Function of Torsin AAA+ ATPases in Pseudorabies Virus Nuclear Egress

 ,
,  ,
,
Abstract
1. Introduction
2. Material and Methods
2.1. Cells and Virus
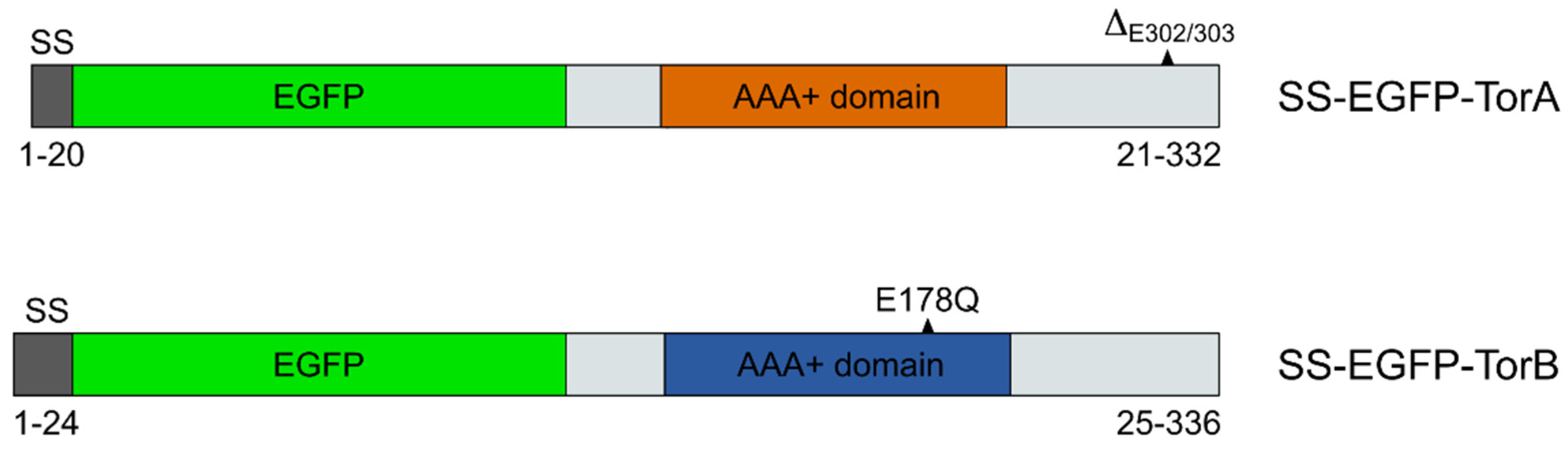
2.2. DNA Constructs
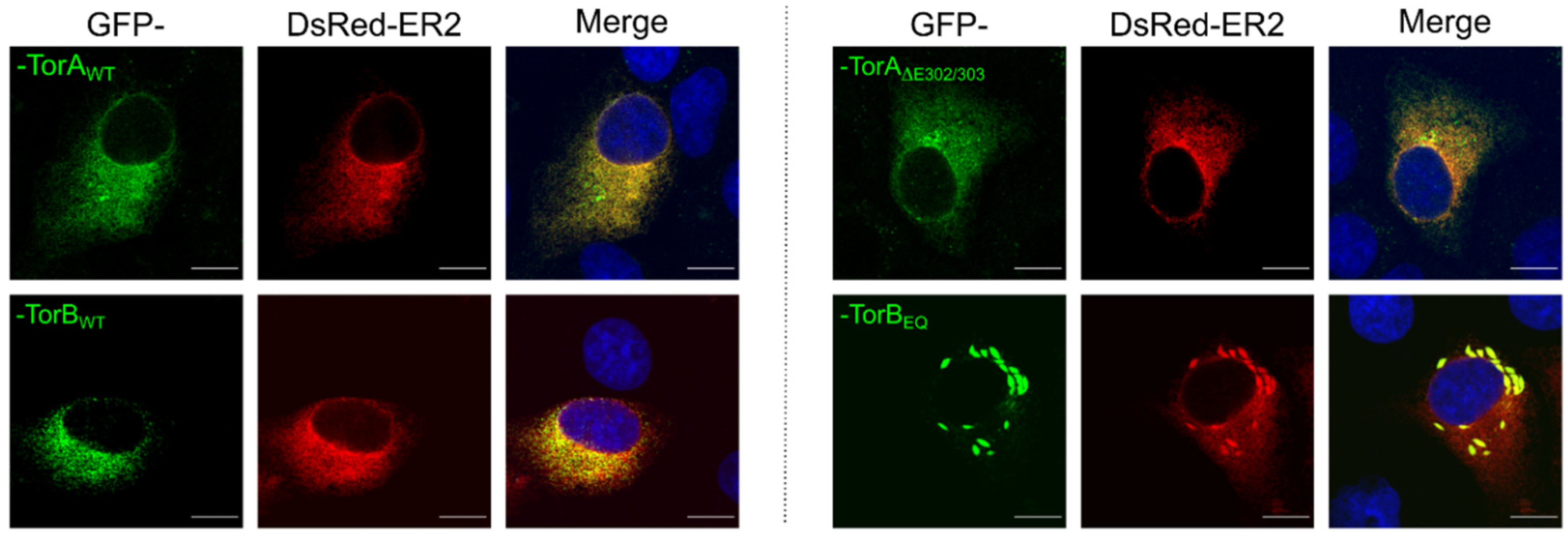
2.3. Transfection of Cells for Co-Localization Studies
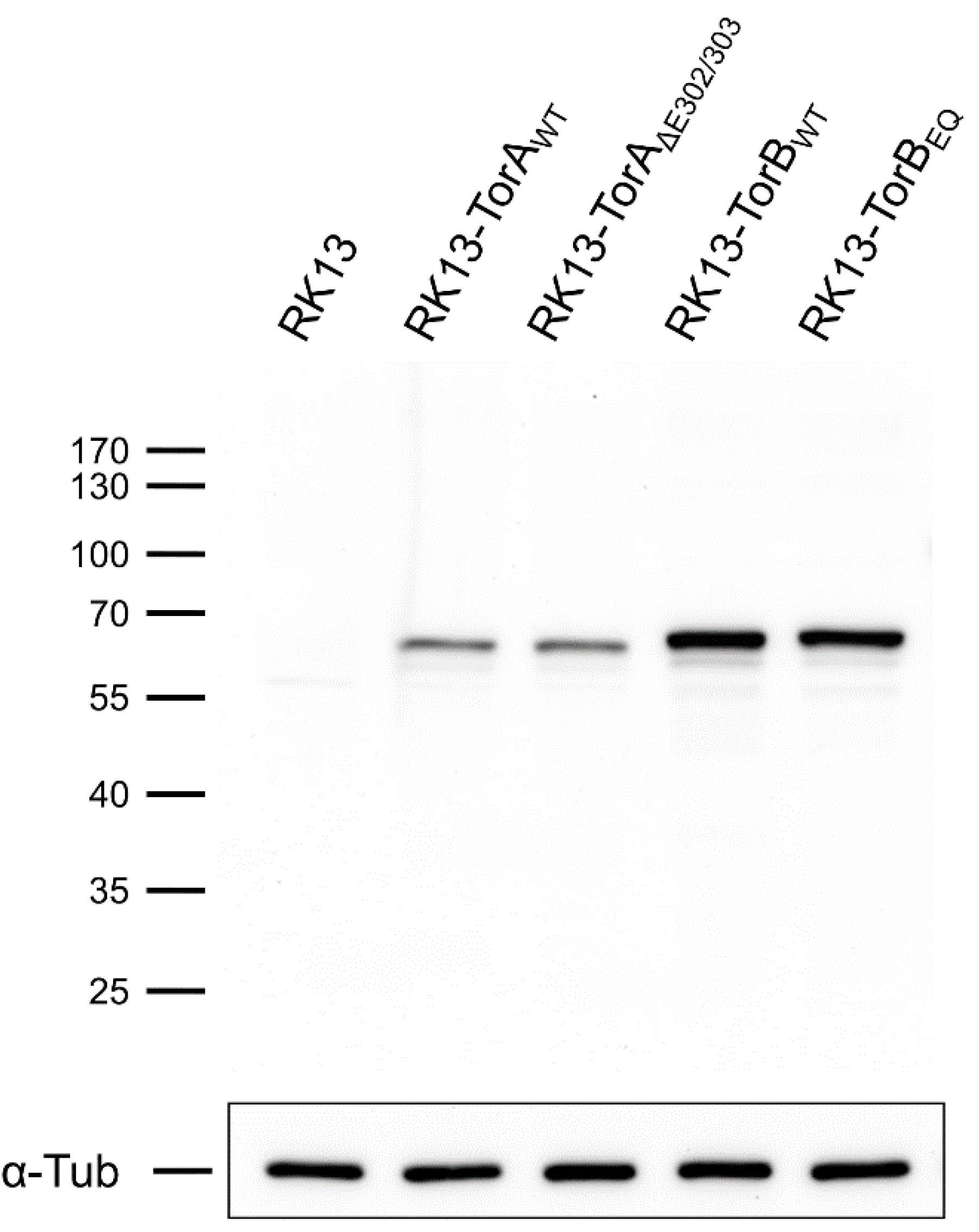
2.4. Immunoblotting
2.5. Generation of Stably Expressing RK13 Cell Lines
2.6. Generation of Stable RK13 Knockout Cell Lines
2.7. Test for Bi-Allelic Gene Knockout
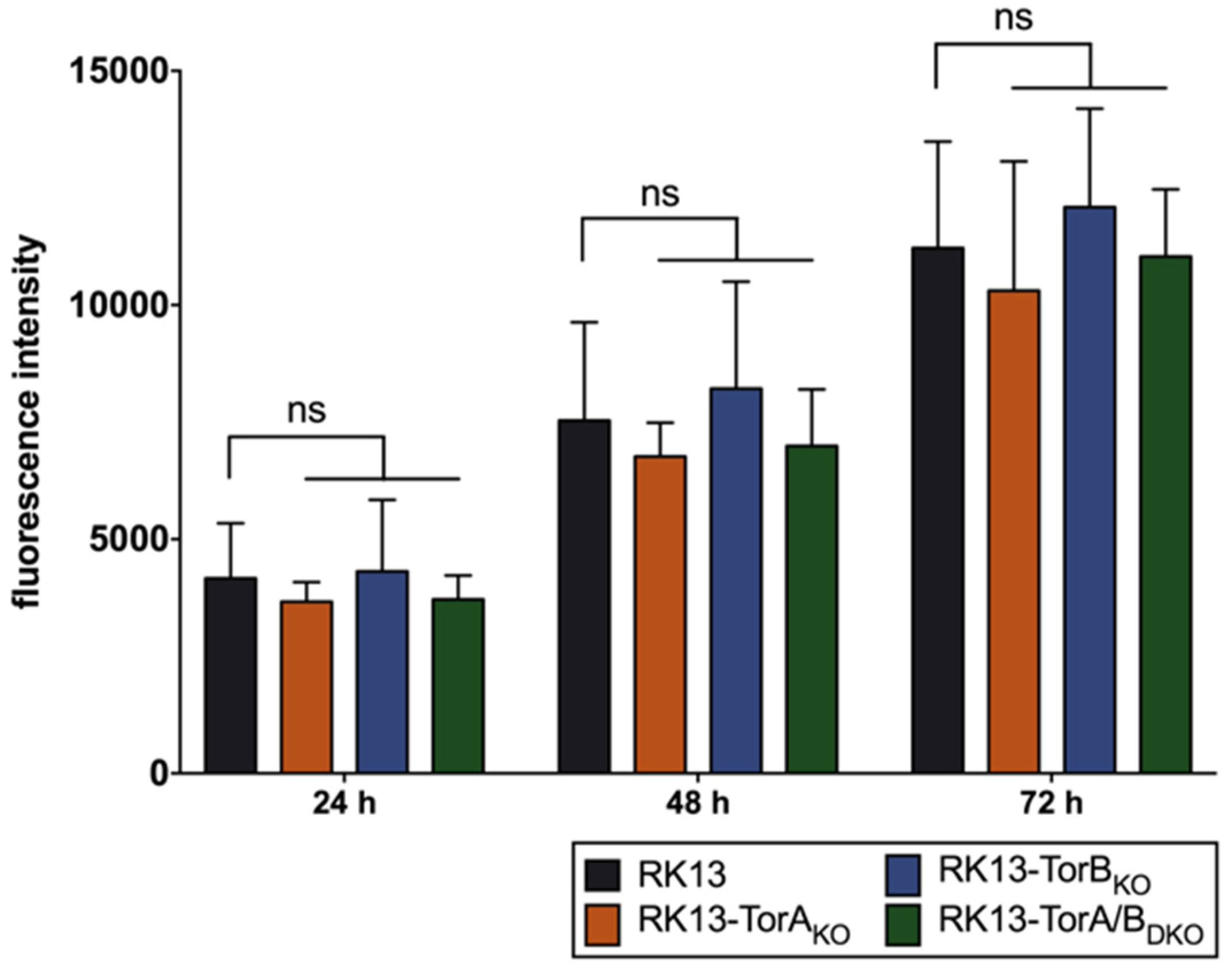
2.8. PrestoBlue Assay
2.9. In Vitro Replication Studies
2.10. Statistics
2.11. Laser Scanning Confocal Microscopy
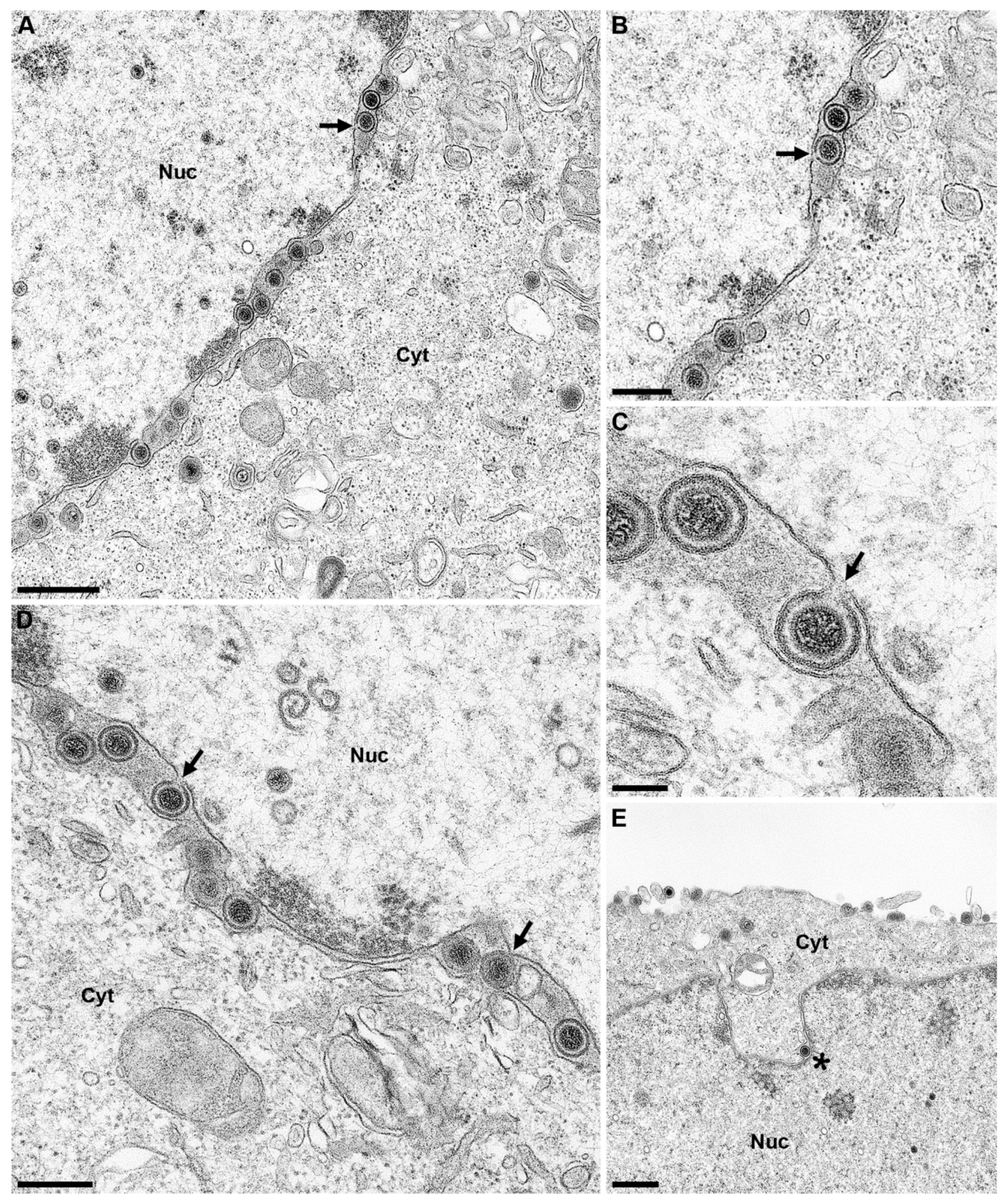
2.12. Ultrastructural Analyses
3. Results
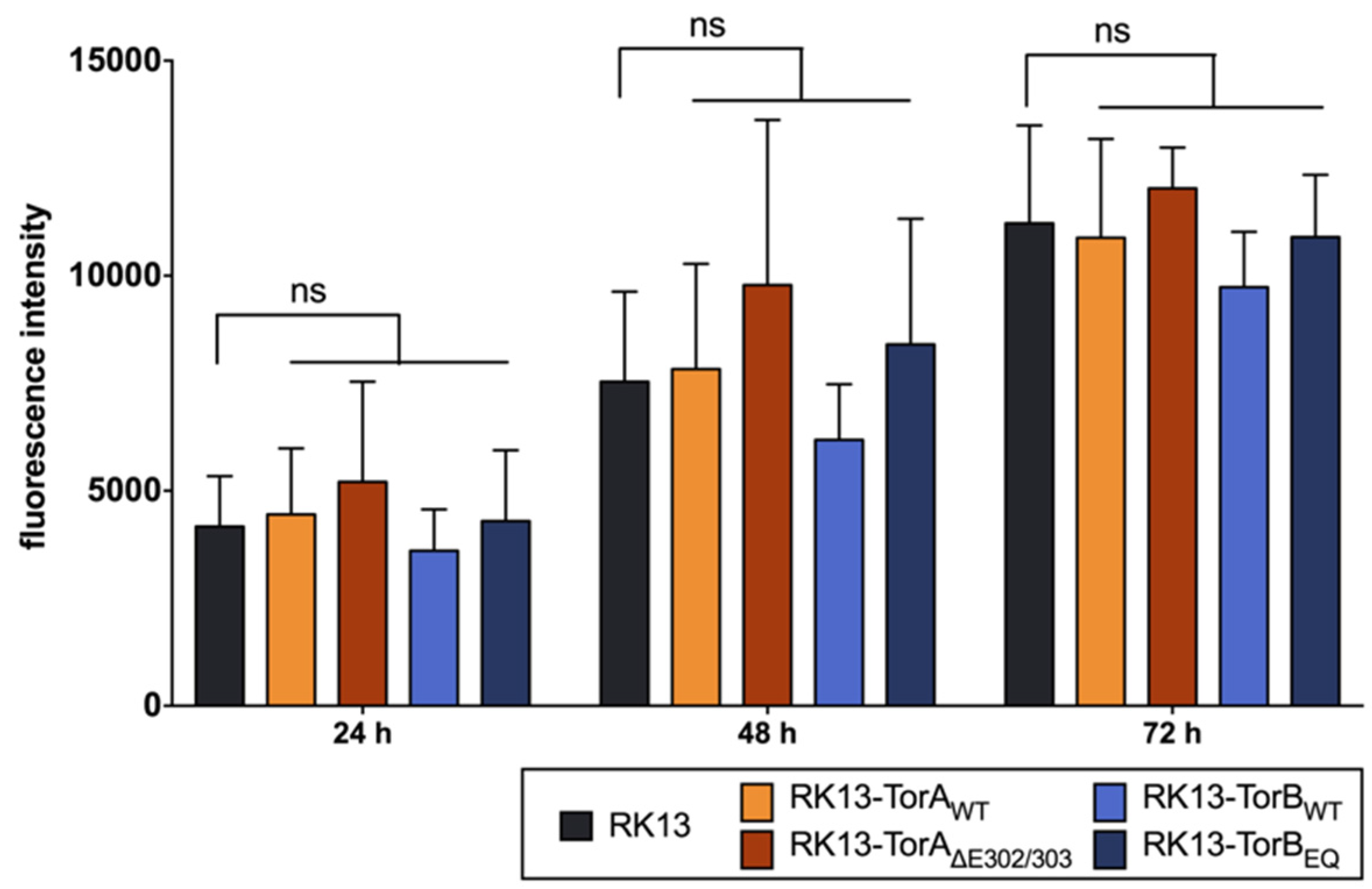
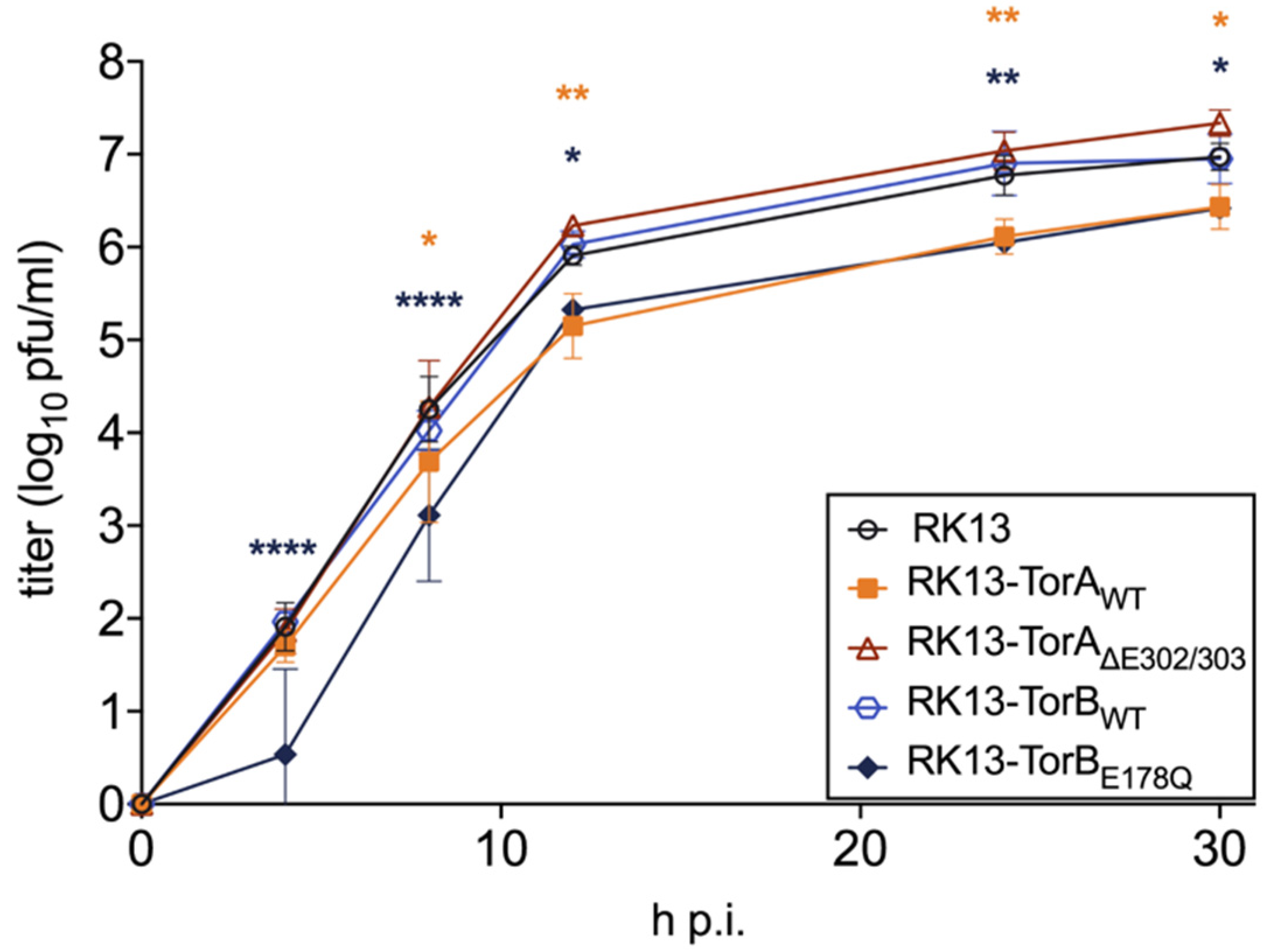
3.1. Influence of Torsin Overexpression on PrV Replication
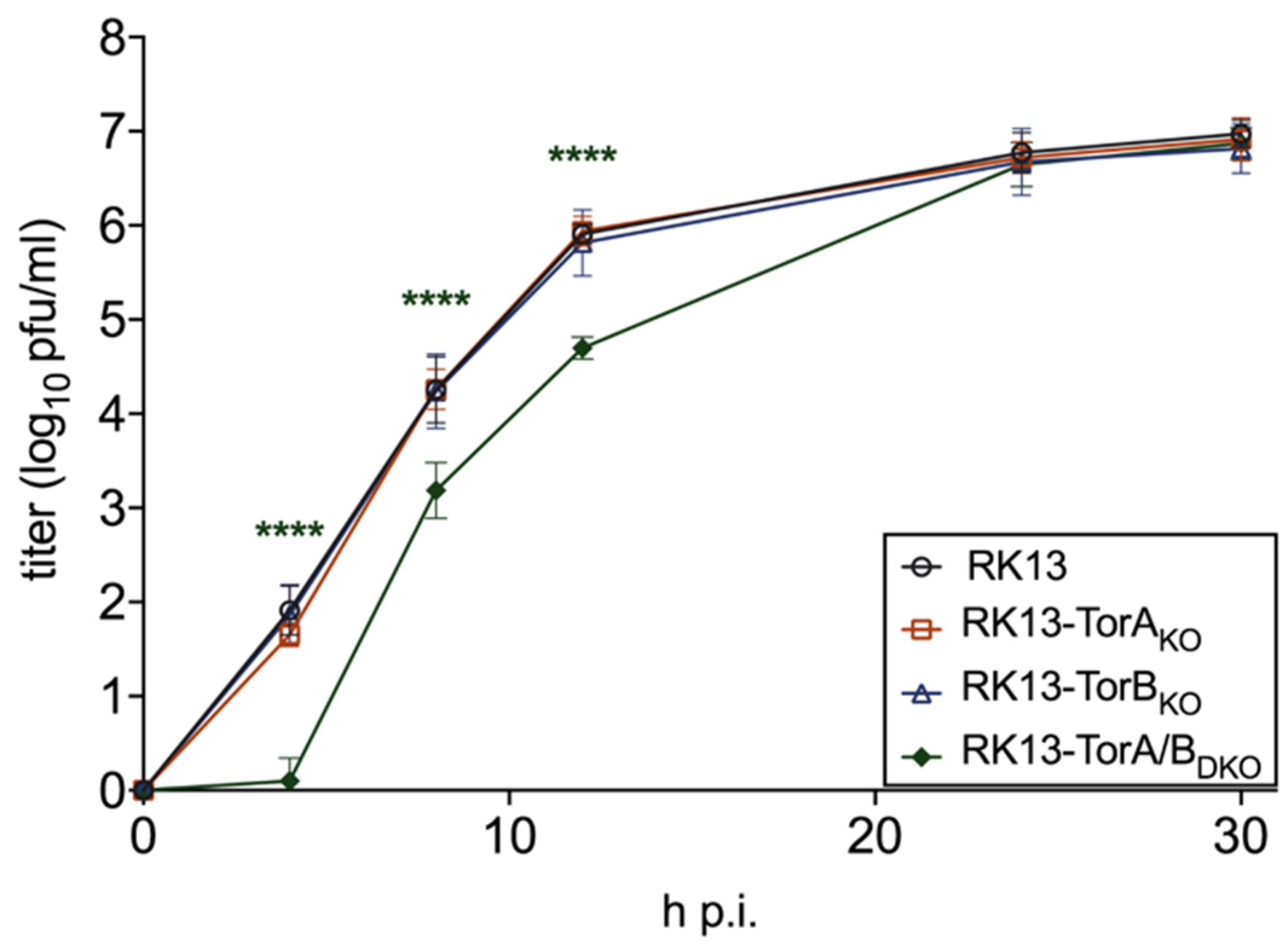
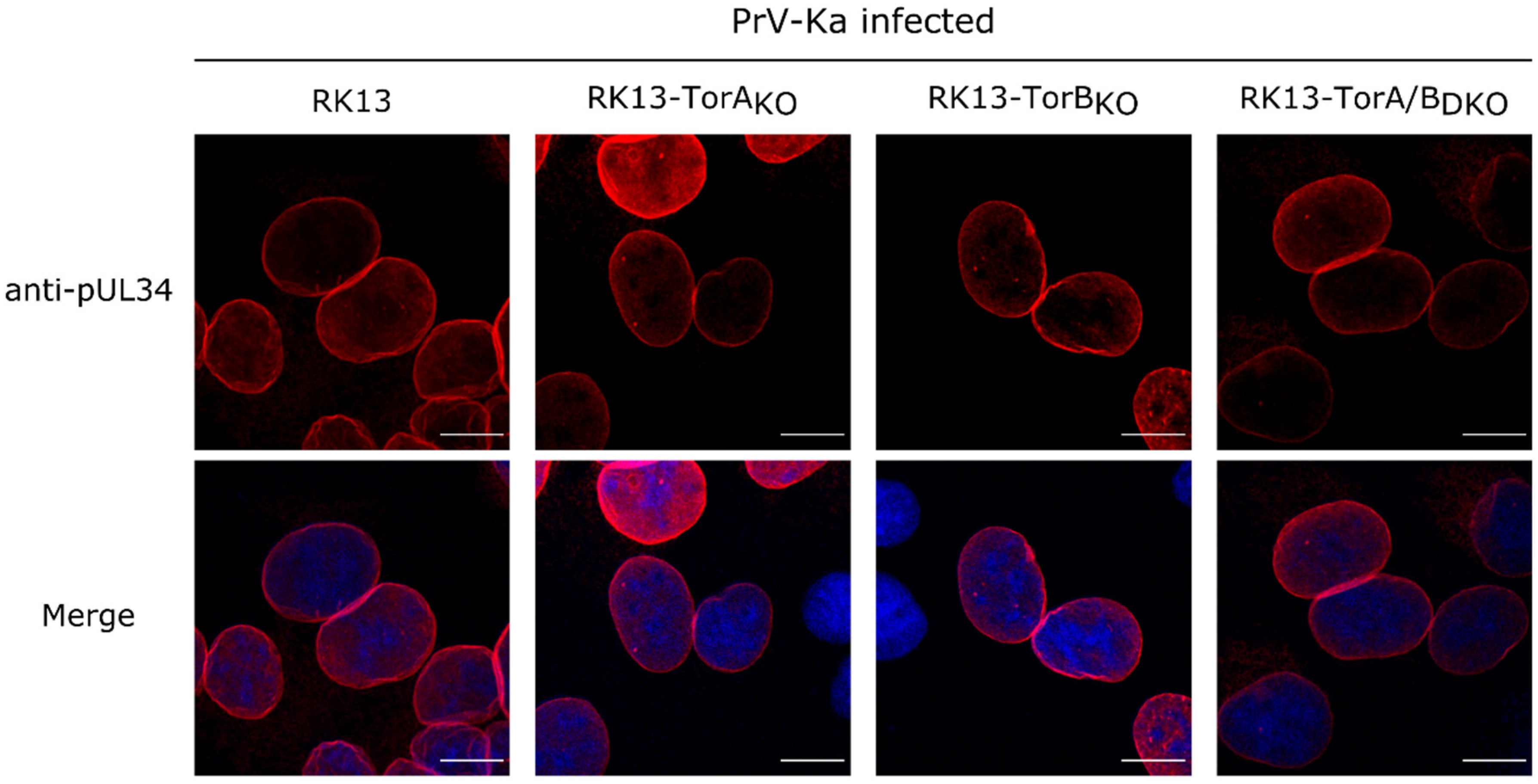
3.2. Torsin A and Torsin B Are Required for Efficient PrV Replication in RK13 Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pante, N.; Kann, M. Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol. Biol. Cell 2002, 13, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Hofemeister, H.; O’Hare, P. Nuclear pore composition and gating in herpes simplex virus-infected cells. J. Virol. 2008, 82, 8392–8399. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.A. The nuclear pore complex. Genome Biol. 2001, 2. [Google Scholar] [CrossRef] [PubMed]
- Knockenhauer, K.E.; Schwartz, T.U. The Nuclear Pore Complex as a Flexible and Dynamic Gate. Cell 2016, 164, 1162–1171. [Google Scholar] [CrossRef]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef]
- Mettenleiter, T.C.; Muller, F.; Granzow, H.; Klupp, B.G. The way out: What we know and do not know about herpesvirus nuclear egress. Cell. Microbiol. 2013, 15, 170–178. [Google Scholar] [CrossRef]
- Klupp, B.G.; Granzow, H.; Fuchs, W.; Keil, G.M.; Finke, S.; Mettenleiter, T.C. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 7241–7246. [Google Scholar] [CrossRef]
- Bigalke, J.M.; Heuser, T.; Nicastro, D.; Heldwein, E.E. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat. Commun. 2014, 5, 4131. [Google Scholar] [CrossRef]
- Desai, P.J.; Pryce, E.N.; Henson, B.W.; Luitweiler, E.M.; Cothran, J. Reconstitution of the Kaposi’s sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J. Virol. 2012, 86, 594–598. [Google Scholar] [CrossRef]
- Lorenz, M.; Vollmer, B.; Unsay, J.D.; Klupp, B.G.; Garcia-Saez, A.J.; Mettenleiter, T.C.; Antonin, W. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J. Biol. Chem. 2015, 290, 6962–6974. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C. Herpesvirus assembly and egress. J. Virol. 2002, 76, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, A.; Wisner, T.W.; Webb, M.; Roller, R.; Cohen, G.; Eisenberg, R.; Johnson, D.C. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. USA 2007, 104, 10187–10192. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.; Altenschmidt, J.; Granzow, H.; Fuchs, W.; Mettenleiter, T.C. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J. Virol. 2008, 82, 6299–6309. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Effect of the pseudorabies virus US3 protein on nuclear membrane localization of the UL34 protein and virus egress from the nucleus. J. Gen. Virol. 2001, 82, 2363–2371. [Google Scholar] [CrossRef]
- Wagenaar, F.; Pol, J.M.; Peeters, B.; Gielkens, A.L.; de Wind, N.; Kimman, T.G. The US3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J. Gen. Virol. 1995, 76 Pt 7, 1851–1859. [Google Scholar] [CrossRef]
- Reynolds, A.E.; Wills, E.G.; Roller, R.J.; Ryckman, B.J.; Baines, J.D. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 2002, 76, 8939–8952. [Google Scholar] [CrossRef]
- Schumacher, D.; McKinney, C.; Kaufer, B.B.; Osterrieder, N. Enzymatically inactive U(S)3 protein kinase of Marek’s disease virus (MDV) is capable of depolymerizing F-actin but results in accumulation of virions in perinuclear invaginations and reduced virus growth. Virology 2008, 375, 37–47. [Google Scholar] [CrossRef]
- Kato, A.; Liu, Z.; Minowa, A.; Imai, T.; Tanaka, M.; Sugimoto, K.; Nishiyama, Y.; Arii, J.; Kawaguchi, Y. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J. Virol. 2011, 85, 9599–9613. [Google Scholar] [CrossRef]
- Sehl, J.; Portner, S.; Klupp, B.G.; Granzow, H.; Franzke, K.; Teifke, J.P.; Mettenleiter, T.C. Roles of the different isoforms of the pseudorabies virus protein kinase pUS3 in nuclear egress. J. Virol. 2020. [Google Scholar] [CrossRef]
- Otsuka, S.; Ellenberg, J. Mechanisms of nuclear pore complex assembly—Two different ways of building one molecular machine. FEBS Lett. 2018, 592, 475–488. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.A.; Anderson, D.J.; Richard, E.; Hetzer, M.W. Nuclear pores form de novo from both sides of the nuclear envelope. Science 2006, 312, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Dultz, E.; Ellenberg, J. Live imaging of single nuclear pores reveals unique assembly kinetics and mechanism in interphase. J. Cell Biol. 2010, 191, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Arii, J.; Watanabe, M.; Maeda, F.; Tokai-Nishizumi, N.; Chihara, T.; Miura, M.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat. Commun. 2018, 9, 3379. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.P.; Liu, G.T.; Kung, H.N.; Liu, P.T.; Liao, Y.T.; Chow, L.P.; Chang, L.S.; Chang, Y.H.; Chang, C.W.; Shu, W.C.; et al. The Ubiquitin Ligase Itch and Ubiquitination Regulate BFRF1-Mediated Nuclear Envelope Modification for Epstein-Barr Virus Maturation. J. Virol. 2016, 90, 8994–9007. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.P.; Liu, P.T.; Kung, H.N.; Su, M.T.; Chua, H.H.; Chang, Y.H.; Chang, C.W.; Tsai, C.H.; Liu, F.T.; Chen, M.R. The ESCRT machinery is recruited by the viral BFRF1 protein to the nucleus-associated membrane for the maturation of Epstein-Barr Virus. PLoS Pathog. 2012, 8, e1002904. [Google Scholar] [CrossRef]
- Crump, C.M.; Yates, C.; Minson, T. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 2007, 81, 7380–7387. [Google Scholar] [CrossRef]
- Kharkwal, H.; Smith, C.G.; Wilson, D.W. Blocking ESCRT-mediated envelopment inhibits microtubule-dependent trafficking of alphaherpesviruses in vitro. J. Virol. 2014, 88, 14467–14478. [Google Scholar] [CrossRef]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar]
- Erzberger, J.P.; Berger, J.M. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 93–114. [Google Scholar] [CrossRef]
- Laudermilch, E.; Schlieker, C. Torsin ATPases: Structural insights and functional perspectives. Curr. Opin. Cell Biol. 2016, 40, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Breakefield, X.O.; Kamm, C.; Hanson, P.I. TorsinA: Movement at many levels. Neuron 2001, 31, 9–12. [Google Scholar] [CrossRef]
- Hanson, P.I.; Whiteheart, S.W. AAA+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 2005, 6, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.E.; Brown, R.S.; Schlieker, C. Torsins: Not your typical AAA+ ATPases. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 532–549. [Google Scholar] [CrossRef]
- Sosa, B.A.; Demircioglu, F.E.; Chen, J.Z.; Ingram, J.; Ploegh, H.L.; Schwartz, T.U. How lamina-associated polypeptide 1 (LAP1) activates Torsin. eLife 2014, 3, e03239. [Google Scholar] [CrossRef]
- Nagy, M.; Wu, H.C.; Liu, Z.; Kedzierska-Mieszkowska, S.; Zolkiewski, M. Walker-A threonine couples nucleotide occupancy with the chaperone activity of the AAA+ ATPase ClpB. Protein Sci. 2009, 18, 287–293. [Google Scholar] [CrossRef]
- Jungwirth, M.; Dear, M.L.; Brown, P.; Holbrook, K.; Goodchild, R. Relative tissue expression of homologous torsinB correlates with the neuronal specific importance of DYT1 dystonia-associated torsinA. Hum. Mol. Genet. 2010, 19, 888–900. [Google Scholar] [CrossRef]
- Kustedjo, K.; Bracey, M.H.; Cravatt, B.F. Torsin A and its torsion dystonia-associated mutant forms are lumenal glycoproteins that exhibit distinct subcellular localizations. J. Biol. Chem. 2000, 275, 27933–27939. [Google Scholar] [CrossRef]
- Vander Heyden, A.B.; Naismith, T.V.; Snapp, E.L.; Hodzic, D.; Hanson, P.I. LULL1 retargets TorsinA to the nuclear envelope revealing an activity that is impaired by the DYT1 dystonia mutation. Mol. Biol. Cell 2009, 20, 2661–2672. [Google Scholar] [CrossRef]
- Brown, R.S.; Zhao, C.; Chase, A.R.; Wang, J.; Schlieker, C. The mechanism of Torsin ATPase activation. Proc. Natl. Acad. Sci. USA 2014, 111, E4822–E4831. [Google Scholar] [CrossRef]
- Zhao, C.; Brown, R.S.; Chase, A.R.; Eisele, M.R.; Schlieker, C. Regulation of Torsin ATPases by LAP1 and LULL1. Proc. Natl. Acad. Sci. USA 2013, 110, E1545–E1554. [Google Scholar] [CrossRef] [PubMed]
- Ozelius, L.J.; Hewett, J.W.; Page, C.E.; Bressman, S.B.; Kramer, P.L.; Shalish, C.; de Leon, D.; Brin, M.F.; Raymond, D.; Corey, D.P.; et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 1997, 17, 40–48. [Google Scholar] [CrossRef]
- Gonzalez-Alegre, P. Advances in molecular and cell biology of dystonia: Focus on torsinA. Neurobiol. Dis. 2019, 127, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, R.E.; Dauer, W.T. Mislocalization to the nuclear envelope: An effect of the dystonia-causing torsinA mutation. Proc. Natl. Acad. Sci. USA 2004, 101, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Naismith, T.V.; Heuser, J.E.; Breakefield, X.O.; Hanson, P.I. TorsinA in the nuclear envelope. Proc. Natl. Acad. Sci. USA 2004, 101, 7612–7617. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.E.; Zhao, C.; Turner, E.M.; Steyer, A.M.; Schlieker, C. Arresting a Torsin ATPase reshapes the endoplasmic reticulum. J. Biol. Chem. 2014, 289, 552–564. [Google Scholar] [CrossRef]
- Hewett, J.; Gonzalez-Agosti, C.; Slater, D.; Ziefer, P.; Li, S.; Bergeron, D.; Jacoby, D.J.; Ozelius, L.J.; Ramesh, V.; Breakefield, X.O. Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum. Mol. Genet. 2000, 9, 1403–1413. [Google Scholar] [CrossRef]
- Chalfant, M.; Barber, K.W.; Borah, S.; Thaller, D.; Lusk, C.P. Expression of TorsinA in a heterologous yeast system reveals interactions with lumenal domains of LINC and nuclear pore complex components. Mol. Biol. Cell 2019, 30, 530–541. [Google Scholar] [CrossRef]
- Pappas, S.S.; Liang, C.C.; Kim, S.; Rivera, C.O.; Dauer, W.T. TorsinA dysfunction causes persistent neuronal nuclear pore defects. Hum. Mol. Genet. 2018, 27, 407–420. [Google Scholar] [CrossRef]
- Laudermilch, E.; Tsai, P.L.; Graham, M.; Turner, E.; Zhao, C.; Schlieker, C. Dissecting Torsin/cofactor function at the nuclear envelope: A genetic study. Mol. Biol. Cell 2016, 27, 3964–3971. [Google Scholar] [CrossRef]
- Goodchild, R.E.; Kim, C.E.; Dauer, W.T. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron 2005, 48, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.E.; Perez, A.; Perkins, G.; Ellisman, M.H.; Dauer, W.T. A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9861–9866. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, L.M.; Liang, C.C.; Dauer, W.T. Neuronal Nuclear Membrane Budding Occurs during a Developmental Window Modulated by Torsin Paralogs. Cell Rep. 2016, 16, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Maric, M.; Shao, J.; Ryan, R.J.; Wong, C.S.; Gonzalez-Alegre, P.; Roller, R.J. A functional role for TorsinA in herpes simplex virus 1 nuclear egress. J. Virol. 2011, 85, 9667–9679. [Google Scholar] [CrossRef]
- Turner, E.M.; Brown, R.S.; Laudermilch, E.; Tsai, P.L.; Schlieker, C. The Torsin Activator LULL1 Is Required for Efficient Growth of Herpes Simplex Virus 1. J. Virol. 2015, 89, 8444–8452. [Google Scholar] [CrossRef]
- Nery, F.C.; Zeng, J.; Niland, B.P.; Hewett, J.; Farley, J.; Irimia, D.; Li, Y.; Wiche, G.; Sonnenberg, A.; Breakefield, X.O. TorsinA binds the KASH domain of nesprins and participates in linkage between nuclear envelope and cytoskeleton. J. Cell Sci. 2008, 121, 3476–3486. [Google Scholar] [CrossRef]
- Saunders, C.A.; Harris, N.J.; Willey, P.T.; Woolums, B.M.; Wang, Y.; McQuown, A.J.; Schoenhofen, A.; Worman, H.J.; Dauer, W.T.; Gundersen, G.G.; et al. TorsinA controls TAN line assembly and the retrograde flow of dorsal perinuclear actin cables during rearward nuclear movement. J. Cell Biol. 2017, 216, 657–674. [Google Scholar] [CrossRef]
- Gill, N.K.; Ly, C.; Kim, P.H.; Saunders, C.A.; Fong, L.G.; Young, S.G.; Luxton, G.W.G.; Rowat, A.C. DYT1 Dystonia Patient-Derived Fibroblasts Have Increased Deformability and Susceptibility to Damage by Mechanical Forces. Front. Cell. Dev. Biol. 2019, 7, 103. [Google Scholar] [CrossRef]
- Dominguez Gonzalez, B.; Billion, K.; Rous, S.; Pavie, B.; Lange, C.; Goodchild, R. Excess LINC complexes impair brain morphogenesis in a mouse model of recessive TOR1A disease. Hum. Mol. Genet. 2018, 27, 2154–2170. [Google Scholar] [CrossRef]
- Zhou, X.; Graumann, K.; Evans, D.E.; Meier, I. Novel plant SUN-KASH bridges are involved in RanGAP anchoring and nuclear shape determination. J. Cell Biol. 2012, 196, 203–211. [Google Scholar] [CrossRef]
- Razafsky, D.; Hodzic, D. Bringing KASH under the SUN: The many faces of nucleo-cytoskeletal connections. J. Cell Biol. 2009, 186, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Meinke, P.; Schirmer, E.C. LINC’ing form and function at the nuclear envelope. FEBS Lett. 2015, 589, 2514–2521. [Google Scholar] [CrossRef] [PubMed]
- Starr, D.A.; Fridolfsson, H.N. Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annu. Rev. Cell Dev. Biol. 2010, 26, 421–444. [Google Scholar] [CrossRef] [PubMed]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef]
- Jungwirth, M.T.; Kumar, D.; Jeong, D.Y.; Goodchild, R.E. The nuclear envelope localization of DYT1 dystonia torsinA-DeltaE requires the SUN1 LINC complex component. BMC Cell Biol. 2011, 12, 24. [Google Scholar] [CrossRef]
- Saunders, C.A.; Luxton, G.W. LINCing defective nuclear-cytoskeletal coupling and DYT1 dystonia. Cell. Mol. Bioeng. 2016, 9, 207–216. [Google Scholar] [CrossRef]
- Jokhi, V.; Ashley, J.; Nunnari, J.; Noma, A.; Ito, N.; Wakabayashi-Ito, N.; Moore, M.J.; Budnik, V. Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep. 2013, 3, 988–995. [Google Scholar] [CrossRef]
- Speese, S.D.; Ashley, J.; Jokhi, V.; Nunnari, J.; Barria, R.; Li, Y.; Ataman, B.; Koon, A.; Chang, Y.T.; Li, Q.; et al. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 2012, 149, 832–846. [Google Scholar] [CrossRef]
- Klupp, B.G.; Hellberg, T.; Granzow, H.; Franzke, K.; Dominguez Gonzalez, B.; Goodchild, R.E.; Mettenleiter, T.C. Integrity of the Linker of Nucleoskeleton and Cytoskeleton Is Required for Efficient Herpesvirus Nuclear Egress. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Kaplan, A.S.; Vatter, A.E. A comparison of herpes simplex and pseudorabies viruses. Virology 1959, 7, 394–407. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Hübner, A.; Petersen, B.; Keil, G.M.; Niemann, H.; Mettenleiter, T.C.; Fuchs, W. Efficient inhibition of African swine fever virus replication by CRISPR/Cas9 targeting of the viral p30 gene (CP204L). Sci. Rep. 2018, 8, 1449. [Google Scholar] [CrossRef] [PubMed]
- Graham, F.L.; van der Eb, A.J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 1973, 52, 456–467. [Google Scholar] [CrossRef]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Qiao, J.; Hu, S.; Zhao, X.; Regouski, M.; Yang, M.; Polejaeva, I.A.; Chen, C. Efficient gene knockout in goats using CRISPR/Cas9 system. PLoS ONE 2014, 9, e106718. [Google Scholar] [CrossRef]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 2000, 74, 10063–10073. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′–3′) |
|---|---|
| TorA_gRNA#1_Fwd | CACCCTGGCGGTAGCGCCGGTCGG |
| TorA_gRNA#1_Rev | AAACCCGACCGGCGCTACCGCCAG |
| TorA_gRNA#2_Fwd | CACCTGTCTGGCGGTAGCGCCGGT |
| TorA_gRNA#2_Rev | AAACACCGGCGCTACCGCCAGACA |
| TorA_gRNA#3_Fwd | CACCCGCCGGTCGGTGGTCAGCGC |
| TorA_gRNA#3_Rev | AAACCGCTGACCACCGACCGGCGG |
| TorA_gRNA#4_Fwd | CACCGTTCCTGCGCTGACCACCGA |
| TorA_gRNA#4_Rev | AAACGTCGGTGGTCAGCGCAGGAA |
| TorB_gRNA #1_Fwd | CACCGTGATTCTGAAGGCGCTGAC |
| TorB_gRNA #1_Rev | AAACGTCAGCGCCTTCAGAATCAC |
| TorB_gRNA #2_Fwd | CACCCGCCTTCAGAATCACTTCCG |
| TorB_gRNA #2_Rev | AAACCGGAAGTGATTCTGAAGGCG |
| TorB_gRNA #3_Fwd | CACCTTTTTGGTTTTTGGTAACGA |
| TorB_gRNA #3_Rev | AAACTCGTTACCAAAAACCAAAAA |
| TorB_gRNA #4_Fwd | CACCGAAGCTGTTCGGACAGCATC |
| TorB_gRNA #4_Rev | AAACGATGCTGTCCGAACAGCTTC |
| Name | Sequence (5′–3′) |
|---|---|
| TorA_seq_Fwd | CACCGGAGACAGCTATAGCC |
| TorA_seq_Rev | GACCTTCTTGGCCAGATGCT |
| TorB_seq_Fwd | CCGCGCGAATGTGAAGTGCGCCCCCGTGGAAC |
| TorB_seq_Rev | GTCTTGTGCTCATGCGGGAAGTGCAGTGTG |
| Mutant | Genotype | Sequence | Mutations | |
|---|---|---|---|---|
| TorAKO | wild type | GAACCGGAAGAGCGTGTCTGGCGGTAGCGCCG..GTCGGTGGTCAGCGCAGGAAGGCGCGGGGAGGCG | ||
| knockout | GAACCGGAAGAGCGTGTCTGGCGGTAGCGCGGTGGT----GGTCAGCGCAGGAAGGCGCGGGGAGGCG (3) GAACCGGAAGAGCGTGTCTGGCGGTAGCGCCG..GT----GGTCAGCGCAGGAAGGCGCGGGGAGGCG (7) | 1 nt Ex, 2 bp In, 4 bp Del4 bp Del | ||
| TorBKO | wild type | CTTGGAGAAGCTGTTCGGACAGCATCTGGCCACGGAAGTGATTCTGAAGGCGCTGACCGGCTTCAAGA | ||
| knockout | CTTGGAGAAGCTGTTCGGACAGC-----------------------------CTGACCGGCTTCAAGA (15) | 29 bp Del | ||
| TorA/BDKO | TorA | wild type | GAACCGGAAGAGCGTGTCTGGCGGTAGCGCCGGTCGGTGGTCAGCGCAGGAAGGCGCGGGGAGGCGCG | |
| knockout | GAACCGGAAGAGCGTGTCTGGCGGTAGCGCCCG-------TCAGCGCAGGAAGGCGCGGGGAGGCGCG (15) | 1 nt Ex, 7 bp Del | ||
| TorB | wild type | CTTGGAGAAGCTGTTCGGACAGCATCTGGCCACGGAAGTGATTCTGAAGGCGCTGACCGGCTTCAAGA | ||
| knockout | CTTGGAGAAGCTGTTCGGACA------------------------------GCTGACCGGCTTCAAGA (15) | 30 bp Del | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hölper, J.E.; Klupp, B.G.; Luxton, G.W.G.; Franzke, K.; Mettenleiter, T.C. Function of Torsin AAA+ ATPases in Pseudorabies Virus Nuclear Egress. Cells 2020, 9, 738. https://doi.org/10.3390/cells9030738
Hölper JE, Klupp BG, Luxton GWG, Franzke K, Mettenleiter TC. Function of Torsin AAA+ ATPases in Pseudorabies Virus Nuclear Egress. Cells. 2020; 9(3):738. https://doi.org/10.3390/cells9030738
Chicago/Turabian StyleHölper, Julia E., Barbara G. Klupp, G. W. Gant Luxton, Kati Franzke, and Thomas C. Mettenleiter. 2020. "Function of Torsin AAA+ ATPases in Pseudorabies Virus Nuclear Egress" Cells 9, no. 3: 738. https://doi.org/10.3390/cells9030738
APA StyleHölper, J. E., Klupp, B. G., Luxton, G. W. G., Franzke, K., & Mettenleiter, T. C. (2020). Function of Torsin AAA+ ATPases in Pseudorabies Virus Nuclear Egress. Cells, 9(3), 738. https://doi.org/10.3390/cells9030738