A Signaling Crosstalk between BMP9 and HGF/c-Met Regulates Mouse Adult Liver Progenitor Cell Survival



 and
and 
Abstract
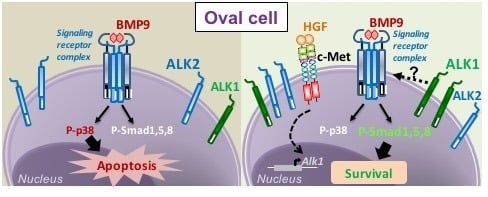
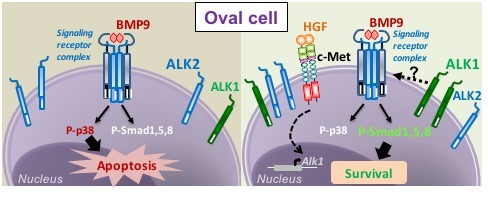{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Lines and Culture Conditions
2.3. Analysis of Cell Number
2.4. Measurement of Caspase-3-like Enzymatic Activity
2.5. Measurement of Apoptotic Index
2.6. Protein Isolation and Western Blot Analysis
2.7. RNA Isolation and Reverse Transcription–quantitative Polymerase Chain Reaction (RT–qPCR)
2.8. Gene Silencing by shRNA and siRNA
2.9. Transcriptional Reporter Assay
2.10. Statistical Analysis
3. Results
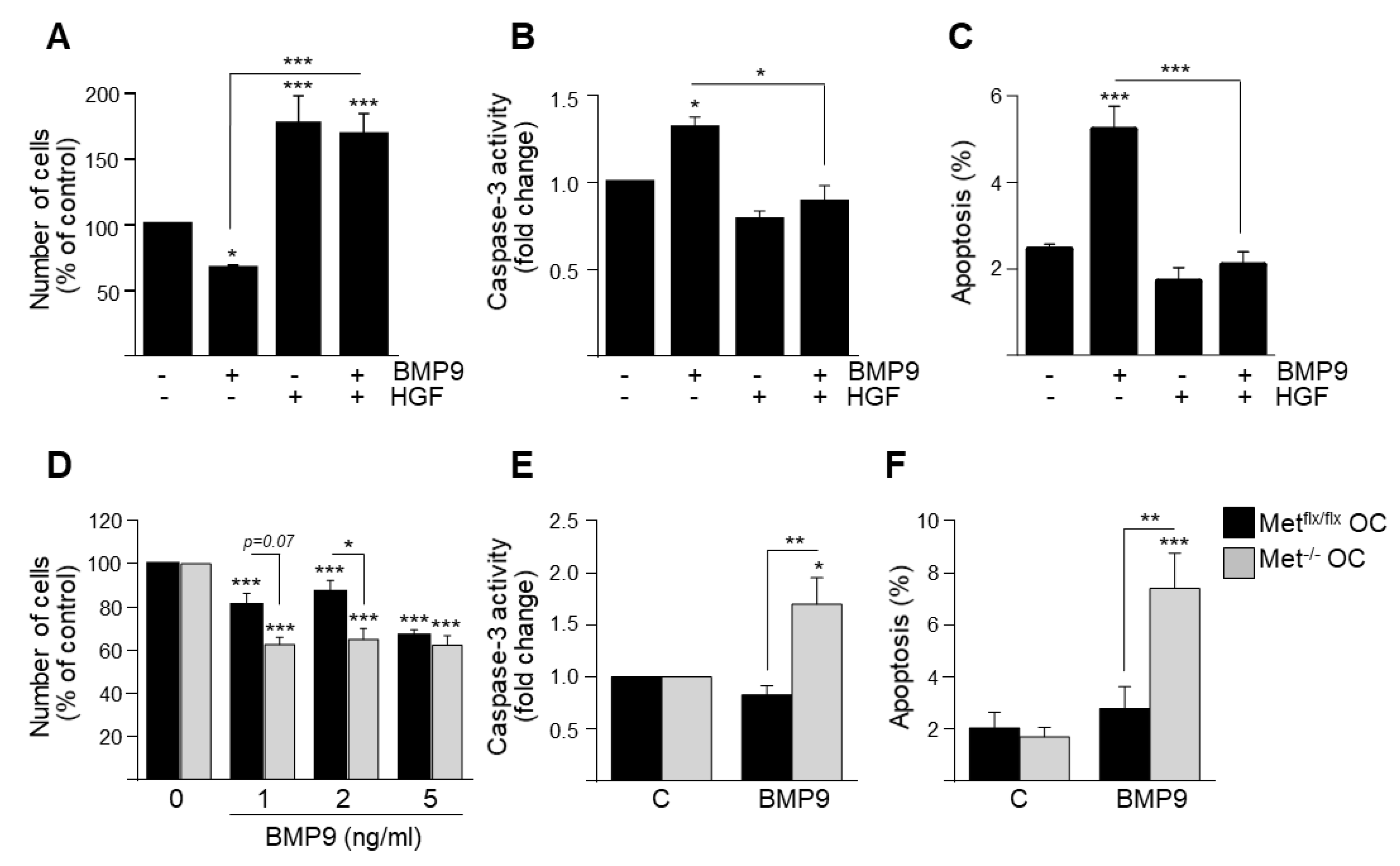
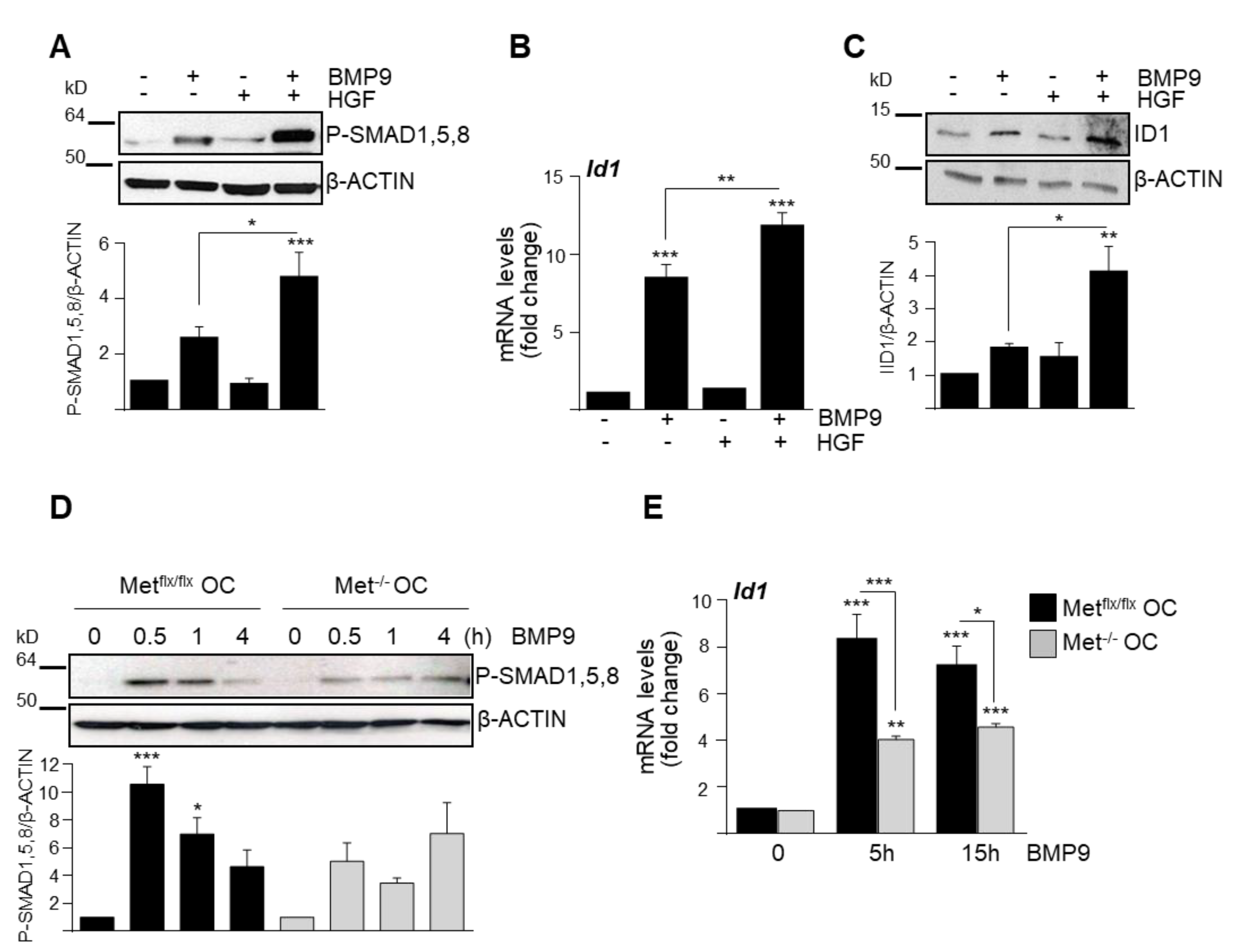
3.1. HGF/c-Met Signaling Inhibits BMP9-Triggered Apoptosis in Oval Cells while Potentiating SMAD Signaling
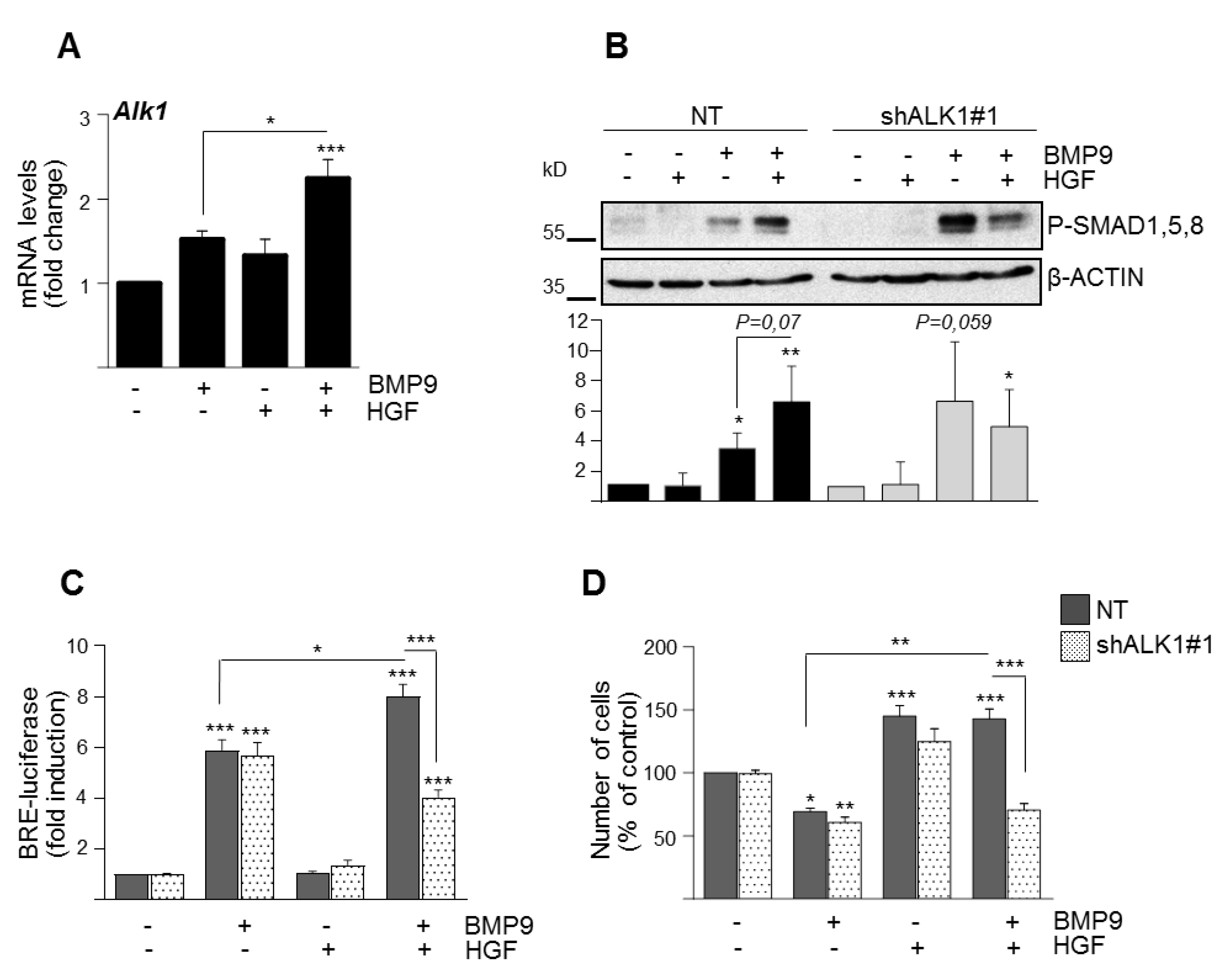
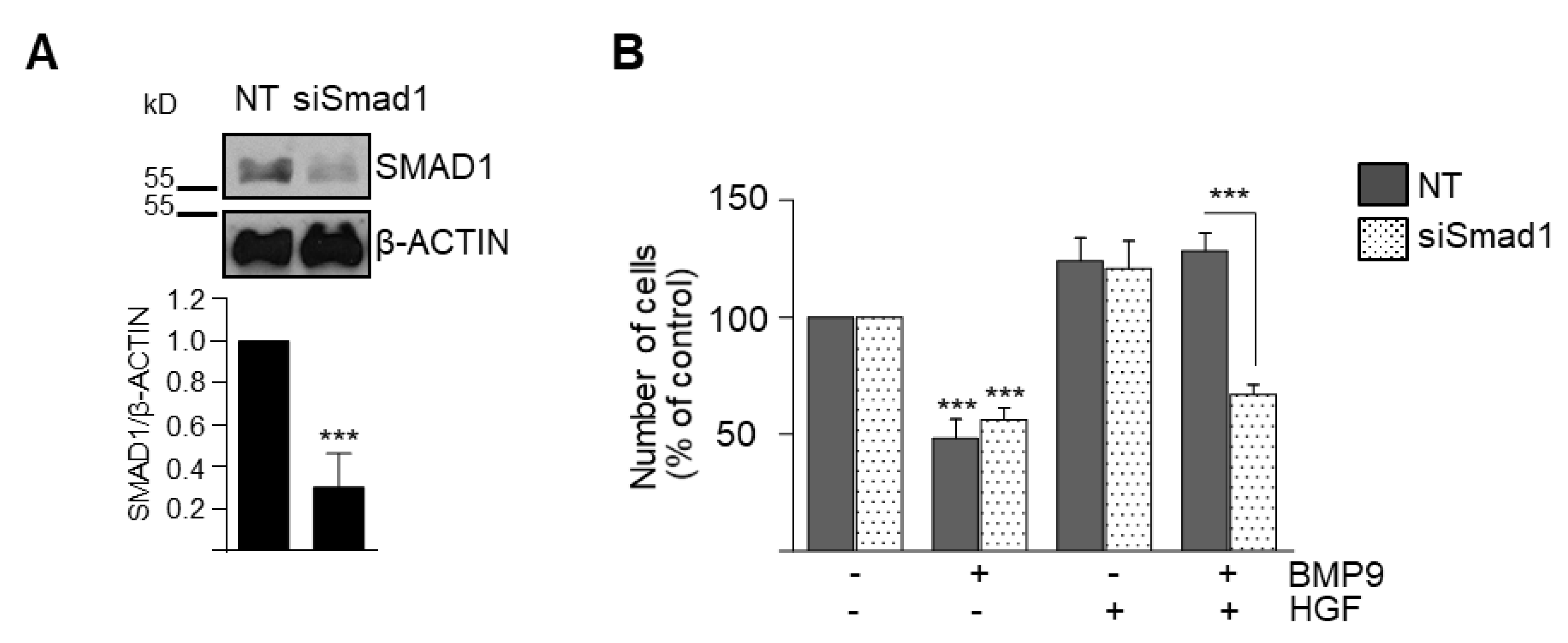
3.2. BMP9 and HGF/c-Met Signaling Crosstalk in Oval Cells is Dependent on ALK1/SMAD1 Signaling Axis
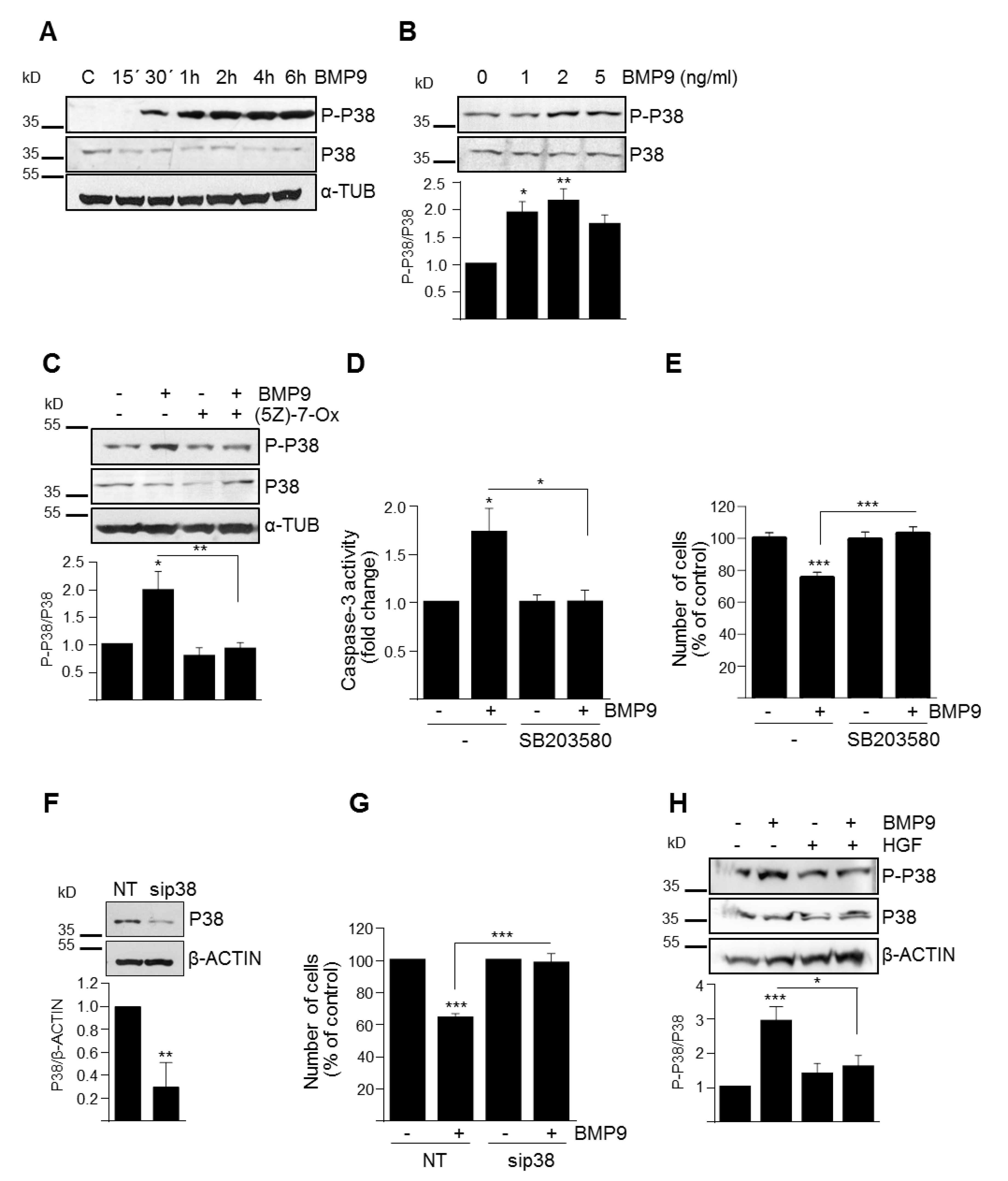
3.3. p38MAPK Activation Mediates BMP9-Induced Apoptosis in Oval Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Itoh, T.; Miyajima, A. Liver regeneration by stem/progenitor cells. Hepatology 2014, 59, 1617–1626. [Google Scholar] [CrossRef]
- Kohn-Gaone, J.; Gogoi-Tiwari, J.; Ramm, G.A.; Olynyk, J.K.; Tirnitz-Parker, J.E. The role of liver progenitor cells during liver regeneration, fibrogenesis, and carcinogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G143–G154. [Google Scholar] [CrossRef] [PubMed]
- Clouston, A.D.; Powell, E.E.; Walsh, M.J.; Richardson, M.M.; Demetris, A.J.; Jonsson, J.R. Fibrosis correlates with a ductular reaction in hepatitis C: roles of impaired replication, progenitor cells and steatosis. Hepatology 2005, 41, 809–818. [Google Scholar] [CrossRef]
- Kuramitsu, K.; Sverdlov, D.Y.; Liu, S.B.; Csizmadia, E.; Burkly, L.; Schuppan, D.; Hanto, D.W.; Otterbein, L.E.; Popov, Y. Failure of fibrotic liver regeneration in mice is linked to a severe fibrogenic response driven by hepatic progenitor cell activation. Am. J. Pathol. 2013, 183, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Castilho, A.; Ma, S.; Ng, I.O. Liver cancer stem cells: implications for a new therapeutic target. Liver Int. 2009, 29, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Factor, V.M.; Marquardt, J.U.; Raggi, C.; Seo, D.; Kitade, M.; Conner, E.A.; Thorgeirsson, S.S. Hepatocyte growth factor/c-met signaling is required for stem-cell-mediated liver regeneration in mice. Hepatology 2012, 55, 1215–1226. [Google Scholar] [CrossRef]
- Almalé, L.; García-Álvaro, M.; Martínez-Palacián, A.; García-Bravo, M.; Lazcanoiturburu, N.; Addante, A.; Roncero, C.; Sanz, J.; de la O López, M.; Bragado, P.; et al. c-Met Signaling Is Essential for Mouse Adult Liver Progenitor Cells Expansion After Transforming Growth Factor-beta-Induced Epithelial-Mesenchymal Transition and Regulates Cell Phenotypic Switch. Stem Cells 2019, 37, 1108–1118. [Google Scholar] [CrossRef]
- Gomez-Puerto, M.C.; Iyengar, P.V.; Garcia de Vinuesa, A.; Ten Dijke, P.; Sanchez-Duffhues, G. Bone morphogenetic protein receptor signal transduction in human disease. J. Pathol. 2019, 247, 9–20. [Google Scholar] [CrossRef]
- Herrera, B.; Addante, A.; Sanchez, A. BMP Signalling at the Crossroad of Liver Fibrosis and Regeneration. Int. J. Mol. Sci. 2017, 19. [Google Scholar] [CrossRef]
- Herrera, B.; Sanchez, A.; Fabregat, I. BMPs And Liver: More Questions Than Answers. Curr. Pharm. Des. 2012, 18, 4114–4125. [Google Scholar] [CrossRef]
- Breitkopf-Heinlein, K.; Meyer, C.; Konig, C.; Gaitantzi, H.; Addante, A.; Thomas, M.; Wiercinska, E.; Cai, C.; Li, Q.; Wan, F.; et al. BMP-9 interferes with liver regeneration and promotes liver fibrosis. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.; Garcia-Alvaro, M.; Cruz, S.; Walsh, P.; Fernandez, M.; Roncero, C.; Fabregat, I.; Sanchez, A.; Inman, G.J. BMP9 is a proliferative and survival factor for human hepatocellular carcinoma cells. PLoS ONE 2013, 8, e69535. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, Y.; Zhu, L.; Yang, Z.; He, J.; Wang, L.; Shang, Q.; Pan, H.; Wang, H.; Ma, X.; et al. Targeting secreted cytokine BMP9 gates the attenuation of hepatic fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gu, X.; Weng, H.; Ghafoory, S.; Liu, Y.; Feng, T.; Dzieran, J.; Li, L.; Ilkavets, I.; Kruithof-de Julio, M.; et al. Bone morphogenetic protein-9 (BMP-9) induces epithelial to mesenchymal transition (EMT) in hepatocellular carcinoma cells. Cancer Sci. 2013, 104, 398–408. [Google Scholar] [CrossRef]
- Addante, A.; Roncero, C.; Almale, L.; Lazcanoiturburu, N.; Garcia-Alvaro, M.; Fernandez, M.; Sanz, J.; Hammad, S.; Nwosu, Z.C.; Lee, S.J.; et al. Bone morphogenetic protein 9 as a key regulator of liver progenitor cells in DDC-induced cholestatic liver injury. Liver Int. 2018, 38, 1664–1675. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, G.; Factor, V.M.; Fernandez, M.; Alvarez-Barrientos, A.; Fabregat, I.; Thorgeirsson, S.S.; Sanchez, A. Deletion of the Met tyrosine kinase in liver progenitor oval cells increases sensitivity to apoptosis in vitro. Am. J. Pathol. 2008, 172, 1238–1247. [Google Scholar] [CrossRef]
- Herrera, B.; Alvarez, A.M.; Sanchez, A.; Fernandez, M.; Roncero, C.; Benito, M.; Fabregat, I. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor (beta) in fetal hepatocytes. FASEB J. 2001, 15, 741–751. [Google Scholar] [CrossRef]
- Martinez-Palacian, A.; del Castillo, G.; Suarez-Causado, A.; Garcia-Alvaro, M.; de Morena-Frutos, D.; Fernandez, M.; Roncero, C.; Fabregat, I.; Herrera, B.; Sanchez, A. Mouse hepatic oval cells require Met-dependent PI3K to impair TGF-beta-induced oxidative stress and apoptosis. PLoS ONE 2013, 8, e53108. [Google Scholar] [CrossRef]
- Li, Y.; Drabsch, Y.; Pujuguet, P.; Ren, J.; van Laar, T.; Zhang, L.; van Dam, H.; Clement-Lacroix, P.; Ten Dijke, P. Genetic depletion and pharmacological targeting of alphav integrin in breast cancer cells impairs metastasis in zebrafish and mouse xenograft models. Breast Cancer Res. 2015, 17, 28. [Google Scholar] [CrossRef]
- Korchynskyi, O.; ten Dijke, P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 2002, 277, 4883–4891. [Google Scholar] [CrossRef]
- Garcia-Alvaro, M.; Addante, A.; Roncero, C.; Fernandez, M.; Fabregat, I.; Sanchez, A.; Herrera, B. BMP9-Induced Survival Effect in Liver Tumor Cells Requires p38MAPK Activation. Int. J. Mol. Sci. 2015, 16, 20431–20448. [Google Scholar] [CrossRef]
- Van Caam, A.; Madej, W.; Garcia de Vinuesa, A.; Goumans, M.J.; Ten Dijke, P.; Blaney Davidson, E.; van der Kraan, P. TGFbeta1-induced SMAD2/3 and SMAD1/5 phosphorylation are both ALK5-kinase-dependent in primary chondrocytes and mediated by TAK1 kinase activity. Arthr. Res. Ther. 2017, 19, 112. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, S.; Kitano, S.; Karasaki, M.; Tsunemi, S.; Sano, H.; Iwasaki, T. Blocking c-Met signaling enhances bone morphogenetic protein-2-induced osteoblast differentiation. FEBS Open Bio 2015, 5, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Huang, Y.L.; Yang, W.H.; Tang, C.H. Hepatocyte growth factor-induced BMP-2 expression is mediated by c-Met receptor, FAK, JNK, Runx2, and p300 pathways in human osteoblasts. Int. Immunopharmacol. 2012, 13, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Terai, H.; Nomura-Furuwatari, C.; Mizuno, S.; Matsumoto, K.; Nakamura, T.; Takaoka, K. Hepatocyte growth factor contributes to fracture repair by upregulating the expression of BMP receptors. J. Bone Miner. Res. 2005, 20, 1723–1730. [Google Scholar] [CrossRef]
- Ye, L.; Lewis-Russell, J.M.; Davies, G.; Sanders, A.J.; Kynaston, H.; Jiang, W.G. Hepatocyte growth factor up-regulates the expression of the bone morphogenetic protein (BMP) receptors, BMPR-IB and BMPR-II, in human prostate cancer cells. Int. J. Oncol. 2007, 30, 521–529. [Google Scholar]
- Ye, L.; Lewis-Russell, J.M.; Sanders, A.J.; Kynaston, H.; Jiang, W.G. HGF/SF up-regulates the expression of bone morphogenetic protein 7 in prostate cancer cells. Urol. Oncol. 2008, 26, 190–197. [Google Scholar] [CrossRef]
- Bollum, L.K.; Huse, K.; Oksvold, M.P.; Bai, B.; Hilden, V.I.; Forfang, L.; Yoon, S.O.; Walchli, S.; Smeland, E.B.; Myklebust, J.H. BMP-7 induces apoptosis in human germinal center B cells and is influenced by TGF-beta receptor type I ALK5. PLoS ONE 2017, 12, e0177188. [Google Scholar] [CrossRef]
- Holien, T.; Vatsveen, T.K.; Hella, H.; Rampa, C.; Brede, G.; Groseth, L.A.; Rekvig, M.; Borset, M.; Standal, T.; Waage, A.; et al. Bone morphogenetic proteins induce apoptosis in multiple myeloma cells by Smad-dependent repression of MYC. Leukemia 2012, 26, 1073–1080. [Google Scholar] [CrossRef]
- Kiyono, M.; Shibuya, M. Bone morphogenetic protein 4 mediates apoptosis of capillary endothelial cells during rat pupillary membrane regression. Mol. Cell. Biol. 2003, 23, 4627–4636. [Google Scholar] [CrossRef]
- Sniegon, I.; Priess, M.; Heger, J.; Schulz, R.; Euler, G. Endothelial Mesenchymal Transition in Hypoxic Microvascular Endothelial Cells and Paracrine Induction of Cardiomyocyte Apoptosis Are Mediated via TGFbeta(1)/SMAD Signaling. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Ueki, Y.; Reh, T.A. Activation of BMP-Smad1/5/8 signaling promotes survival of retinal ganglion cells after damage in vivo. PLoS ONE 2012, 7, e38690. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.Z.; Teng, X.; Zhang, Z.B.; Zheng, C.J.; Chen, S.H. Mangiferin inhibits apoptosis and oxidative stress via BMP2/Smad-1 signaling in dexamethasone-induced MC3T3-E1 cells. Int. J. Mol. Med. 2018, 41, 2517–2526. [Google Scholar] [CrossRef]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Graf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef]
- Yuan, S.X.; Wang, D.X.; Wu, Q.X.; Ren, C.M.; Li, Y.; Chen, Q.Z.; Zeng, Y.H.; Shao, Y.; Yang, J.Q.; Bai, Y.; et al. BMP9/p38 MAPK is essential for the antiproliferative effect of resveratrol on human colon cancer. Oncol. Rep. 2016, 35, 939–947. [Google Scholar] [CrossRef]
- Mitrofan, C.G.; Appleby, S.L.; Nash, G.B.; Mallat, Z.; Chilvers, E.R.; Upton, P.D.; Morrell, N.W. Bone morphogenetic protein 9 (BMP9) and BMP10 enhance tumor necrosis factor-alpha-induced monocyte recruitment to the vascular endothelium mainly via activin receptor-like kinase 2. J. Biol. Chem. 2017, 292, 13714–13726. [Google Scholar] [CrossRef]
- Saremba, S.; Nickel, J.; Seher, A.; Kotzsch, A.; Sebald, W.; Mueller, T.D. Type I receptor binding of bone morphogenetic protein 6 is dependent on N-glycosylation of the ligand. FEBS J. 2008, 275, 172–183. [Google Scholar] [CrossRef]
- Hassel, S.; Schmitt, S.; Hartung, A.; Roth, M.; Nohe, A.; Petersen, N.; Ehrlich, M.; Henis, Y.I.; Sebald, W.; Knaus, P. Initiation of Smad-dependent and Smad-independent signaling via distinct BMP-receptor complexes. J. Bone Jt. Surg. Am. 2003, 85, 44–51. [Google Scholar] [CrossRef]
- Yadin, D.; Knaus, P.; Mueller, T.D. Structural insights into BMP receptors: Specificity, activation and inhibition. Cytokine Growth Factor Rev. 2016, 27, 13–34. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Addante, A.; Roncero, C.; Lazcanoiturburu, N.; Méndez, R.; Almalé, L.; García-Álvaro, M.; ten Dijke, P.; Fabregat, I.; Herrera, B.; Sánchez, A. A Signaling Crosstalk between BMP9 and HGF/c-Met Regulates Mouse Adult Liver Progenitor Cell Survival. Cells 2020, 9, 752. https://doi.org/10.3390/cells9030752
Addante A, Roncero C, Lazcanoiturburu N, Méndez R, Almalé L, García-Álvaro M, ten Dijke P, Fabregat I, Herrera B, Sánchez A. A Signaling Crosstalk between BMP9 and HGF/c-Met Regulates Mouse Adult Liver Progenitor Cell Survival. Cells. 2020; 9(3):752. https://doi.org/10.3390/cells9030752
Chicago/Turabian StyleAddante, Annalisa, Cesáreo Roncero, Nerea Lazcanoiturburu, Rebeca Méndez, Laura Almalé, María García-Álvaro, Peter ten Dijke, Isabel Fabregat, Blanca Herrera, and Aránzazu Sánchez. 2020. "A Signaling Crosstalk between BMP9 and HGF/c-Met Regulates Mouse Adult Liver Progenitor Cell Survival" Cells 9, no. 3: 752. https://doi.org/10.3390/cells9030752
APA StyleAddante, A., Roncero, C., Lazcanoiturburu, N., Méndez, R., Almalé, L., García-Álvaro, M., ten Dijke, P., Fabregat, I., Herrera, B., & Sánchez, A. (2020). A Signaling Crosstalk between BMP9 and HGF/c-Met Regulates Mouse Adult Liver Progenitor Cell Survival. Cells, 9(3), 752. https://doi.org/10.3390/cells9030752