The Role of Autophagy in Pancreatic Cancer: From Bench to the Dark Bedside

 , ,
, ,
Abstract
1. Introduction
2. Autophagy and Molecular Mechanisms
2.1. Execution of Autophagy
2.2. Stimulation of Autophagy
2.3. Cargo Selection
3. Autophagy and Pancreatic Cancer
3.1. The Role of Autophagy in Pancreatic Cancer
3.1.1. Macroautophagy and Pancreatic Cancer
3.1.2. Other Autophagic Machineries and Pancreatic Cancer
3.2. The Role of Autophagy in the Micro/Macroenvironment of Pancreatic Cancer
3.2.1. Autophagy and Cancer Associated Fibroblasts
3.2.2. Autophagy and Immune Cells
3.2.3. Autophagy and the Macroenvironment of Pancreatic Cancer
4. Targeting Autophagy as a Therapeutic Approach for Pancreatic Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers (Basel) 2017, 9, 42. [Google Scholar] [CrossRef]
- Abada, A.; Elazar, Z. Getting ready for building: Signaling and autophagosome biogenesis. EMBO Rep. 2014, 15, 839–852. [Google Scholar] [CrossRef]
- Noda, N.N.; Inagaki, F. Mechanisms of Autophagy. Annu. Rev. Biophys. 2015, 44, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Flaks, B.; Moore, M.A.; Flaks, A. Ultrastructural analysis of pancreatic carcinogenesis. IV. Pseudoductular transformation of acini in the hamster pancreas during N-nitroso-bis(2-hydroxypropyl)amine carcinogenesis. Carcinogenesis 1981, 2, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Invest. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S. Biological Roles of Alternative Autophagy. Mol. Cells 2018, 41, 50–54. [Google Scholar] [PubMed]
- Honda, S.; Arakawa, S.; Nishida, Y.; Yamaguchi, H.; Ishii, E.; Shimizu, S. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat. Commun. 2014, 5, 4004. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Arakawa, S.; Kanaseki, T.; Miyatsuka, T.; Fujitani, Y.; Watada, H.; Tsujimoto, Y.; Shimizu, S. Golgi membrane-associated degradation pathway in yeast and mammals. EMBO J. 2016, 35, 1991–2007. [Google Scholar] [CrossRef]
- Ra, E.A.; Lee, T.A.; Won Kim, S.; Park, A.; Choi, H.J.; Jang, I.; Kang, S.; Hee Cheon, J.; Cho, J.W.; Eun Lee, J.; et al. TRIM31 promotes Atg5/Atg7-independent autophagy in intestinal cells. Nat. Commun. 2016, 7, 11726. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef]
- Sasaki, Y.; Odan, M.; Yamamoto, S.; Kida, S.; Ueyama, A.; Shimizu, M.; Haruna, T.; Watanabe, A.; Okuno, T. Discovery of a potent orally bioavailable retinoic acid receptor-related orphan receptor-gamma-t (RORgammat) inhibitor, S18-000003. Bioorg. Med. Chem. Lett. 2018, 28, 3549–3553. [Google Scholar] [CrossRef]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; Tiano, S.; Diaz, A.; Bartholdy, B.A.; Will, B.; Cuervo, A.M. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2019, 17, e3000301. [Google Scholar] [CrossRef] [PubMed]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, L.; Jiao, H.; Guan, L.; Zha, J.; Li, X.; Wu, M.; Wang, Z.; Han, J.; You, H. Chaperone-mediated autophagy prevents apoptosis by degrading BBC3/PUMA. Autophagy 2015, 11, 1623–1635. [Google Scholar] [CrossRef]
- Cheng, Z. The FoxO-Autophagy Axis in Health and Disease. Trends Endocrinol. Metab. 2019, 30, 658–671. [Google Scholar] [CrossRef]
- Czarny, P.; Pawlowska, E.; Bialkowska-Warzecha, J.; Kaarniranta, K.; Blasiak, J. Autophagy in DNA damage response. Int. J. Mol. Sci. 2015, 16, 2641–2662. [Google Scholar] [CrossRef]
- Fasano, C.; Disciglio, V.; Bertora, S.; Lepore Signorile, M.; Simone, C. FOXO3a from the Nucleus to the Mitochondria: A Round Trip in Cellular Stress Response. Cells 2019, 8, 1110. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2019, 432, 80–103. [Google Scholar] [CrossRef]
- Perera, R.M.; Bardeesy, N. Pancreatic Cancer Metabolism: Breaking It Down to Build It Back Up. Cancer Discov. 2015, 5, 1247–1261. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.R.; Yaffee, P.M.; Jamil, L.H.; Lo, S.K.; Nissen, N.; Pandol, S.J.; Tuli, R.; Hendifar, A.E. Pancreatic cancer cachexia: A review of mechanisms and therapeutics. Front. Physiol. 2014, 5, 88. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Réz, G.; Tóth, S.; Pálfia, Z. Cellular autophagic capacity is highly increased in azaserine-induced premalignant atypical acinar nodule cells. Carcinogenesis 1999, 20, 1893–1898. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Akar, U.; Ozpolat, B.; Mehta, K.; Fok, J.; Kondo, Y.; Lopez-Berestein, G. Tissue transglutaminase inhibits autophagy in pancreatic cancer cells. Mol. Cancer Res. 2007, 5, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Ozpolat, B.; Akar, U.; Mehta, K.; Lopez-Berestein, G. PKC delta and tissue transglutaminase are novel inhibitors of autophagy in pancreatic cancer cells. Autophagy 2007, 3, 480–483. [Google Scholar] [CrossRef]
- Fujii, S.; Mitsunaga, S.; Yamazaki, M.; Hasebe, T.; Ishii, G.; Kojima, M.; Kinoshita, T.; Ueno, T.; Esumi, H.; Ochiai, A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008, 99, 1813–1819. [Google Scholar] [CrossRef]
- Ko, Y.H.; Cho, Y.S.; Won, H.S.; Jeon, E.K.; An, H.J.; Hong, S.U.; Park, J.H.; Lee, M.A. Prognostic significance of autophagy-related protein expression in resected pancreatic ductal adenocarcinoma. Pancreas 2013, 42, 829–835. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D.; Schapiro, N.E.; Livesey, K.M.; Farkas, A.; Loughran, P.; Bierhaus, A.; Lotze, M.T.; Zeh, H.J. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010, 17, 666–676. [Google Scholar] [CrossRef]
- Kang, R.; Livesey, K.M.; Zeh, H.J.; Loze, M.T.; Tang, D. HMGB1: A novel Beclin 1-binding protein active in autophagy. Autophagy 2010, 6, 1209–1211. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Rausch, V.; Liu, L.; Apel, A.; Rettig, T.; Gladkich, J.; Labsch, S.; Kallifatidis, G.; Kaczorowski, A.; Groth, A.; Gross, W.; et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J. Pathol. 2012, 227, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, D.; Liu, Y.; Su, Z.; Zhang, L.; Chen, F.; Zhou, Y.; Wu, Y.; Yu, M.; Zhang, Z.; et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int. 2013, 13, 119. [Google Scholar] [CrossRef] [PubMed]
- Balic, A.; Sørensen, M.D.; Trabulo, S.M.; Sainz, B., Jr.; Cioffi, M.; Vieira, C.R.; Miranda-Lorenzo, I.; Hidalgo, M.; Kleeff, J.; Erkan, M.; et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, C.; Menegazzi, M.; Padroni, C.; Dando, I.; Dalla Pozza, E.; Gregorelli, A.; Costanzo, C.; Palmieri, M.; Donadelli, M. Autophagy induced by p53-reactivating molecules protects pancreatic cancer cells from apoptosis. Apoptosis 2013, 18, 337–346. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Yang, A.; Kimmelman, A.C. Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status. Autophagy 2014, 10, 1683–1684. [Google Scholar] [CrossRef]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef]
- Morgan, M.J.; Gamez, G.; Menke, C.; Hernandez, A.; Thorburn, J.; Gidan, F.; Staskiewicz, L.; Morgan, S.; Cummings, C.; Maycotte, P.; et al. Regulation of autophagy and chloroquine sensitivity by oncogenic RAS in vitro is context-dependent. Autophagy 2014, 10, 1814–1826. [Google Scholar] [CrossRef]
- Görgülü, K.; Diakopoulos, K.N.; Ai, J.; Schoeps, B.; Kabacaoglu, D.; Karpathaki, A.F.; Ciecielski, K.J.; Kaya-Aksoy, E.; Ruess, D.A.; Berninger, A.; et al. Levels of the Autophagy-Related 5 Protein Affect Progression and Metastasis of Pancreatic Tumors in Mice. Gastroenterology 2019, 156, 203.e20–217.e20. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Herter-Sprie, G.; Zhang, H.; Lin, E.Y.; Biancur, D.; Wang, X.; Deng, J.; Hai, J.; Yang, S.; Wong, K.K.; et al. Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discov. 2018, 8, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Manent, J.; Banerjee, S.; de Matos Simoes, R.; Zoranovic, T.; Mitsiades, C.; Penninger, J.M.; Simpson, K.J.; Humbert, P.O.; Richardson, H.E. Autophagy suppresses Ras-driven epithelial tumourigenesis by limiting the accumulation of reactive oxygen species. Oncogene 2017, 36, 5658–5660. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiong, H.; Liu, D.; Hill, C.; Ertay, A.; Li, J.; Zou, Y.; Miller, P.; White, E.; Downward, J.; et al. Autophagy inhibition specifically promotes epithelial-mesenchymal transition and invasion in RAS-mutated cancer cells. Autophagy 2019, 15, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Todoric, J.; Antonucci, L.; Di Caro, G.; Li, N.; Wu, X.; Lytle, N.K.; Dhar, D.; Banerjee, S.; Fagman, J.B.; Browne, C.D.; et al. Stress-Activated NRF2-MDM2 Cascade Controls Neoplastic Progression in Pancreas. Cancer Cell 2017, 32, 824.e8–839.e8. [Google Scholar] [CrossRef]
- Liang, C.; Xu, J.; Meng, Q.; Zhang, B.; Liu, J.; Hua, J.; Zhang, Y.; Shi, S.; Yu, X. TGFB1-induced autophagy affects the pattern of pancreatic cancer progression in distinct ways depending on SMAD4 status. Autophagy 2019, 1–15. [Google Scholar] [CrossRef]
- Lee, C.S.; Lee, L.C.; Yuan, T.L.; Chakka, S.; Fellmann, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4508–4517. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef]
- Mareninova, O.A.; Sendler, M.; Malla, S.R.; Yakubov, I.; French, S.W.; Tokhtaeva, E.; Vagin, O.; Oorschot, V.; Lüllmann-Rauch, R.; Blanz, J.; et al. Lysosome associated membrane proteins maintain pancreatic acinar cell homeostasis: LAMP-2 deficient mice develop pancreatitis. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 678–694. [Google Scholar] [CrossRef]
- Welsch, T.; Endlich, K.; Giese, T.; Büchler, M.W.; Schmidt, J. Eps8 is increased in pancreatic cancer and required for dynamic actin-based cell protrusions and intercellular cytoskeletal organization. Cancer Lett. 2007, 255, 205–218. [Google Scholar] [CrossRef]
- Welsch, T.; Younsi, A.; Disanza, A.; Rodriguez, J.A.; Cuervo, A.M.; Scita, G.; Schmidt, J. Eps8 is recruited to lysosomes and subjected to chaperone-mediated autophagy in cancer cells. Exp. Cell Res. 2010, 316, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Zuiderweg, E.R.; Hightower, L.E.; Gestwicki, J.E. The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones 2017, 22, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.J.; Lee, H.S.; Keum, B.; Seo, Y.S.; Jeen, Y.T.; Chun, H.J.; Um, S.H.; Kim, C.D. Expression of heat shock protein 70 modulates the chemoresponsiveness of pancreatic cancer. Gut Liver 2013, 7, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Ali, D.M.; Ansari, S.S.; Zepp, M.; Knapp-Mohammady, M.; Berger, M.R. Optineurin downregulation induces endoplasmic reticulum stress, chaperone-mediated autophagy, and apoptosis in pancreatic cancer cells. Cell Death Discov. 2019, 5, 128. [Google Scholar] [CrossRef]
- Springer, M.Z.; Macleod, K.F. In Brief: Mitophagy: Mechanisms and role in human disease. J. Pathol. 2016, 240, 253–255. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- Humpton, T.J.; Alagesan, B.; DeNicola, G.M.; Lu, D.; Yordanov, G.N.; Leonhardt, C.S.; Yao, M.A.; Alagesan, P.; Zaatari, M.N.; Park, Y.; et al. Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer. Cancer Discov. 2019, 9, 1268–1287. [Google Scholar] [CrossRef]
- Akada, M.; Crnogorac-Jurcevic, T.; Lattimore, S.; Mahon, P.; Lopes, R.; Sunamura, M.; Matsuno, S.; Lemoine, N.R. Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clin. Cancer Res. 2005, 11, 3094–3101. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Esposito, I.; Giese, T.; Ketterer, K.; Büchler, M.W.; Giese, N.A.; Friess, H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 2005, 24, 4421–4432. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Wen, Q.; Xie, Y.; Liu, J.; Kroemer, G.; et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 2018, 46, 441.e8–455.e8. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.J.; Drake, J.C.; Cui, D.; Zhang, M.; Perry, H.M.; Kashatus, J.A.; Kusminski, C.M.; Scherer, P.E.; Kashatus, D.F.; Okusa, M.D.; et al. Conditional MitoTimer reporter mice for assessment of mitochondrial structure, oxidative stress, and mitophagy. Mitochondrion 2019, 44, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, J.I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 2017, 66, 517.e9–532.e9. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Siciliano, D.; Calcagni, A.; Monfregola, J.; Punzi, S.; Pastore, N.; Eastes, A.N.; Davis, O.; De Cegli, R.; Zampelli, A.; et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 2017, 356, 1188–1192. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.J.; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef]
- Bynigeri, R.R.; Jakkampudi, A.; Jangala, R.; Subramanyam, C.; Sasikala, M.; Rao, G.V.; Reddy, D.N.; Talukdar, R. Pancreatic stellate cell: Pandora’s box for pancreatic disease biology. World J. Gastroenterol. 2017, 23, 382–405. [Google Scholar] [CrossRef] [PubMed]
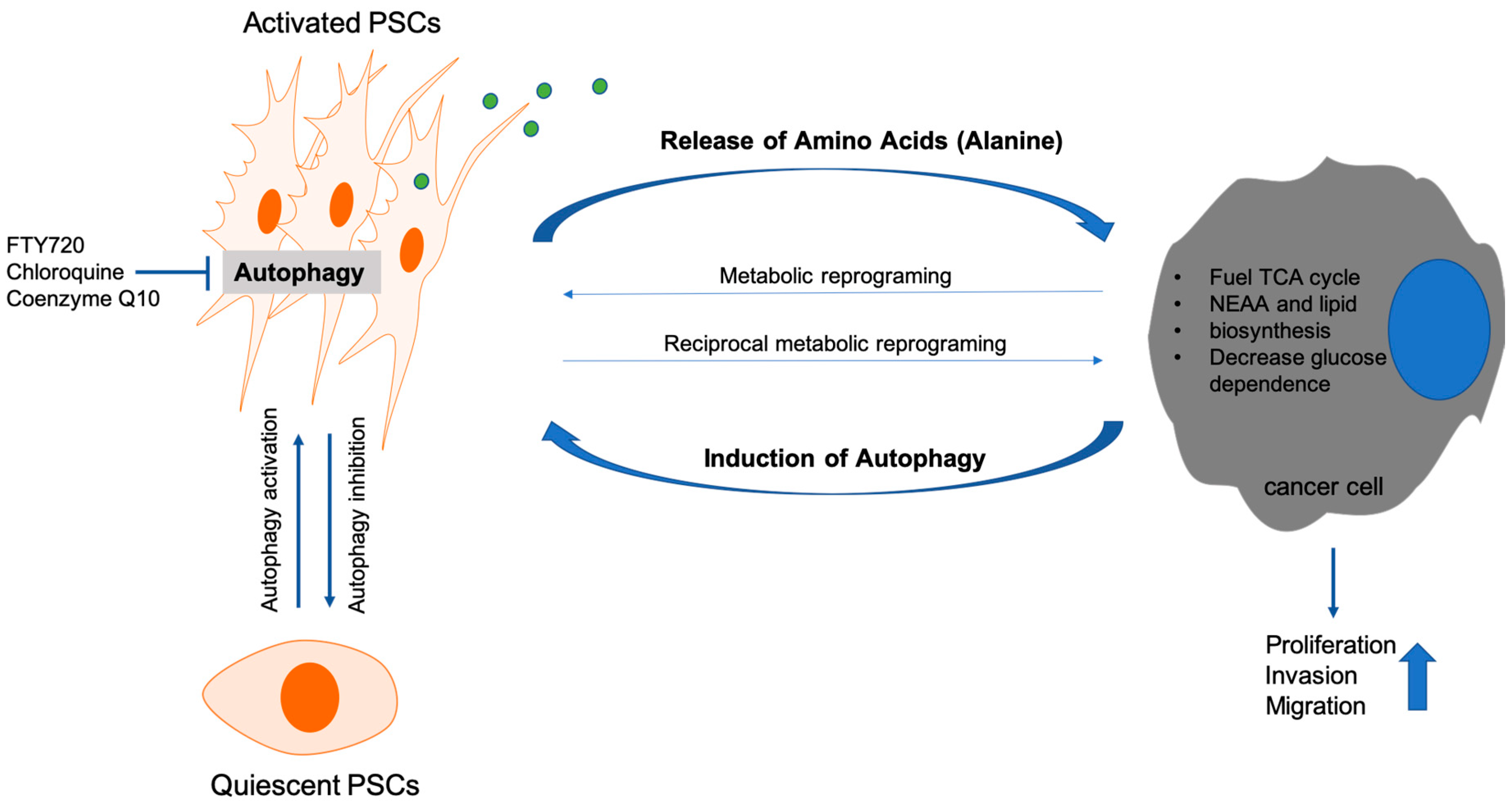
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492.e24–1506.e24. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Yang, J.; Wu, J.; Meng, Q.; Hao, J. Coenzyme Q10 inhibits the activation of pancreatic stellate cells through PI3K/AKT/mTOR signaling pathway. Oncotarget 2017, 8, 92300–92311. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Li, C.; Gao, G.; Zhuo, Y.; Yang, L.; Cui, N.; Zhang, S. FTY720 inhibits the activation of pancreatic stellate cells by promoting apoptosis and suppressing autophagy via the AMPK/mTOR pathway. Life Sci. 2019, 217, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Cui, L.H.; Zhuo, Y.Z.; Hu, J.G.; Cui, N.Q.; Zhang, S.K. Inhibiting autophagy promotes collagen degradation by regulating matrix metalloproteinases in pancreatic stellate cells. Life Sci. 2018, 208, 276–283. [Google Scholar] [CrossRef]
- Su, H.Y.; Waldron, R.T.; Gong, R.; Ramanujan, V.K.; Pandol, S.J.; Lugea, A. The Unfolded Protein Response Plays a Predominant Homeostatic Role in Response to Mitochondrial Stress in Pancreatic Stellate Cells. PLoS ONE 2016, 11, e0148999. [Google Scholar] [CrossRef]
- Molejon, M.I.; Swayden, M.; Fanale, D.; Bintz, J.; Gayet, O.; Soubeyran, P.; Iovanna, J. Chloroquine plays a cell-dependent role in the response to treatment of pancreatic adenocarcinoma. Oncotarget 2018, 9, 30837–30846. [Google Scholar] [CrossRef]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.N.; Vidal, N.; Berthezène, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef]
- Guillaumond, F.; Vasseur, S. The metabolic facet of pancreatic cancer: How hypoxia shapes fatal cancer cells. Cell Cycle 2013, 12, 1155–1156. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef]
- Serrao, E.M.; Kettunen, M.I.; Rodrigues, T.B.; Dzien, P.; Wright, A.J.; Gopinathan, A.; Gallagher, F.A.; Lewis, D.Y.; Frese, K.K.; Almeida, J.; et al. MRI with hyperpolarised [1-13C]pyruvate detects advanced pancreatic preneoplasia prior to invasive disease in a mouse model. Gut 2016, 65, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2019, 39, 517–560. [Google Scholar] [CrossRef] [PubMed]
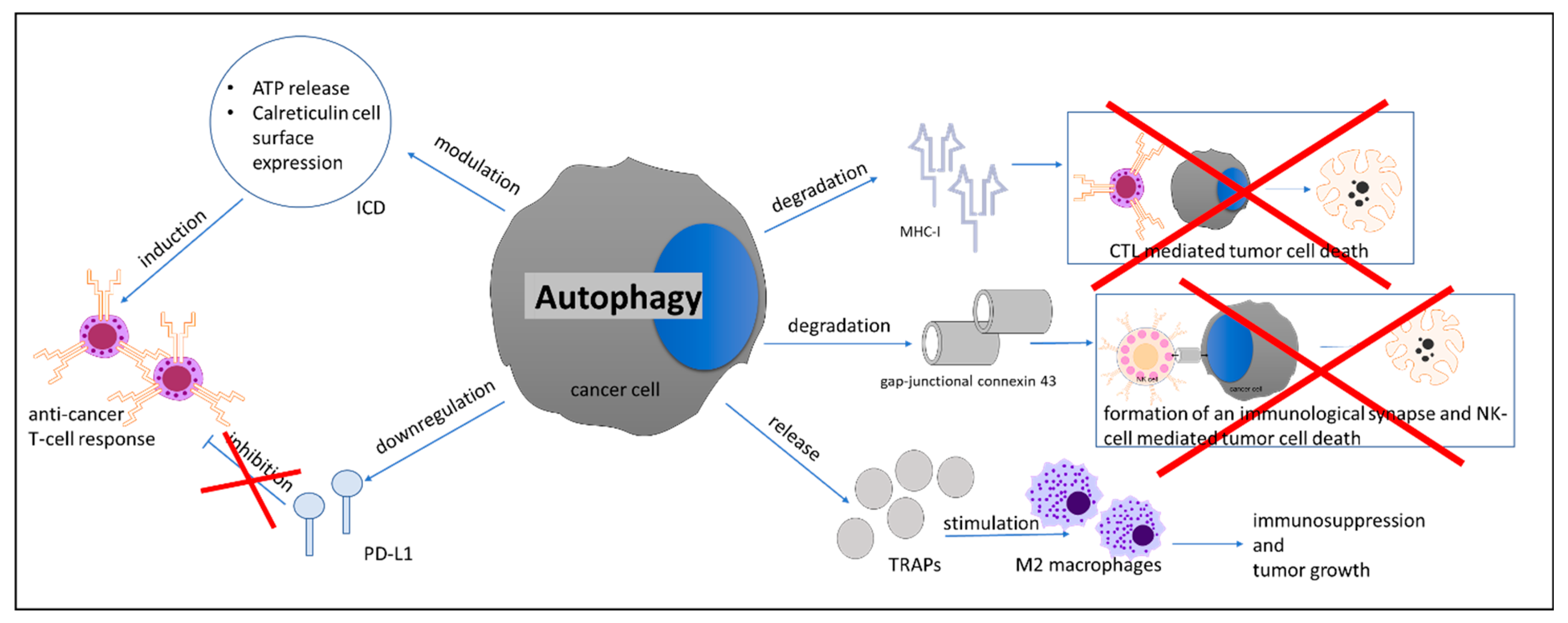
- Fruci, D.; Benevolo, M.; Cifaldi, L.; Lorenzi, S.; Lo Monaco, E.; Tremante, E.; Giacomini, P. Major histocompatibility complex class i and tumour immuno-evasion: How to fool T cells and natural killer cells at one time. Curr. Oncol. 2012, 19, 39–41. [Google Scholar] [CrossRef][Green Version]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef]
- Li, B.; Lei, Z.; Lichty, B.D.; Li, D.; Zhang, G.M.; Feng, Z.H.; Wan, Y.; Huang, B. Autophagy facilitates major histocompatibility complex class I expression induced by IFN-gamma in B16 melanoma cells. Cancer Immunol. Immunother. 2010, 59, 313–321. [Google Scholar] [CrossRef]
- Zeng, H.; Zhang, W.; Gong, Y.; Xie, C. Radiotherapy activates autophagy to increase CD8(+) T cell infiltration by modulating major histocompatibility complex class-I expression in non-small cell lung cancer. J. Int. Med. Res. 2019, 47, 3818–3830. [Google Scholar] [CrossRef]
- Tittarelli, A.; Janji, B.; Van Moer, K.; Noman, M.Z.; Chouaib, S. The Selective Degradation of Synaptic Connexin 43 Protein by Hypoxia-induced Autophagy Impairs Natural Killer Cell-mediated Tumor Cell Killing. J. Biol. Chem. 2015, 290, 23670–23679. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking Hypoxia-Induced Autophagy in Tumors Restores Cytotoxic T-Cell Activity and Promotes Regression. Cancer Res. 2011, 71, 5976–5986. [Google Scholar] [CrossRef]
- Wen, Z.F.; Liu, H.; Gao, R.; Zhou, M.; Ma, J.; Zhang, Y.; Zhao, J.; Chen, Y.; Zhang, T.; Huang, F.; et al. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J. Immunother. Cancer 2018, 6, 151. [Google Scholar] [CrossRef]
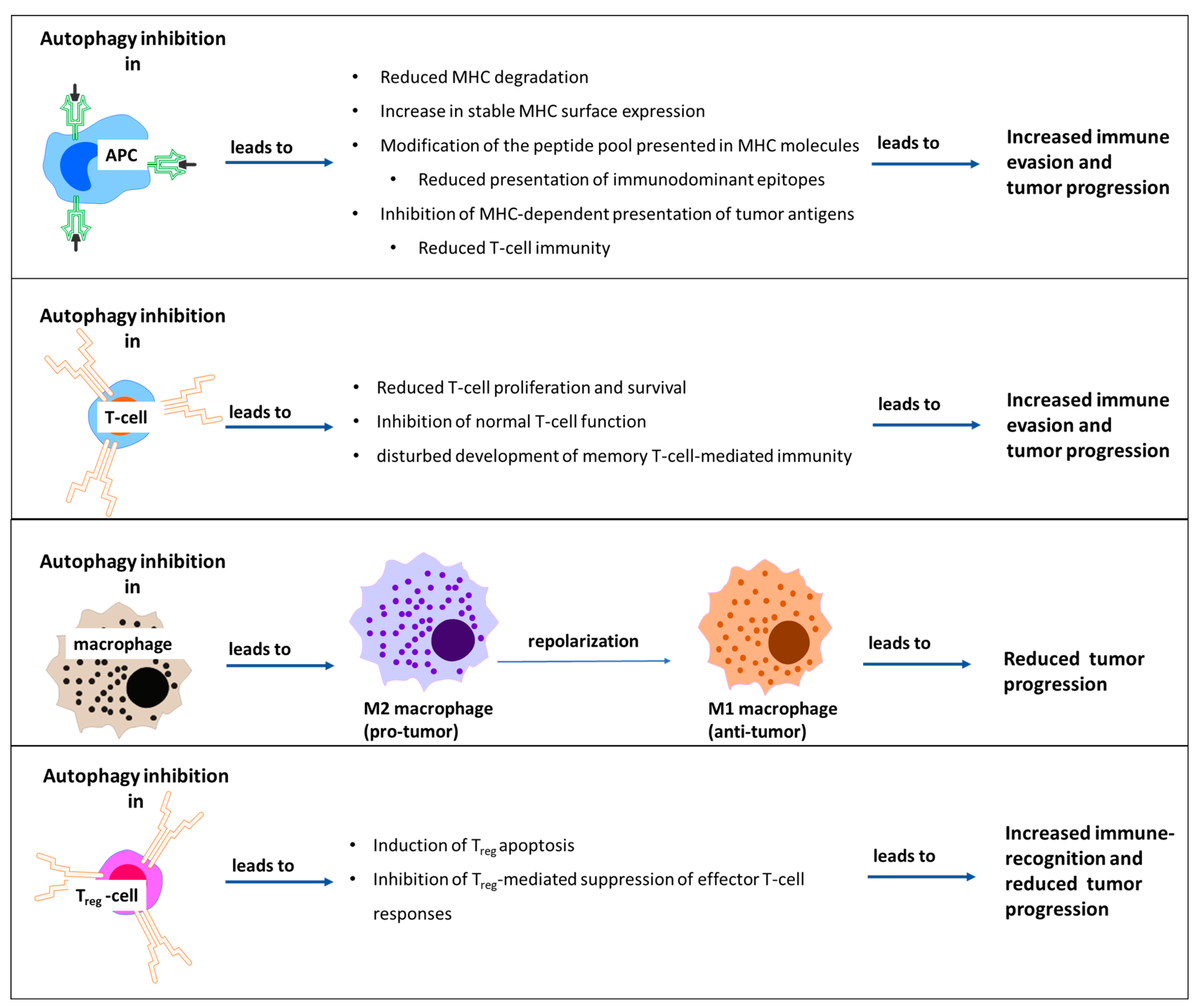
- Loi, M.; Müller, A.; Steinbach, K.; Niven, J.; Barreira da Silva, R.; Paul, P.; Ligeon, L.A.; Caruso, A.; Albrecht, R.A.; Becker, A.C.; et al. Macroautophagy Proteins Control MHC Class I Levels on Dendritic Cells and Shape Anti-viral CD8(+) T Cell Responses. Cell Rep. 2016, 15, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Vidyarthi, A.; Pahari, S.; Negi, S.; Aqdas, M.; Nadeem, S.; Agnihotri, T.; Agrewala, J.N. Signaling through NOD-2 and TLR-4 Bolsters the T cell Priming Capability of Dendritic cells by Inducing Autophagy. Sci. Rep. 2016, 6, 19084. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Feng, Y.; Cui, X.; Wang, Q.; Pan, X. Autophagy inhibition induces the repolarisation of tumour-associated macrophages and enhances chemosensitivity of laryngeal cancer cells to cisplatin in mice. Cancer Immunol. Immunother. 2019, 68, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef]
- Kabacaoglu, D.; Ciecielski, K.J.; Ruess, D.A.; Algül, H. Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front. Immunol. 2018, 9, 1878. [Google Scholar] [CrossRef]
- Henderson, S.E.; Makhijani, N.; Mace, T.A. Pancreatic Cancer-Induced Cachexia and Relevant Mouse Models. Pancreas 2018, 47, 937–945. [Google Scholar] [CrossRef]
- Egerman, M.A.; Glass, D.J. Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 59–68. [Google Scholar] [CrossRef]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef]
- Johns, N.; Hatakeyama, S.; Stephens, N.A.; Degen, M.; Degen, S.; Frieauff, W.; Lambert, C.; Ross, J.A.; Roubenoff, R.; Glass, D.J.; et al. Clinical classification of cancer cachexia: Phenotypic correlates in human skeletal muscle. PLoS ONE 2014, 9, e83618. [Google Scholar] [CrossRef]
- Aversa, Z.; Pin, F.; Lucia, S.; Penna, F.; Verzaro, R.; Fazi, M.; Colasante, G.; Tirone, A.; Fanelli, F.R.; Ramaccini, C.; et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 2016, 6, 30340. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- de Castro, G.S.; Simoes, E.; Lima, J.D.C.C.; Ortiz-Silva, M.; Festuccia, W.T.; Tokeshi, F.; Alcântara, P.S.; Otoch, J.P.; Coletti, D.; Seelaender, M. Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers 2019, 11, 1264. [Google Scholar] [CrossRef] [PubMed]
- Ashour, A.A.; Abdel-Aziz, A.A.; Mansour, A.M.; Alpay, S.N.; Huo, L.; Ozpolat, B. Targeting elongation factor-2 kinase (eEF-2K) induces apoptosis in human pancreatic cancer cells. Apoptosis 2014, 19, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef]
- Hrzenjak, A.; Kremser, M.L.; Strohmeier, B.; Moinfar, F.; Zatloukal, K.; Denk, H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 2008, 216, 495–504. [Google Scholar] [CrossRef]
- Chun, S.G.; Zhou, W.; Yee, N.S. Combined targeting of histone deacetylases and hedgehog signaling enhances cytoxicity in pancreatic cancer. Cancer Biol. Ther. 2009, 8, 1328–1339. [Google Scholar] [CrossRef]
- Mujumdar, N.; Mackenzie, T.N.; Dudeja, V.; Chugh, R.; Antonoff, M.B.; Borja-Cacho, D.; Sangwan, V.; Dawra, R.; Vickers, S.M.; Saluja, A.K. Triptolide induces cell death in pancreatic cancer cells by apoptotic and autophagic pathways. Gastroenterology 2010, 139, 598–608. [Google Scholar] [CrossRef]
- Udelnow, A.; Kreyes, A.; Ellinger, S.; Landfester, K.; Walther, P.; Klapperstueck, T.; Wohlrab, J.; Henne-Bruns, D.; Knippschild, U.; Würl, P. Omeprazole inhibits proliferation and modulates autophagy in pancreatic cancer cells. PLoS ONE 2011, 6, e20143. [Google Scholar] [CrossRef]
- Hashimoto, D.; Bläuer, M.; Hirota, M.; Ikonen, N.H.; Sand, J.; Laukkarinen, J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur. J. Cancer 2014, 50, 1382–1390. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial ID | Clinical Trial | Treatment | Phase |
|---|---|---|---|
| NCT01777477 | Adjuvant Effect of Chloroquine on Gemcitabine | Chloroquine + Gemcitabine | Phase 1 |
| NCT01273805 | Hydroxychloroquine in Previously Treated Patients With Metastatic Pancreatic Cancer | Hydroxychloroquine | Phase 2 |
| NCT01494155 | Short Course Radiation Therapy With Proton or Photon Beam Capecitabine and Hydroxychloroquine for Resectable Pancreatic Cancer | Capecitabine + Hydroxychloroquine + Proton or Photon Radiation Therapy | Phase 2 |
| NCT04132505 | Binimetinib and Hydroxychloroquine in Treating Patients With KRAS Mutant Metastatic Pancreatic Cancer | Binimetinib + Hydroxychloroquine | Phase 1 |
| NCT03344172 | Pre-Operative Trial (PGHA vs. PGH) for Resectable Pancreatic Cancer (17-134) | Gemcitabine + Nab-Paclitaxel + Hydroxychloroquine + Avelumab | Phase 2 |
| NCT01978184 | Randomized Phase II Trial of Pre-Operative Gemcitabine and Nab Paclitacel With or With Out Hydroxychloroquine | Gemcitabine + Abraxane + Hydroxychloroquine | Phase 2 |
| NCT03825289 | Trametinib and Hydroxychloroquine in Treating Patients With Pancreatic Cancer (THREAD) | Hydroxychloroquine + Trametinib | Phase 1 |
| NCT01128296 | Study of Pre-surgery Gemcitabine + Hydroxychloroquine (GcHc) in Stage IIb or III Adenocarcinoma of the Pancreas | Hydroxychloroquine + Gemcitabine | Phase 1/2 |
| NCT01506973 | A Phase I/II/Pharmacodynamic Study of Hydroxychloroquine in Combination With Gemcitabine/Abraxane to Inhibit Autophagy in Pancreatic Cancer | Hydroxychloroquine + Gemcitabine | Phase 1/2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Görgülü, K.; Diakopoulos, K.N.; Kaya-Aksoy, E.; Ciecielski, K.J.; Ai, J.; Lesina, M.; Algül, H. The Role of Autophagy in Pancreatic Cancer: From Bench to the Dark Bedside. Cells 2020, 9, 1063. https://doi.org/10.3390/cells9041063
Görgülü K, Diakopoulos KN, Kaya-Aksoy E, Ciecielski KJ, Ai J, Lesina M, Algül H. The Role of Autophagy in Pancreatic Cancer: From Bench to the Dark Bedside. Cells. 2020; 9(4):1063. https://doi.org/10.3390/cells9041063
Chicago/Turabian StyleGörgülü, Kıvanç, Kalliope N. Diakopoulos, Ezgi Kaya-Aksoy, Katrin J. Ciecielski, Jiaoyu Ai, Marina Lesina, and Hana Algül. 2020. "The Role of Autophagy in Pancreatic Cancer: From Bench to the Dark Bedside" Cells 9, no. 4: 1063. https://doi.org/10.3390/cells9041063
APA StyleGörgülü, K., Diakopoulos, K. N., Kaya-Aksoy, E., Ciecielski, K. J., Ai, J., Lesina, M., & Algül, H. (2020). The Role of Autophagy in Pancreatic Cancer: From Bench to the Dark Bedside. Cells, 9(4), 1063. https://doi.org/10.3390/cells9041063