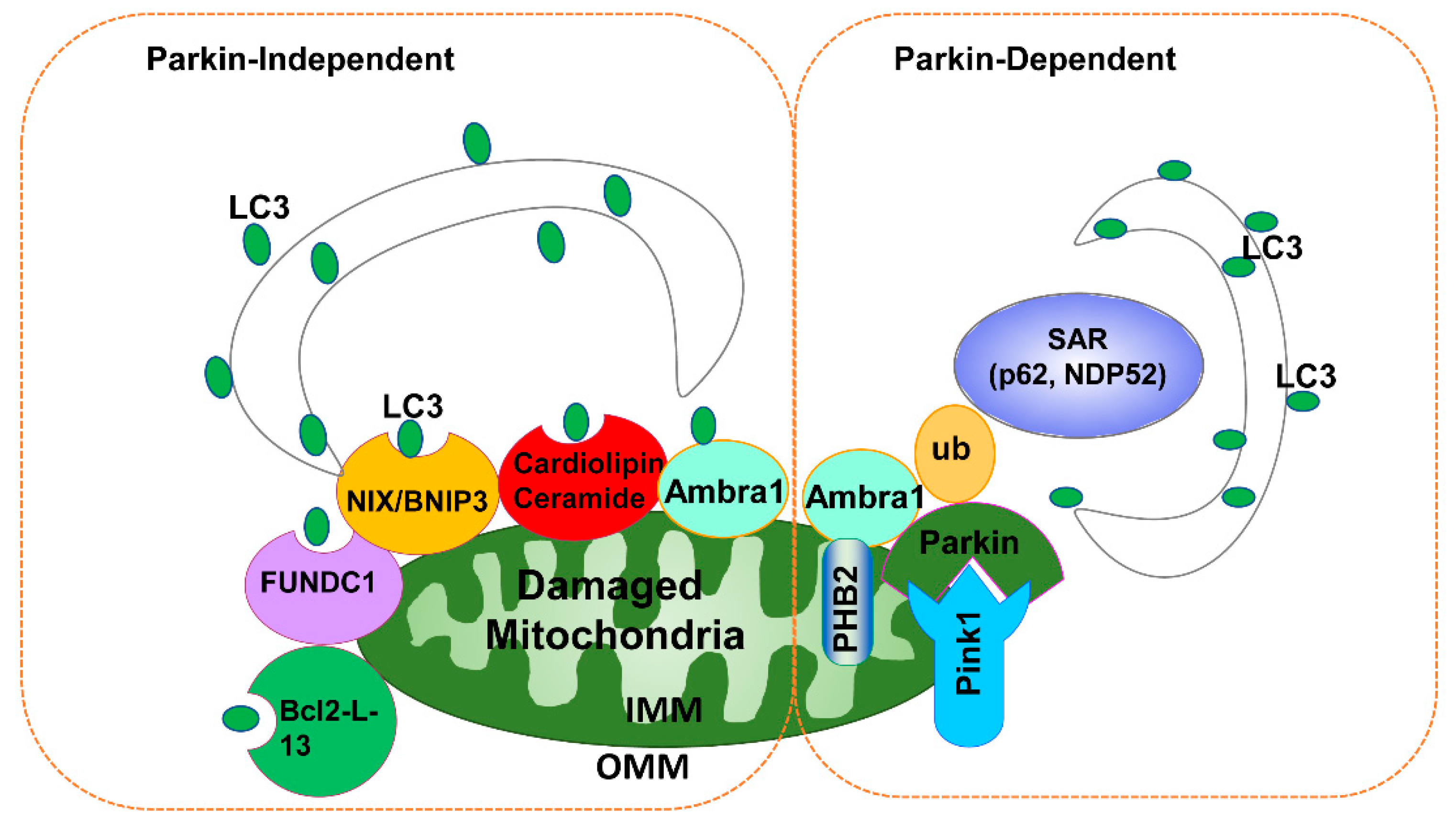
3.2. Parkin-Independent Mitophagy
3.3. Other Mitochondrial Quality Control Mechanisms
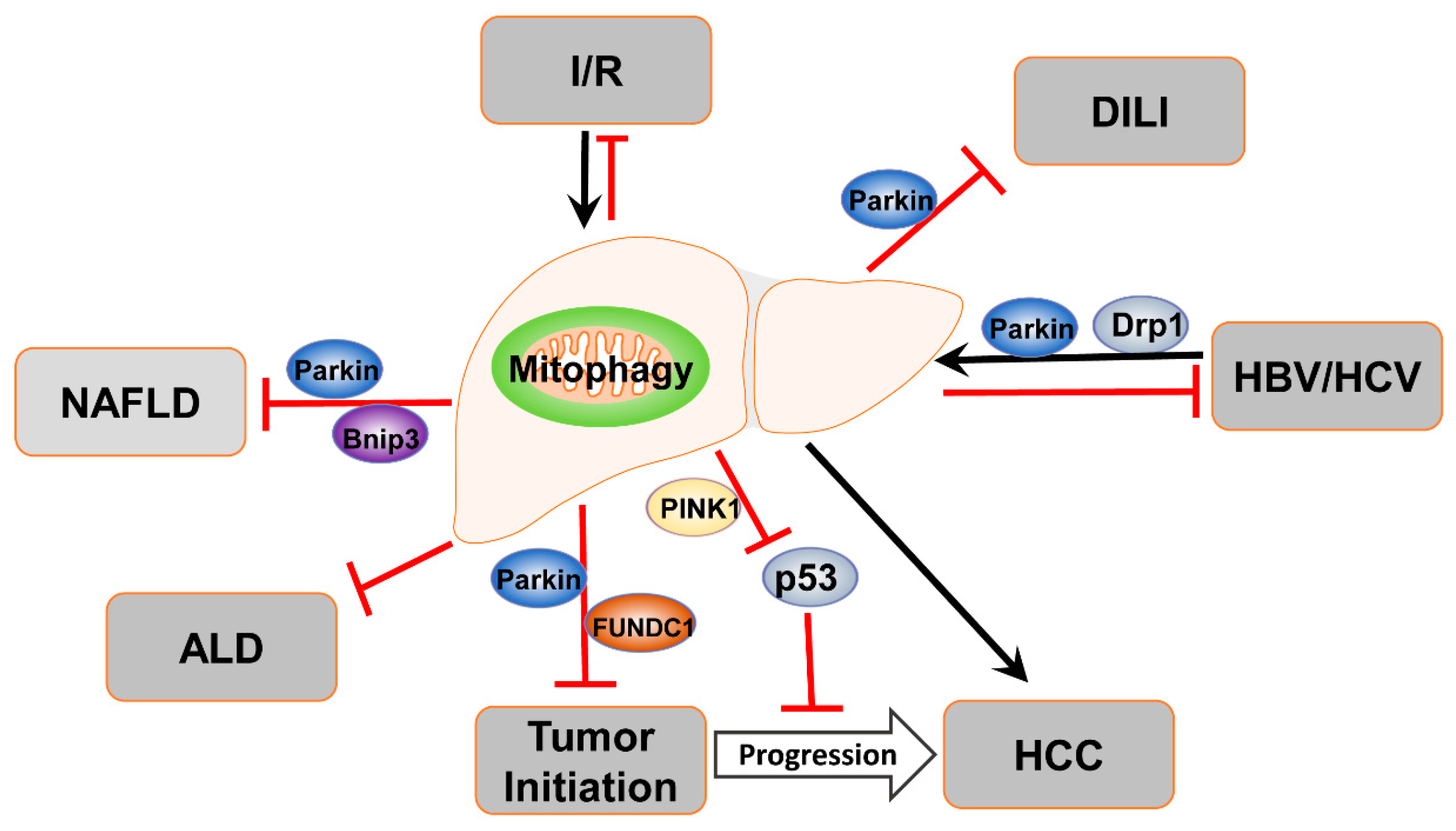
4. Mitophagy in Liver Diseases
4.1. Mitophagy in ALD
4.2. Mitophagy in NAFLD
4.3. Mitophagy in Drug-Induced Liver Injury
4.4. Mitophagy in Liver Ischemia/Reperfusion Injury
4.5. Mitophagy in Viral Hepatitis
4.6. Mitophagy in Liver Cancer
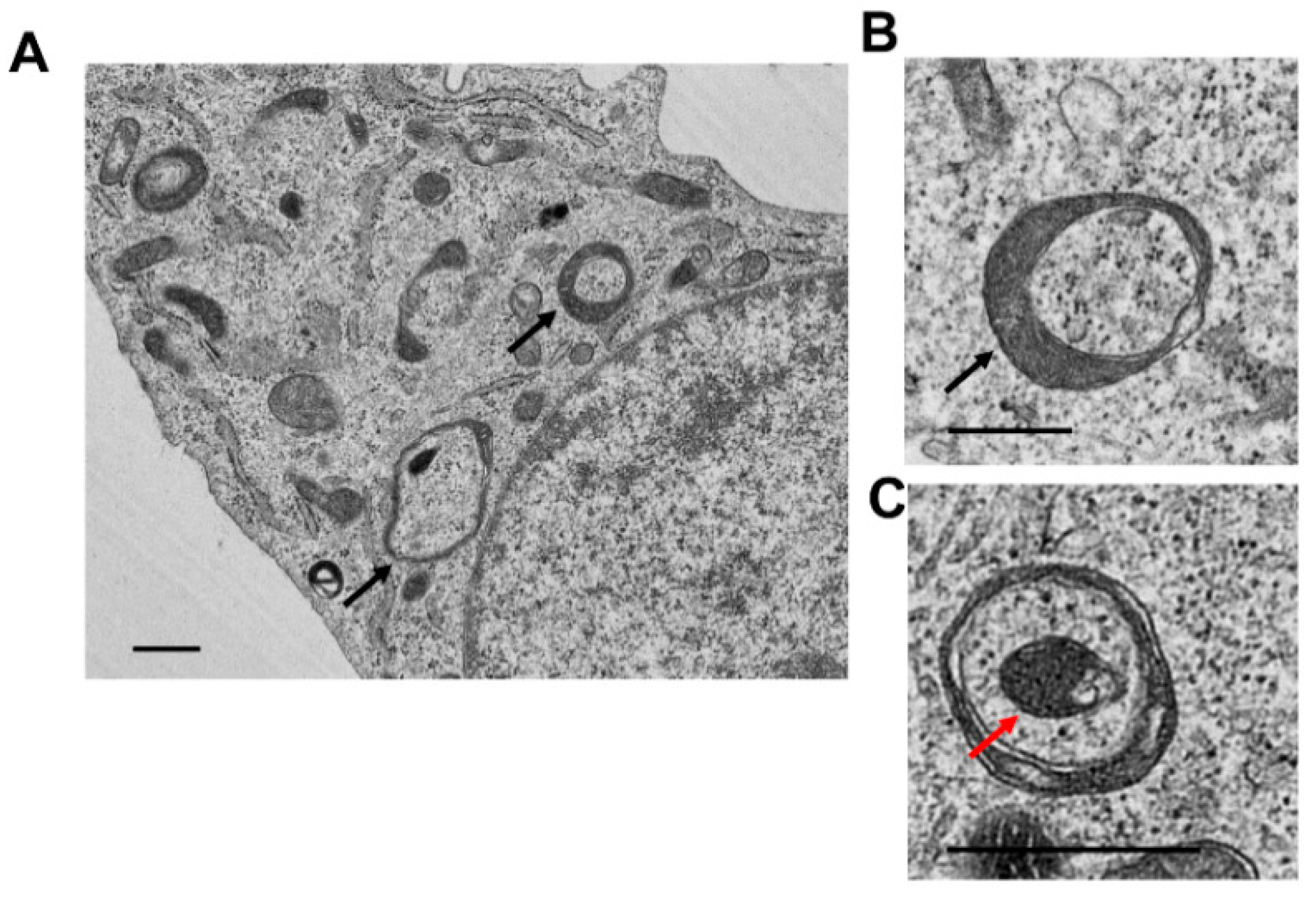
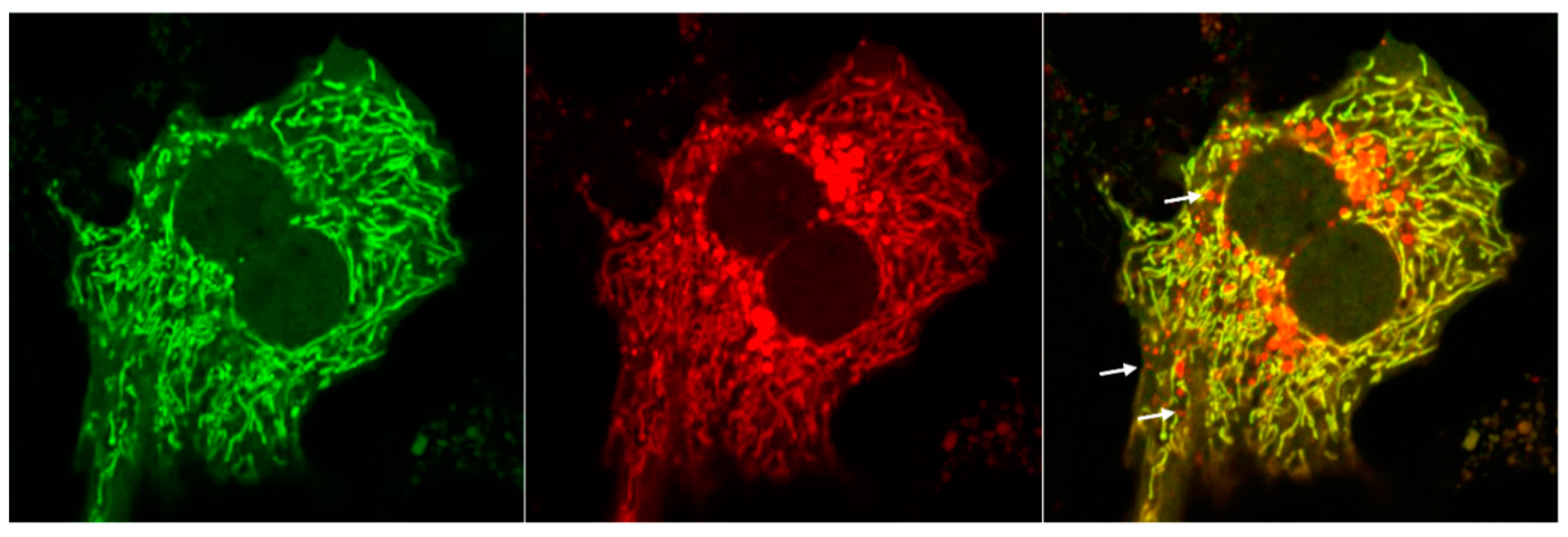
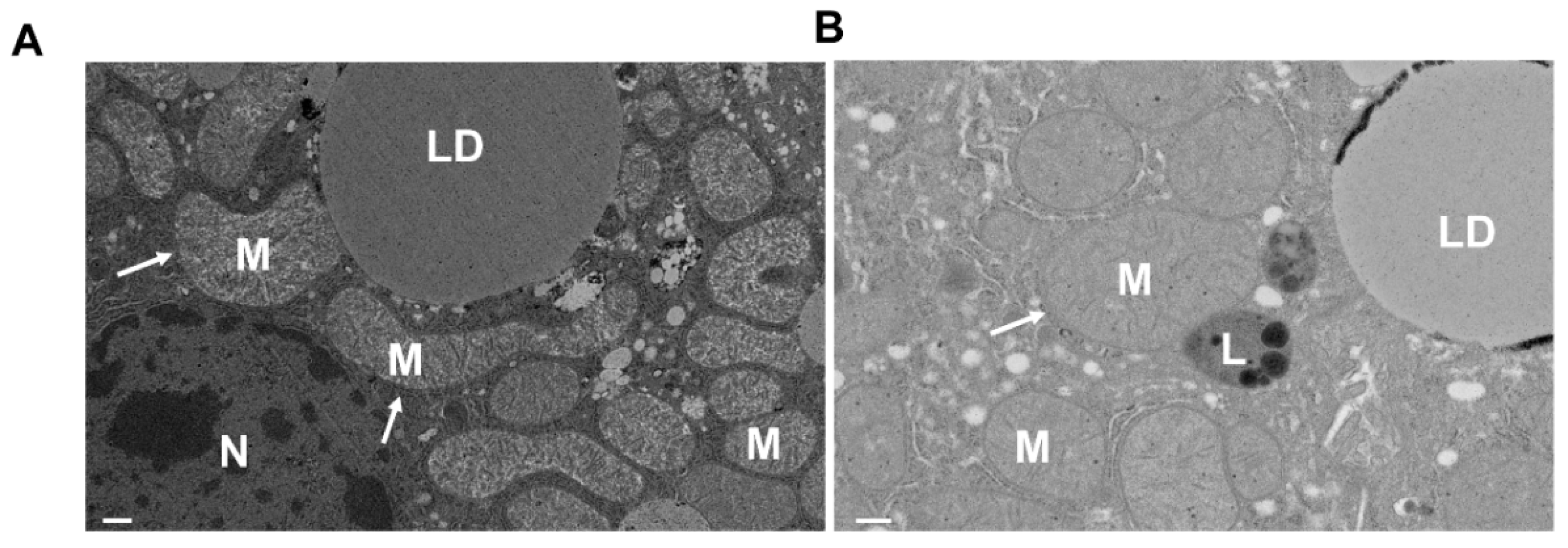
5. Analysis of Mitophagy in the Liver
6. Summary and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
List of Abbreviations
| ALCAT1 | Acyl-CoA:lysocardiolipin acyltransferase-1; |
| Ambra1 | autophagy/beclin-1 regulator-1; |
| ALD | alcoholic liver disease; |
| AMPK | AMP-regulated kinase; |
| APAP | acetaminophen; |
| ATF5 | Activating transcription factor 5; |
| Atg8 | autophagy-related protein 8; |
| ATL3 | atlastin GTPase 3; |
| Bcl2L13 | Bcl2 like 13; |
| BNIP3 | Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3; |
| CCPG1 | cell cycle progression 1; |
| CL | cardiolipin; |
| CSCs | cancer stem cells; |
| DAMPs | damage-associated molecular patterns; |
| DEN | diethylnitrosamine; |
| Drp1 | dynamin-related protein; |
| ETC | electron transport chain; |
| FFA | free fatty acid; |
| FAM134B | family with sequence similarity 134, member B; |
| FUNDC1 | FUN14 domain containing 1; |
| Gp78 | glycoprotein 78; |
| G-Rg3 | ginsenoside Rg3; |
| GSH | glutathione; |
| HBx | HBV-encoded X protein; |
| HBV | Hepatitis B virus; |
| HCV | hepatitis C virus; |
| HFD | high-fat diet; |
| HSP70 | heat shock protein 70; I |
| R | ischemia-reperfusion injury; |
| LC3 | microtubule-associated protein 1A/1B light chain3; |
| LIR | LC3 interacting region; |
| MCD | methionine- and choline-deficient; |
| MDVs | mitochondria-derived vesicles; |
| Mfn1 | mitochondrial fusion protein 1; |
| Mfn2 | mitochondrial fusion protein 2; |
| MPT | mitochondrial permeability transition; |
| Mst1 | macrophage stimulating 1; |
| mtDNA | mitochondrial DNA; |
| NAFLD | non-alcoholic fatty liver disease; |
| NAPQI | N-acetyl-p-benzoquinone imine; |
| NASH | nonalcoholic steatohepatitis; |
| NBR1 | BRCA1 gene 1; |
| NDP52 | nuclear domain 10 protein 52 kDa; |
| NS5A | non-structural protein 5A; |
| Opa1 | optic atrophy 1; |
| 8-OHdG | 8-hydroxydeoxyguanosine; |
| OA | oleic acid; |
| PA | palmitic acid; |
| PARL | presenilin associated, rhomboid-like; |
| PGAM5 | mitochondrial phosphatase phosphoglycerate mutase family member 5; |
| PINK1 | phosphatase and tensin homolog-induced putative kinase 1; |
| PHB2 | prohibitin 2; |
| PRDX6 | Peroxiredoxin 6; |
| ROS | reactive oxygen species; |
| RTN3 | reticulon 3; |
| SARs | soluble autophagy receptors; |
| SQSTM1 | Sequestosome 1 (SQSTM1); |
| TAX1BP1 | TAX1 binding protein 1; |
| TEX264 | testis expressed 264; |
| TH | thyroid hormone; |
| TOM20 | translocase of outer mitochondrial membrane 20; |
| USP30 | ubiquitin-specific peptidase 30; |
| USP15 | ubiquitin-specific peptidase 15; |
| UPRmt | mitochondrial unfolded protein response; |
| VDAC | voltage-dependent anion channel. |
References
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ding, W.-X. Adipose tissue autophagy and homeostasis in alcohol-induced liver injury. Liver Res. 2017, 1, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J.; Ding, W.-X.; Donohue, T.M.; Friedman, S.L.; Kim, J.-S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.-H.J.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.-M.; Ding, W.-X.; Gao, W. Autophagy in the liver. Hepatology 2008, 47, 1773–1785. [Google Scholar] [CrossRef]
- Ding, W.-X. Role of autophagy in liver physiology and pathophysiology. World J. Boil. Chem. 2010, 1, 3–12. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Allaire, M.; Rautou, P.-E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterol. 2015, 150, 328–339. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterol. 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ding, W.-X. Mechanisms, pathophysiological roles and methods for analyzing mitophagy – recent insights. Boil. Chem. 2018, 399, 147–178. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A Role for Ubiquitin in Selective Autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Williams, J.A.; Ding, W.X. Ding, Role of p62/SQSTM1 in liver physiology and pathogenesis. Exp. Biol. Med. 2013, 238, 525–538. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell Sci. 2018, 131, jcs217364. [Google Scholar] [CrossRef]
- Wilkinson, S. Emerging Principles of Selective ER Autophagy. J. Mol. Boil. 2019, 432, 185–205. [Google Scholar] [CrossRef]
- Ma, X.; Parson, C.; Ding, W.-X. Regulation of the homeostasis of hepatic endoplasmic reticulum and cytochrome P450 enzymes by autophagy. Liver Res. 2018, 2, 138–145. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Boil. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Kirkin, V.; Rogov, V.V. A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cell 2019, 76, 268–285. [Google Scholar] [CrossRef]
- Mizushima, N. A Dual Binding Receptor for ER-phagy. Dev. Cell 2018, 44, 133–135. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Nieminen, A.-L.; Qian, T.; Trost, L.C.; Elmore, S.P.; Nishimura, Y.; Crowe, R.A.; E Cascio, W.; Bradham, C.A.; Brenner, D.A.; et al. The mitochondrial permeability transition in cell death: A common mechanism in necrosis, apoptosis and autophagy. Biochim. et Biophys. Acta (BBA) Bioenerg. 1998, 1366, 177–196. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective Mitochondrial Autophagy, or Mitophagy, as a Targeted Defense Against Oxidative Stress, Mitochondrial Dysfunction, and Aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Boil. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Kawajiri, S.; Saiki, S.; Sato, S.; Sato, F.; Hatano, T.; Eguchi, H.; Hattori, N. PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett. 2010, 584, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Boil. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Boil. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Boil. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef]
- Fedorowicz, M.A.; De Vries-Schneider, R.L.A.; Rüb, C.; Becker, D.; Huang, Y.; Zhou, C.; Wolken, D.M.A.; Voos, W.; Liu, Y.; Przedborski, S. Cytosolic cleaved PINK 1 represses P arkin translocation to mitochondria and mitophagy. EMBO Rep. 2013, 15, 86–93. [Google Scholar] [CrossRef]
- Iguchi, M.; Kujuro, Y.; Okatsu, K.; Koyano, F.; Kosako, H.; Kimura, M.; Suzuki, N.; Uchiyama, S.; Tanaka, K.; Matsuda, N. Parkin-catalyzed Ubiquitin-Ester Transfer Is Triggered by PINK1-dependent Phosphorylation*. J. Boil. Chem. 2013, 288, 22019–22032. [Google Scholar] [CrossRef]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deák, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Boil. 2012, 2, 120080. [Google Scholar] [CrossRef]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef]
- Sarraf, S.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.; Hess, S.; Chan, D. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J. Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Yu, S.; Vincow, E.S.; Pallanck, L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 2010, 5, e10054. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Lemasters, J.J. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am. J. Physiol. Physiol. 2010, 300, C308-17. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; A Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; E Haigh, S.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Boil. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef]
- Yoshii, S.; Kishi, C.; Ishihara, N.; Mizushima, N. Parkin Mediates Proteasome-dependent Protein Degradation and Rupture of the Outer Mitochondrial Membrane*♦. J. Boil. Chem. 2011, 286, 19630–19640. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Ni, H.-M.; Li, M.; Liao, Y.; Chen, X.; Stolz, N.B.; Dorn, G.W.; Yin, X.-M. Nix Is Critical to Two Distinct Phases of Mitophagy, Reactive Oxygen Species-mediated Autophagy Induction and Parkin-Ubiquitin-p62-mediated Mitochondrial Priming*. J. Boil. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Saisho, K.; Shimanuki, M.; Nakada, K.; Shitara, H.; Sou, Y.S.; Kimura, M.; Sato, S.; Hattori, N.; Komatsu, M.; et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010, 15, 887–900. [Google Scholar] [CrossRef]
- Gao, F.; Chen, D.; Si, J.; Hu, Q.; Qin, Z.; Fang, M.; Wang, G. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum. Mol. Genet. 2015, 24, 2528–2538. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.-C.; Sumpter, R.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2016, 168, 224–238.e10. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin Interacts with Ambra1 to Induce Mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; Lavoie, M.J.; Schwarz, T.L. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef]
- Bingol, B.; Tea, J.; Phu, L.; Reichelt, M.; Bakalarski, C.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Cornelissen, T.; Haddad, D.; Wauters, F.; Van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; De Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of Serines 17 and 24 in the LC3-interacting Region of Bnip3 Determines Pro-survival Mitophagy versus Apoptosis*. J. Boil. Chem. 2012, 288, 1099–1113. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, Å.B. Microtubule-associated Protein 1 Light Chain 3 (LC3) Interacts with Bnip3 Protein to Selectively Remove Endoplasmic Reticulum and Mitochondria via Autophagy*. J. Boil. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2009, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Rogov, V.V.; Suzuki, H.; Marinković, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubasic, M.; Šprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef]
- Murakawa, T.; Yamaguchi, O.; Hashimoto, A.; Hikoso, S.; Takeda, T.; Oka, T.; Yasui, H.; Ueda, H.; Akazawa, Y.; Nakayama, H.; et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015, 6, 7527. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A Regulatory Signaling Loop Comprising the PGAM5 Phosphatase and CK2 Controls Receptor-Mediated Mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK 1 translocates to mitochondria and phosphorylates FUNDC 1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, L.; Cheng, Q.; Li, Y.; Wu, H.; Zhang, W.; Wang, Y.; Sehgal, S.A.; Siraj, S.; Wang, X.; et al. Mitochondrial E3 ligase MARCH 5 regulates FUNDC 1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017, 18, 495–509. [Google Scholar] [CrossRef]
- Chu, C.; Ji, J.; Dagda, R.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, O.; Tyurin, V.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Jiang, J.; Huang, Z.; Tyurina, Y.Y.; Desbourdes, C.; Cottet-Rousselle, C.; Dar, H.H.; Verma, M.; Tyurin, V.; Kapralov, O.; et al. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ. 2016, 23, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Methods 2012, 8, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Puri, R.; Yang, H.; Lizzio, M.A.; Wu, C.; Sheng, Z.H.; Guo, M. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife. 2014, 3, e01958. [Google Scholar] [CrossRef]
- Orvedahl, A.; Sumpter, R.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.G.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef]
- Fu, M.; St-Pierre, P.; Shankar, J.; Wang, P.T.C.; Joshi, B.; Nabi, I.R. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol. Boil. Cell 2013, 24, 1153–1162. [Google Scholar] [CrossRef]
- Di Rita, A.; Peschiaroli, A.; D Acunzo, P.; Strobbe, D.; Hu, Z.; Gruber, J.; Nygaard, M.; Lambrughi, M.; Melino, G.; Papaleo, E.; et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKalpha. Nat. Commun. 2018, 9, 3755. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Villa, E.; Marchetti, S.; Ricci, J.-E. No Parkin Zone: Mitophagy without Parkin. Trends Cell Boil. 2018, 28, 882–895. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Boil. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- McWilliams, T.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Boil. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Boil. 2017, 19, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Fiorese, C.; Schulz, A.M.; Lin, Y.-F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Boil. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.-L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; A Fon, E. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of Mitochondria Derived Vesicle Formation Demonstrates Selective Enrichment of Oxidized Cargo. PLOS ONE 2012, 7, e52830. [Google Scholar] [CrossRef]
- Abuaita, B.H.; Schultz, T.L.; O’Riordan, M. Mitochondria-Derived Vesicles Deliver Antimicrobial Reactive Oxygen Species to Control Phagosome-Localized Staphylococcus aureus. Cell Host Microbe 2018, 24, 625–636.e5. [Google Scholar] [CrossRef]
- Ding, W.-X.; Guo, F.; Ni, H.-M.; Bockus, A.; Manley, S.; Stolz, N.B.; Eskelinen, E.-L.; Jaeschke, H.; Yin, X.-M. Parkin and Mitofusins Reciprocally Regulate Mitophagy and Mitochondrial Spheroid Formation*. J. Boil. Chem. 2012, 287, 42379–42388. [Google Scholar] [CrossRef]
- Ding, W.-X.; Li, M.; Biazik, J.M.; Morgan, D.G.; Guo, F.; Ni, H.-M.; Goheen, M.; Eskelinen, E.-L.; Yin, X.-M. Electron Microscopic Analysis of a Spherical Mitochondrial Structure*. J. Boil. Chem. 2012, 287, 42373–42378. [Google Scholar] [CrossRef]
- Yin, X.-M.; Ding, W.-X. The reciprocal roles of PARK2 and mitofusins in mitophagy and mitochondrial spheroid formation. Autophagy 2013, 9, 1687–1692. [Google Scholar] [CrossRef]
- Manley, S.; Ni, H.-M.; Williams, J.A.; Kong, B.; DiTacchio, L.; Guo, G.; Ding, W.-X. Farnesoid X receptor regulates forkhead Box O3a activation in ethanol-induced autophagy and hepatotoxicity. Redox Boil. 2014, 2, 991–1002. [Google Scholar] [CrossRef]
- Gautam, M.; Xie, E.F.; Kocak, N.; Ozdinler, P.H. Mitoautophagy: A Unique Self-Destructive Path Mitochondria of Upper Motor Neurons With TDP-43 Pathology Take, Very Early in ALS. Front. Cell. Neurosci. 2019, 13, 489. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.-M.; A Williams, J.; Jaeschke, H.; Ding, W.-X. Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox Boil. 2013, 1, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Boil. 2014, 4, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Yamashita, S.; Kim, H.; Na, D.; Lee, H.; Kim, S.J.; Cho, D.; Kanki, T.; Jung, Y.-K. FKBP8 LIRL-dependent mitochondrial fragmentation facilitates mitophagy under stress conditions. FASEB J. 2019, 34, 2944–2957.e5. [Google Scholar] [CrossRef]
- Yamada, T.; Murata, D.; Adachi, Y.; Itoh, K.; Kameoka, S.; Igarashi, A.; Kato, T.; Araki, Y.; Huganir, R.L.; Dawson, T.M.; et al. Mitochondrial Stasis Reveals p62-Mediated Ubiquitination in Parkin-Independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab. 2018, 28, 588–604. [Google Scholar] [CrossRef]
- Kageyama, Y.; Hoshijima, M.; Seo, K.; Bedja, D.; Sysa-Shah, P.; A Andrabi, S.; Chen, W.; Hoke, A.; Dawson, V.L.; Dawson, T.M.; et al. Parkin-independent mitophagy requires D rp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014, 33, 2798–2813. [Google Scholar] [CrossRef]
- Yoshii, S.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Yamashita, S.-I.; Jin, X.; Furukawa, K.; Hamasaki, M.; Nezu, A.; Otera, H.; Saigusa, T.; Yoshimori, T.; Sakai, Y.; Mihara, K.; et al. Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J. Cell Boil. 2016, 215, 649–665. [Google Scholar] [CrossRef]
- Mendl, N.; Occhipinti, A.; Müller, M.; Wild, P.; Dikic, I.; Reichert, A.S. Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2. J. Cell Sci. 2011, 124, 1339–1350. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Shaid, S.; Vakhrusheva, O.; Koschade, S.E.; Klann, K.; Thölken, M.; Baker, F.; Zhang, J.; Oellerich, T.; Sürün, D.; et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood 2019, 133, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Murakawa, T.; Otsu, K. 1182The novel mitophagic receptor protein bcl2-like protein 13: New insights for the molecular mechanisms of the pathogenesis of heart disease. Eur. Hear. J. 2019, 40, 658. [Google Scholar] [CrossRef]
- Bhujabal, Z.; Birgisdottir, A.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef]
- Abudu, Y.P.; Pankiv, S.; Mathai, B.J.; Lamark, T.; Johansen, T.; Simonsen, A. NIPSNAP1 and NIPSNAP2 act as "eat me" signals to allow sustained recruitment of autophagy receptors during mitophagy. Autophagy 2019, 15, 1845–1847. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 517. [Google Scholar] [CrossRef]
- Chen, Y.; Ii, G.W.D. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, Å.B. Parkin Protein Deficiency Exacerbates Cardiac Injury and Reduces Survival following Myocardial Infarction*♦. J. Boil. Chem. 2012, 288, 915–926. [Google Scholar] [CrossRef]
- Tang, C.; Han, H.; Yan, M.; Zhu, S.; Liu, J.; Liu, Z.; He, L.; Tan, J.; Liu, Y.; Liu, H.; et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018, 14, 880–897. [Google Scholar] [CrossRef]
- Kobayashi, K.; Araya, J.; Minagawa, S.; Hara, H.; Saito, N.; Kadota, T.; Sato, N.; Yoshida, M.; Tsubouchi, K.; Kurita, Y.; et al. Involvement of PARK2-Mediated Mitophagy in Idiopathic Pulmonary Fibrosis Pathogenesis. J. Immunol. 2016, 197, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schlehe, J.S.; Lavoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Boil. 2014, 206, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterol. 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.C.; Coleman, W.B.; Spach, P.I. The Effects of Chronic Ethanol Consumption on Hepatic Mitochondrial Energy Metabolism. Alcohol Alcohol. 1990, 25, 127–136. [Google Scholar] [CrossRef]
- Falkenberg, M.; Larsson, N.-G.; Gustafsson, C.M. DNA Replication and Transcription in Mammalian Mitochondria. Annu. Rev. Biochem. 2007, 76, 679–699. [Google Scholar] [CrossRef]
- Harman, D. The Biologic Clock: The Mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Mansouri, A.; Gaou, I.; De Kerguenec, C.; Amsellem, S.; Haouzi, D.; Berson, A.; Moreau‡, A.; Feldmann‡, G.; Lettéron, P.; Pessayre, D.; et al. An alcoholic binge causes massive degradation of hepatic mitochondrial DNA in mice. Gastroenterology 1999, 117, 181–190. [Google Scholar] [CrossRef]
- Cahill, A.; Stabley, G.J.; Wang, X.; Hoek, J. Chronic ethanol consumption causes alterations in the structural integrity of mitochondrial DNA in aged rats. Hepatology 1999, 30, 881–888. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction, and Aging. J. Signal Transduct. 2011, 2012, 1–13. [Google Scholar] [CrossRef]
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Eccleston, H.B.; Bailey, S.M. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free. Radic. Boil. Med. 2008, 44, 1259–1272. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Kaplowitz, N.; Fernándezcheca, J.C. Role of Mitochondria in Alcoholic Liver Disease. Curr. Pathobiol. Rep. 2013, 1, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Li, M.; Yin, X.-M. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy 2011, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Eid, N.; Ito, Y.; Maemura, K.; Otsuki, Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: An immunohistochemical and electron microscopic study. J. Mol. Histol. 2013, 44, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ni, H.-M.; Ding, Y.; Ding, W.-X. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am. J. Physiol. Liver Physiol. 2015, 309, G324–G340. [Google Scholar] [CrossRef]
- Yu, X.; Xu, Y.; Zhang, S.; Sun, J.; Liu, P.; Xiao, L.; Tang, Y.; Liu, L.; Yao, P. Quercetin Attenuates Chronic Ethanol-Induced Hepatic Mitochondrial Damage through Enhanced Mitophagy. Nutrients 2016, 8, 27. [Google Scholar] [CrossRef]
- Gao, H.; Lv, Y.; Liu, Y.; Li, J.; Wang, X.; Zhou, Z.; Tipoe, G.L.; Ouyang, S.; Guo, Y.; Zhang, J.; et al. Wolfberry-Derived Zeaxanthin Dipalmitate Attenuates Ethanol-Induced Hepatic Damage. Mol. Nutr. Food Res. 2019, 63, e1801339. [Google Scholar] [CrossRef]
- Williams, J.A.; Ding, W.-X. Targeting Pink1-Parkin-mediated mitophagy for treating liver injury. Pharmacol. Res. 2015, 102, 264–269. [Google Scholar] [CrossRef]
- Williams, J.A.; Ding, W.-X. A Mechanistic Review of Mitophagy and Its Role in Protection against Alcoholic Liver Disease. Biomolecules 2015, 5, 2619–2642. [Google Scholar] [CrossRef]
- Eid, N.; Ito, Y.; Horibe, A.; Otsuki, Y. Ethanol-induced mitophagy in liver is associated with activation of the PINK1-Parkin pathway triggered by oxidative DNA damage. Histol. Histopathol. 2016, 31, 1143–1159. [Google Scholar]
- Ma, G.-D.; Liu, Y.-H.; Zhang, Q.-L.; Zhang, B.-G.; Zhao, N.; Wang, Q.-L.; Wang, X.-D. Pre-endurance training prevents acute alcoholic liver injury in rats through the regulation of damaged mitochondria accumulation and mitophagy balance. Hepatol. Int. 2014, 8, 425–435. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Zhong, Z. Mitophagy in hepatocytes: Types, initiators and role in adaptive ethanol metabolism. Liver Res. 2018, 2, 125–132. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, P.; Wang, J.; Toan, S.; Ren, J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduct. Target. Ther. 2019, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Kronborg, I.; Peters, R.L. Giant mitochondria in the alcoholic liver diseases--their identification, frequency and pathologic significance. Liver 1984, 4, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Johnson, H.S.; Rao, M.P.; Martin, G.; Sancheti, H.; Silkwood, K.H.; Decker, C.W.; Nguyen, K.T.; Casian, J.G.; Cadenas, E.; et al. Mitochondrial remodeling in the liver following chronic alcohol feeding to rats. Free. Radic. Boil. Med. 2016, 102, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Ma, X.; Riva, A.; Iansante, V.; Dhawan, A.; Wang, S.; Ni, H.-M.; Sesaki, H.; Williams, R.; Ding, W.-X.; et al. Dynamin-1–Like Protein Inhibition Drives Megamitochondria Formation as an Adaptive Response in Alcohol-Induced Hepatotoxicity. Am. J. Pathol. 2019, 189, 580–589. [Google Scholar] [CrossRef]
- Chao, X.; Wang, S.; Zhao, K.; Li, Y.; Williams, J.A.; Li, T.; Chavan, H.; Krishnamurthy, P.; He, X.C.; Li, L.; et al. Impaired TFEB-Mediated Lysosome Biogenesis and Autophagy Promote Chronic Ethanol-Induced Liver Injury and Steatosis in Mice. Gastroenterol. 2018, 155, 865–879.e12. [Google Scholar] [CrossRef]
- Younes, R.; Bugianesi, E. A spotlight on pathogenesis, interactions and novel therapeutic options in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 80–82. [Google Scholar] [CrossRef]
- Lee, J.; Park, J.-S.; Roh, Y.-S. Molecular insights into the role of mitochondria in non-alcoholic fatty liver disease. Arch. Pharmacal Res. 2019, 42, 935–946. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Baulies, A.; Marí, M.; García-Rovés, P.M.; Fernándezcheca, J.C. Mitochondrial dysfunction in non-alcoholic fatty liver disease and insulin resistance: Cause or consequence? Free. Radic. Res. 2013, 47, 854–868. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell–Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterol. 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; E Hespenheide, E.; Parks, J.K.; Parker, W. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chang, L.; Luo, Y.; Zhou, Y.; Zhang, J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Boil. 2019, 21, 101120. [Google Scholar] [CrossRef]
- Lee, N.H.; Jung, Y.Y.; Park, M.H.; Jo, M.R.; Han, S.B.; Yoon, -Y.; Roh, Y.S.; Hong, J.T. Peroxiredoxin 6 Confers Protection Against Nonalcoholic Fatty Liver Disease Through Maintaining Mitochondrial Function. Antioxidants Redox Signal. 2019, 31, 387–402. [Google Scholar] [CrossRef]
- Li, R.; Xin, T.; Li, D.; Wang, C.; Zhu, H.; Zhou, H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Boil. 2018, 18, 229–243. [Google Scholar] [CrossRef]
- Liu, P.; Lin, H.; Xu, Y.; Zhou, F.; Wang, J.; Liu, J.; Zhu, X.; Guo, X.; Tang, Y.; Yao, P. Frataxin-Mediated PINK1-Parkin-Dependent Mitophagy in Hepatic Steatosis: The Protective Effects of Quercetin. Mol. Nutr. Food Res. 2018, 62, e1800164. [Google Scholar] [CrossRef]
- Yu, X.; Hao, M.; Liu, Y.; Ma, X.; Lin, W.; Xu, Q.; Zhou, H.; Shao, N.; Kuang, H. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 2019, 864, 172715. [Google Scholar] [CrossRef]
- Gong, L.-L.; Yang, S.; Zhang, W.; Han, F.-F.; Lv, Y.-L.; Wan, Z.-R.; Liu, H.; Jia, Y.; Xuan, L.-L.; Liu, L.; et al. Akebia saponin D alleviates hepatic steatosis through BNip3 induced mitophagy. J. Pharmacol. Sci. 2018, 136, 189–195. [Google Scholar] [CrossRef]
- Lin, D.; He, H.; Ji, H.; Willis, J.; Willard, L.; Jiang, Y.; Medeiros, D.; Wark, L.; Han, J.; Liu, Y.; et al. Wolfberries potentiate mitophagy and enhance mitochondrial biogenesis leading to prevention of hepatic steatosis in obese mice: The role of AMP-activated protein kinase α2 subunit. Mol. Nutr. Food Res. 2014, 58, 1005–1015. [Google Scholar] [CrossRef]
- Kim, K.-Y.; Stevens, M.V.; Akter, M.H.; Rusk, S.E.; Huang, R.J.; Cohen, A.; Noguchi, A.; Springer, D.; Bocharov, A.V.; Eggerman, T.L.; et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Investig. 2011, 121, 3701–3712. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-Y.; Sack, M.N. Parkin in the regulation of fat uptake and mitochondrial biology. Curr. Opin. Lipidol. 2012, 23, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.K.; Huckestein, B.; Edmunds, L.R.; Petersen, M.C.; Nasiri, A.; Butrico, G.M.; Abulizi, A.; Harmon, D.B.; Lu, C.; Mantell, B.; et al. Reduced intestinal lipid absorption and body weight-independent improvements in insulin sensitivity in high-fat diet-fed Park2 knockout mice. Am. J. Physiol. Metab. 2016, 311, E105–E116. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Zhang, W.; Beaton, M.; Marsboom, G.; Gruber, M.; Simon, M.C.; Hart, J.; Dorn, G.W.; Brady, M.J.; MacLeod, K. BNip3 Regulates Mitochondrial Function and Lipid Metabolism in the Liver. Mol. Cell. Boil. 2012, 32, 2570–2584. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, D.; Liu, S.; Burkewitz, K.; Kory, N.; Knudsen, N.H.; Alexander, R.K.; Ünlütürk, U.; Li, X.; Kong, X.; Hyde, A.L.; et al. Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell Metab. 2015, 22, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B.; Berson, A.; Robin, M.-A.; Letteron, P.; Moreau, R.; Mansouri, A. Central role of mitochondria in drug-induced liver injury. Drug Metab. Rev. 2012, 44, 34–87. [Google Scholar] [CrossRef]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef]
- Jollow, D.; Thorgeirsson, S.; Potter, W.; Hashimoto, M.; Mitchell, J. Acetaminophen-Induced Hepatic Necrosis. Pharmacol. 1974, 12, 251–271. [Google Scholar] [CrossRef]
- Larson, A.M.; Polson, J.; Fontana, R.J.; Davern, T.J.; Lalani, E.; Hynan, L.; Reisch, J.S.; Schiødt, F.V.; Ostapowicz, G.; Shakil, A.O.; et al. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology 2005, 42, 1364–1372. [Google Scholar] [CrossRef]
- McGill, M.R.; Jaeschke, H. Metabolism and disposition of acetaminophen: Recent advances in relation to hepatotoxicity and diagnosis. Pharm. Res. 2013, 30, 2174–2187. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Acetaminophen Toxicity: Novel Insights Into Mechanisms and Future Perspectives. Gene Expr. 2018, 18, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Cover, C.; Mansouri, A.; Knight, T.R.; Bajt, M.L.; Lemasters, J.J.; Pessayre, D.; Jaeschke, H. Peroxynitrite-Induced Mitochondrial and Endonuclease-Mediated Nuclear DNA Damage in Acetaminophen Hepatotoxicity. J. Pharmacol. Exp. Ther. 2005, 315, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Kon, K.; Kim, J.-S.; Jaeschke, H.; Lemasters, J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004, 40, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Lebofsky, M.; Baines, C.P.; Lemasters, J.J.; Jaeschke, H. Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free. Radic. Res. 2010, 45, 156–164. [Google Scholar] [CrossRef]
- Williams, J.A.; Ni, H.-M.; Haynes, A.; Manley, S.; Li, Y.; Jaeschke, H.; Ding, W.-X. Chronic Deletion and Acute Knockdown of Parkin Have Differential Responses to Acetaminophen-induced Mitophagy and Liver Injury in Mice*. J. Boil. Chem. 2015, 290, 10934–10946. [Google Scholar] [CrossRef]
- Ni, H.-M.; Bockus, A.; Boggess, N.; Jaeschke, H.; Ding, W.-X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2011, 55, 222–232. [Google Scholar] [CrossRef]
- Shan, S.; Shen, Z.; Zhang, C.; Kou, R.; Xie, K.; Song, F. Mitophagy protects against acetaminophen-induced acute liver injury in mice through inhibiting NLRP3 inflammasome activation. Biochem. Pharmacol. 2019, 169, 113643. [Google Scholar] [CrossRef]
- Baulies, A.; Ribas, V.; Núñez, S.T.; Torres, S.; Alarcón-Vila, C.; Martinez, L.; Suda, J.; Ybanez, M.D.; Kaplowitz, N.; García-Ruiz, C.; et al. Lysosomal Cholesterol Accumulation Sensitizes To Acetaminophen Hepatotoxicity by Impairing Mitophagy. Sci. Rep. 2015, 5, 18017. [Google Scholar] [CrossRef]
- Wang, H.; Ni, H.-M.; Chao, X.; Ma, X.; Rodriguez, Y.A.; Chavan, H.; Wang, S.; Krishnamurthy, P.; Dobrowsky, R.; Xu, D.-X.; et al. Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Boil. 2019, 22, 101148. [Google Scholar] [CrossRef]
- Kang, S.W.S.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLOS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef]
- Gao, Y.; Chu, S.; Zhang, Z.; Zuo, W.; Xia, C.; Ai, Q.; Luo, P.; Cao, P.; Chen, N. Early Stage Functions of Mitochondrial Autophagy and Oxidative Stress in Acetaminophen-Induced Liver Injury. J. Cell. Biochem. 2017, 118, 3130–3141. [Google Scholar] [CrossRef] [PubMed]
- Go, K.L.; Lee, S.; Zendejas, I.; Behrns, K.E.; Kim, J.-S. Mitochondrial Dysfunction and Autophagy in Hepatic Ischemia/Reperfusion Injury. BioMed Res. Int. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kantrow, S.P.; Gierman, J.L.; Jaligam, V.R.; Zhang, P.; Piantadosi, C.A.; Summer, W.R.; Lancaster, J.R. Regulation of tumor necrosis factor cytotoxicity by calcineurin. FEBS Lett. 2000, 483, 119–124. [Google Scholar] [CrossRef]
- Hong, J.-M.; Kim, S.-J.; Lee, S.-M. Role of necroptosis in autophagy signaling during hepatic ischemia and reperfusion. Toxicol. Appl. Pharmacol. 2016, 308, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Yan, X.; Wang, Y.; Wang, R.; Fan, X.; Zhong, Z.; Ye, Q. Parkin deficiency elevates hepatic ischemia/reperfusion injury accompanying decreased mitochondrial autophagy, increased apoptosis, impaired DNA damage repair and altered cell cycle distribution. Mol. Med. Rep. 2018, 18, 5663–5668. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, R.H.; Weston, C.J.; Velduis, S.; Leuvenink, H.G.D.; Reynolds, G.M.; Davies, S.; Thin, L.N.; Alfaifi, M.; Shepard, E.L.; Boteon, Y.; et al. The Reactive Oxygen Species-Mitophagy Signaling Pathway Regulates Liver Endothelial Cell Survival During Ischemia/Reperfusion Injury. Liver Transplant. 2018, 24, 1437–1452. [Google Scholar] [CrossRef]
- Shin, J.-K.; Lee, S.-M. Genipin protects the liver from ischemia/reperfusion injury by modulating mitochondrial quality control. Toxicol. Appl. Pharmacol. 2017, 328, 25–33. [Google Scholar] [CrossRef]
- Hong, J.-M.; Lee, S.-M. Heme oxygenase-1 protects liver against ischemia/reperfusion injury via phosphoglycerate mutase family member 5-mediated mitochondrial quality control. Life Sci. 2018, 200, 94–104. [Google Scholar] [CrossRef]
- Kang, J.-W.; Choi, H.-S.; Lee, S.-M. Resolvin D1 attenuates liver ischaemia/reperfusion injury through modulating thioredoxin 2-mediated mitochondrial quality control. Br. J. Pharmacol. 2018, 175, 2441–2453. [Google Scholar] [CrossRef]
- Okaya, T.; Blanchard, J.; Schuster, R.; Kuboki, S.; Husted, T.; Caldwell, C.; Zingarelli, B.; Wong, H.R.; Solomkin, J.S.; Lentsch, A.B. Age-Dependent Responses to Hepatic Ischemia/Reperfusion Injury. Shock 2005, 24, 421–427. [Google Scholar] [CrossRef]
- Chun, S.K.; Lee, S.; Flores-Toro, J.; U, R.Y.; Yang, M.-J.; Go, K.L.; Biel, T.G.; Miney, C.E.; Louis, S.P.; Law, B.K.; et al. Loss of sirtuin 1 and mitofusin 2 contributes to enhanced ischemia/reperfusion injury in aged livers. Aging Cell 2018, 17, e12761. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ruan, D.-Y.; Jia, C.-C.; Zheng, J.; Wang, G.-Y.; Zhao, H.; Yang, Q.; Liu, W.; Yi, S.-H.; Li, H.; et al. Aging aggravates hepatic ischemia-reperfusion injury in mice by impairing mitophagy with the involvement of the EIF2α-parkin pathway. Aging 2018, 10, 1902–1920. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-I.; Seo, M.-J.; Lee, S.-M. 2-Methoxyestradiol protects against ischemia/reperfusion injury in alcoholic fatty liver by enhancing sirtuin 1-mediated autophagy. Biochem. Pharmacol. 2017, 131, 40–51. [Google Scholar] [CrossRef] [PubMed]
- The Polaris Observatory Collaborators. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef]
- The Polaris Observatory HCV Collaborators. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 161–176. [Google Scholar] [CrossRef]
- Sir, N.; Tian, Y.; Chen, W.-L.; Ann, D.K.; Yen, T.-S.B.; Ou, J.-H.J. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. 2010, 107, 4383–4388. [Google Scholar] [CrossRef]
- Khan, M.; Imam, H.; Siddiqui, A. Subversion of cellular autophagy during virus infection: Insights from hepatitis B and hepatitis C viruses. Liver Res. 2018, 2, 146–156. [Google Scholar] [CrossRef]
- Kim, S.-J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B Virus Disrupts Mitochondrial Dynamics: Induces Fission and Mitophagy to Attenuate Apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Huang, X.-Y.; Li, D.; Chen, Z.-X.; Huang, Y.-H.; Gao, W.-Y.; Zheng, B.-Y.; Wang, X.-Z. Hepatitis B Virus X protein elevates Parkin-mediated mitophagy through Lon Peptidase in starvation. Exp. Cell Res. 2018, 368, 75–83. [Google Scholar] [CrossRef]
- Yoo, Y.-S.; Park, Y.-J.; Lee, H.-S.; Oanh, N.T.K.; Cho, M.-Y.; Heo, J.; Lee, E.-S.; Cho, H.; Park, Y.-Y.; Cho, H. Mitochondria ubiquitin ligase, MARCH5 resolves hepatitis B virus X protein aggregates in the liver pathogenesis. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Chi, H.C.; Chen, S.L.; Lin, S.L.; Tsai, C.Y.; Chuang, W.Y.; Lin, Y.H.; Huang, Y.H.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Thyroid hormone protects hepatocytes from HBx-induced carcinogenesis by enhancing mitochondrial turnover. Oncogene 2017, 36, 5274–5284. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Genet. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.-L.; Choi, J.; Wakita, T.; Yen, T.B.; Ou, J.-H.J. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Sir, N.; Liang, C.; Chen, W.-L.; Jung, J.U.; Ou, J.-H.J. Perturbation of autophagic pathway by hepatitis C virus. Autophagy 2008, 4, 830–831. [Google Scholar] [CrossRef]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C Virus Genotype 1a Growth and Induction of Autophagy. J. Virol. 2008, 82, 6783. [Google Scholar] [CrossRef]
- Huang, H.; Kang, R.; Wang, J.; Luo, G.; Yang, W.; Zhao, Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy 2013, 9, 175–195. [Google Scholar] [CrossRef]
- Wang, L.; Ou, J.-H.J. Hepatitis C virus and autophagy. Boil. Chem. 2015, 396, 1215–1222. [Google Scholar] [CrossRef]
- Wang, L.; Tian, Y.; Ou, J.-H.J. HCV Induces the Expression of Rubicon and UVRAG to Temporally Regulate the Maturation of Autophagosomes and Viral Replication. PLoS Pathog. 2015, 11, 1004764. [Google Scholar] [CrossRef]
- Kim, S.-J.; Syed, G.; Siddiqui, A. Hepatitis C Virus Induces the Mitochondrial Translocation of Parkin and Subsequent Mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef]
- Hara, Y.; Yanatori, I.; Ikeda, M.; Kiyokage, E.; Nishina, S.; Tomiyama, Y.; Toida, K.; Kishi, F.; Kato, N.; Imamura, M.; et al. Hepatitis C Virus Core Protein Suppresses Mitophagy by Interacting with Parkin in the Context of Mitochondrial Depolarization. Am. J. Pathol. 2014, 184, 3026–3039. [Google Scholar] [CrossRef]
- Kim, S.-J.; Syed, G.; Khan, M.; Chiu, W.-W.; Sohail, M.A.; Gish, R.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed]
- Jassey, A.; Liu, C.-H.; Changou, C.A.; Richardson, C.D.; Hsu, H.-Y.; Lin, L.-T. Hepatitis C Virus Non-Structural Protein 5A (NS5A) Disrupts Mitochondrial Dynamics and Induces Mitophagy. Cells 2019, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Jang, J.Y.; Kim, E.-J.; Cho, E.K.; Ahn, D.-G.; Kim, C.; Park, H.S.; Jeong, S.W.; Lee, S.H.; Kim, S.G.; et al. Ginsenoside Rg3 restores hepatitis C virus-induced aberrant mitochondrial dynamics and inhibits virus propagation. Hepatology 2017, 66, 758–771. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Aita, V.M.; Liang, X.H.; Murty, V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.; Levine, B. Cloning and Genomic Organization of Beclin 1, a Candidate Tumor Suppressor Gene on Chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.-M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.-X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef]
- Lee, Y.A.; Noon, L.A.; Akat, K.M.; Ybanez, M.D.; Lee, T.-F.; Berres, M.-L.; Fujiwara, N.; Goossens, N.; Chou, H.-I.; Parvin-Nejad, F.P.; et al. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat. Commun. 2018, 9, 4962. [Google Scholar] [CrossRef]
- Khambu, B.; Huda, N.; Chen, X.; Antoine, D.J.; Li, Y.; Dai, G.; Köhler, U.A.; Zong, W.-X.; Waguri, S.; Werner, S.; et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J. Clin. Investig. 2018, 128, 2419–2435. [Google Scholar] [CrossRef]
- Ni, H.; Chao, X.; Yang, H.; Deng, F.; Wang, S.; Bai, Q.; Qian, H.; Cui, Y.; Cui, W.; Shi, Y.; et al. Dual Roles of Mammalian Target of Rapamycin in Regulating Liver Injury and Tumorigenesis in Autophagy-Defective Mouse Liver. Hepatology 2019, 70, 2142–2155. [Google Scholar] [CrossRef]
- Yang, H.; Ni, H.-M.; Ding, W.-X. Emerging Players in Autophagy Deficiency-Induced Liver Injury and Tumorigenesis. Gene Expr. 2019, 19, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genome Res. 2007, 21, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genome Res. 2007, 21, 1367–1381. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef]
- Takahashi, Y.; Coppola, M.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nature 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Mariño, G.; Salvador-Montoliu, N.; Fueyo-Silva, A.; Knecht, E.; Mizushima, N.; López-Otín, C. Tissue-specific Autophagy Alterations and Increased Tumorigenesis in Mice Deficient in Atg4C/Autophagin-3. J. Boil. Chem. 2007, 282, 18573–18583. [Google Scholar] [CrossRef]
- Kim, M.-J.; Woo, S.-J.; Yoon, C.-H.; Lee, J.-S.; An, S.; Choi, Y.-H.; Hwang, S.-G.; Yoon, G.; Lee, S.-J. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Boil. Chem. 2011, 286, 12924–19232. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Chen, L.-Y.; Yang, B.; Zhou, L.; Ren, F.; Duan, Z.-P.; Ma, Y.-J. Promotion of mitochondrial energy metabolism during hepatocyte apoptosis in a rat model of acute liver failure. Mol. Med. Rep. 2015, 12, 5035–5041. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Boil. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Severi, T.; Van Malenstein, H.; Verslype, C.; Van Pelt, J.F. Tumor initiation and progression in hepatocellular carcinoma: Risk factors, classification, and therapeutic targets. Acta Pharmacol. Sin. 2010, 31, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Vera-Ramirez, L.; Sanchez-Rovira, P.; Ramirez-Tortosa, M.; Ramírez-Tortosa, C.; Granados-Principal, S.; Lorente, J.A.; Quiles, J.L. Free radicals in breast carcinogenesis, breast cancer progression and cancer stem cells. Biological bases to develop oxidative-based therapies. Crit. Rev. Oncol. 2011, 80, 347–368. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A. The role of inflammation and liver cancer. Adv. Exp. Med. Biol. 2014, 816, 401–435. [Google Scholar]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2010, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Y.; Siraj, S.; Jin, H.; Fan, Y.; Yang, X.; Huang, X.; Wang, X.; Wang, J.; Liu, L.; et al. FUN14 Domain-Containing 1–Mediated Mitophagy Suppresses Hepatocarcinogenesis by Inhibition of Inflammasome Activation in Mice. Hepatology 2019, 69, 604–621. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, B.; Wang, Y.; Zou, C.; Qiao, Q.; Diao, Z.; Mi, Y.; Zhu, D.; Liu, X. Sesamol Induces Human Hepatocellular Carcinoma Cells Apoptosis by Impairing Mitochondrial Function and Suppressing Autophagy. Sci. Rep. 2017, 7, 45728. [Google Scholar] [CrossRef]
- Wei, R.; Cao, J.; Yao, S. Matrine promotes liver cancer cell apoptosis by inhibiting mitophagy and PINK1/Parkin pathways. Cell Stress Chaperones 2018, 23, 1295–1309. [Google Scholar] [CrossRef]
- Kang, X.; Wang, H.; Li, Y.; Xiao, Y.; Zhao, L.; Zhang, T.; Zhou, S.; Zhou, X.; Li, Y.; Shou, Z.; et al. Alantolactone induces apoptosis through ROS-mediated AKT pathway and inhibition of PINK1-mediated mitophagy in human HepG2 cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1961–1970. [Google Scholar] [CrossRef]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.-H.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292.e5. [Google Scholar] [CrossRef]
- Qian, H.; Chao, X.; Ding, W.-X. A PINK1-mediated mitophagy pathway decides the fate of tumors—to be benign or malignant? Autophagy 2018, 14, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Boil. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Boil. 2016, 214, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Rojansky, R.; Cha, M.-Y.; Chan, D. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. eLife 2016, 5, 1144. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Type | Interactor | Activated Conditions | Functions in Mitophagy | Refs |
|---|---|---|---|---|---|
| SQSTM1/p62 | SAR | Ubiquitin | Mitochondrial depolarization | Recruited by Parkin-mediated ubiquitination, favoring mitochondrial cluster and recognition by the autophagy machinery and subsequent elimination | [42,43,44,68,91] |
| NDP52/OPTN | SAR | Ubiquitin | Mitochondrial depolarization | Recruited by PINK1 to mitochondria to activate mitophagy directly, independently of parkin | [68,92,93] |
| BNIP3 | MAR | OMM | Hypoxia | Dual functions in regulating both cell death and mitophagy; Enhanced binding to LC3 when LIR motif is phosphorylated on Ser17 and Ser24 | [51,52] |
| BNIP3L (NIX) | MAR | OMM | Hypoxia; erythrocyte maturation | Binds to LC3 mediating mitochondrial elimination during erythrocyte maturation; Ubiquitinated by Parkin to recruits other SARs (NBR1) | [45,53,54,55] |
| FUNDC1 | MAR | OMM | Hypoxia | Recruits LC3 to initiate mitophagy; Binds to Drp1 to facilitate mitochondrial fission once activated | [57,84] |
| Bcl2-L-13 | MAR | OMM | Mitochondrial depolarization | Stimulates mitochondria fragmentation and induces mitophagy through LC3 binding in HEK293 cells | [56,94] |
| FKBP8 | MAR | OMM | Hypoxia | Recruits LC3A to mediate Parkin-independent mitophagy; Facilitates mitophagy by inducing mitochondrial fragmentation | [85,95] |
| NIPSNAP1/2 | MAR | OMM | Mitochondrial depolarization | Mitochondrial matrix proteins, accumulating on the OMM following mitochondrial depolarization, recruiting autophagy receptors and adaptors | [96] |
| Ambra1 | MAR | OMM | Mitochondrial toxins | Collaborates with E3 ligase HUWE1, binding to LC3 to induce mitochondrial clearance | [67,97] |
| PHB2 | MAR | IMM | Mitochondrial depolarization | Activated upon proteasome-dependent OMM rupture | [46] |
| Cardiolipin | Lipid | OMM | Mitochondrial toxins | Externalizes to OMM and interacts with LC3 under mitochondrial stress in neuron cells | [61] |
| Ceramide | Lipid | OMM | Unknown | Binds LC3 to recruit autophagosomes to the mitochondria resulting in lethal mitophagy in cancer cells | [63] |
| Methods | Pros | Cons | Applications in Liver and Liver Disease Study |
|---|---|---|---|
| Electron Microscopy (EM) | Provides mitochondria-containing autophagosome and autolysosome ultrastructure | Limitations in quantification, steady-state rather than detecting flux | ALD [113,114,115,119] DILI [155,159] I/R [168] HBV/HCV [189] |
| Immunoelectron Microscopy (IEM) | Provides mitochondria-containing autophagosome and autolysosome ultrastructure and related proteins | Not quantitative | ALD [113,119] HBV/HCV [189] |
| Co-localization of LC3 with a Mitochondrial Protein | Large number of cells | The fluorescence-labeled LC3 aggregates may be misleading Not objective and robust Will not be able to detect LC3-independent mitophagy, microautophagy or MDVs | ALD [119,122] NAFLD [86,133,134,136,137,138,139] DILI [158] I/R [166] HBV/HCV [178,179,189] Cancer [217,218] |
| Autophagy/Mitophagy Marker Proteins | Objective Quantitative | Non-specific The intracellular distribution of marker proteins is more important than its total amount, total amount does not equal activity, only steady state | ALD [114,115,119,120,122] NAFLD [133,134,136,137,138,139,140] I/R [165,167,168,169,172,173] HBV/HCV [178,179,189,190,192] Cancer [216,217,218] |
| Mitochondrial Mass | Objective Quantitative | Only reflect steady state, rather than flux, nor the degradation or the initiation process of mitophagy. Mitochondrial outer membrane proteins are also degraded by proteasome. | ALD [114,116,119,122] NAFLD [134,140] DILI [155,157,159] HBV/HCV [192] Cancer [216] |
| pH-Sensitive Fluorescent Probe | Specific High image quality Apply in vivo and in vitro | The expression level of fluorescent proteins varies in different cells/tissues Not robust and easy to be dependent on individuals who are performing the quantification. Also, the half-life of the red puncta in the lysosomes may be dependent on cellular context and conditions, e.g., the activities of the lysosomal proteases | NAFLD [86] DILI [159] Cancer [216] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; McKeen, T.; Zhang, J.; Ding, W.-X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. https://doi.org/10.3390/cells9040837
Ma X, McKeen T, Zhang J, Ding W-X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells. 2020; 9(4):837. https://doi.org/10.3390/cells9040837
Chicago/Turabian StyleMa, Xiaowen, Tara McKeen, Jianhua Zhang, and Wen-Xing Ding. 2020. "Role and Mechanisms of Mitophagy in Liver Diseases" Cells 9, no. 4: 837. https://doi.org/10.3390/cells9040837
APA StyleMa, X., McKeen, T., Zhang, J., & Ding, W.-X. (2020). Role and Mechanisms of Mitophagy in Liver Diseases. Cells, 9(4), 837. https://doi.org/10.3390/cells9040837