VEGF Triggers Transient Induction of Autophagy in Endothelial Cells via AMPKα1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Antibodies
2.3. Cell Culture
2.4. Cell Treatment
2.5. siRNA Transfection
2.6. Cell Lysis and Western Blot
2.7. Cytokine Measurement
2.8. GSH Measurement
2.9. Cell Cycle Analysis
2.10. Mitochondrial ROS Measurement
2.11. ATP Measurement
2.12. Spheroid Assay
2.13. qRT-PCR
2.14. LC3B Immunofluorescence
2.15. Statistics
3. Results
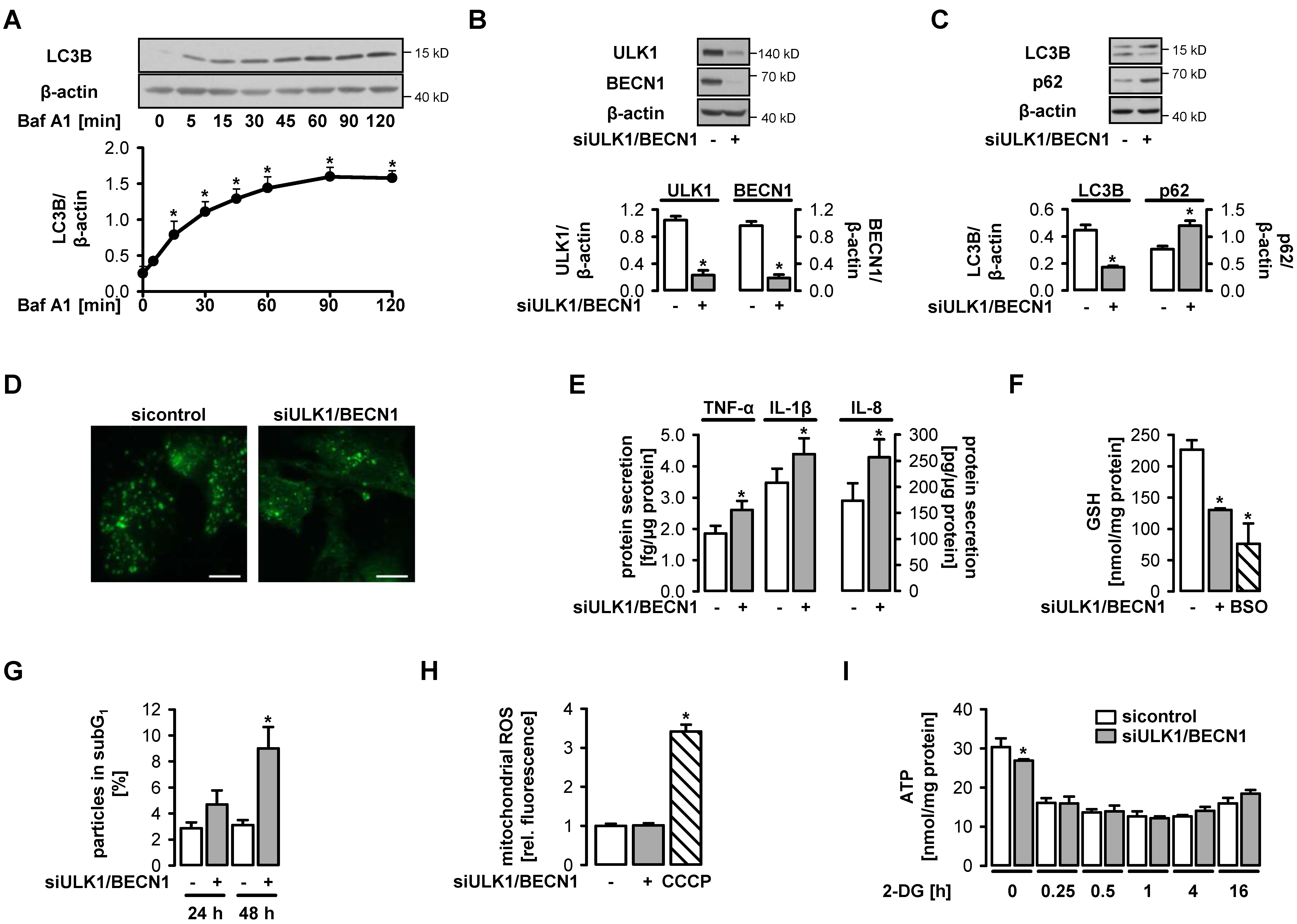
3.1. Functional Autophagy is a Prerequisite for Endothelial Homeostasis
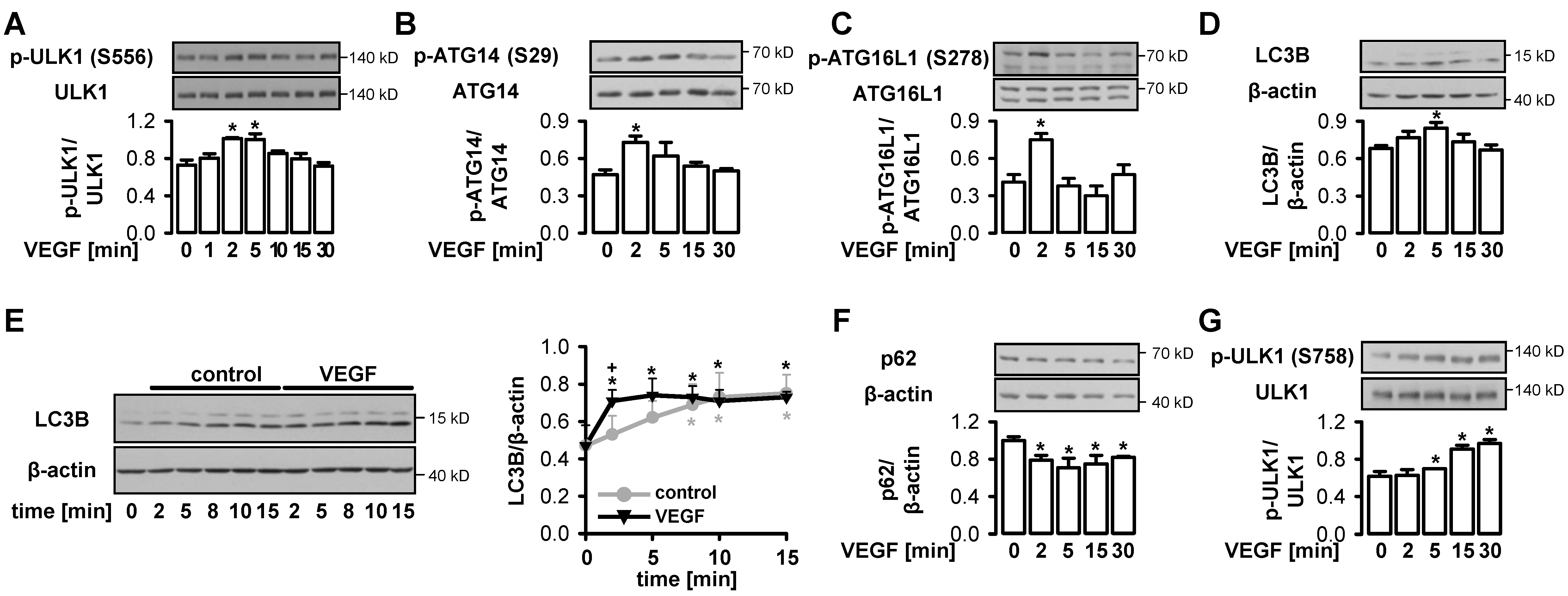
3.2. VEGF Initiates Functional Autophagy in Endothelial Cells via Phosphorylation of ULK1 at S556
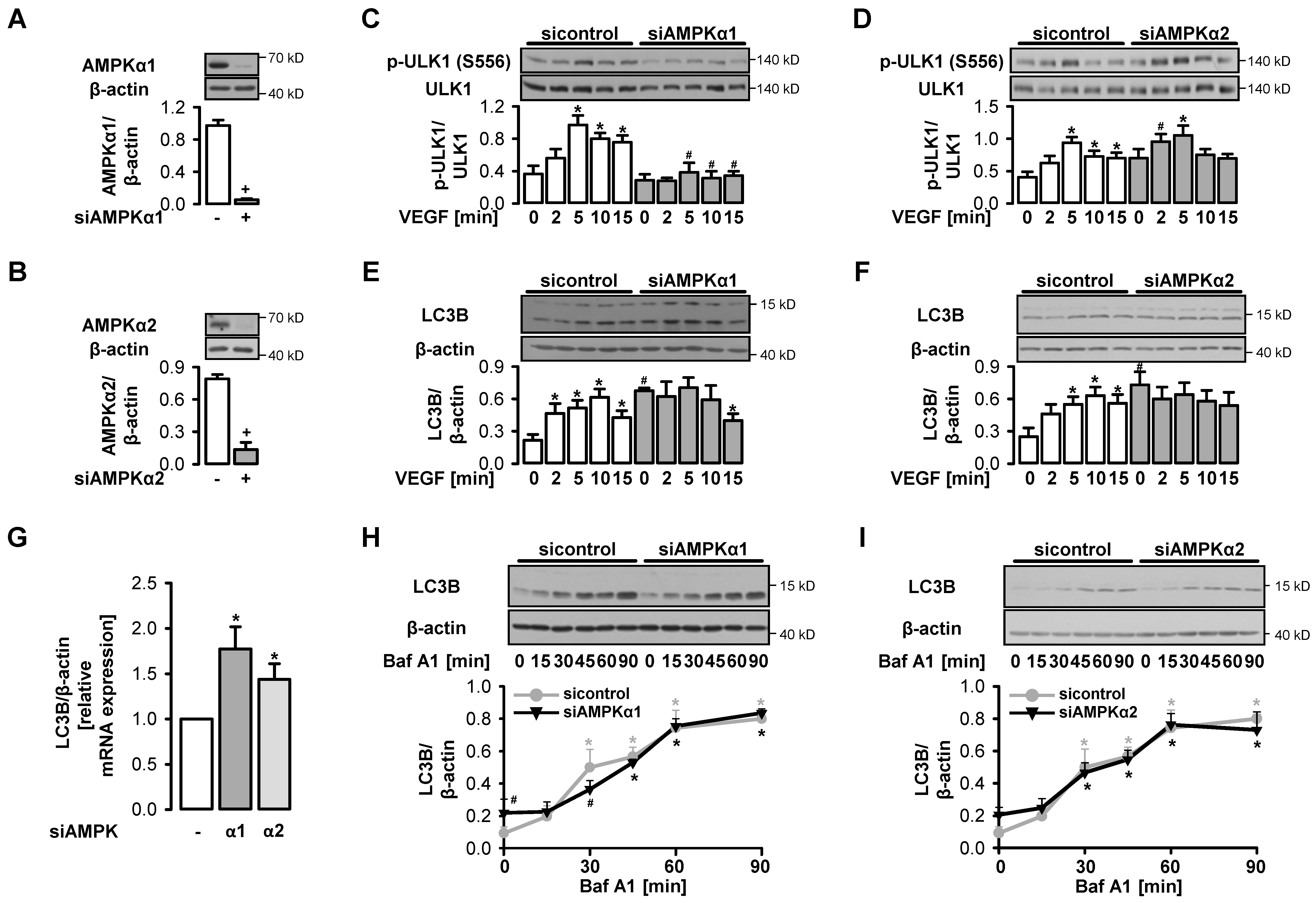
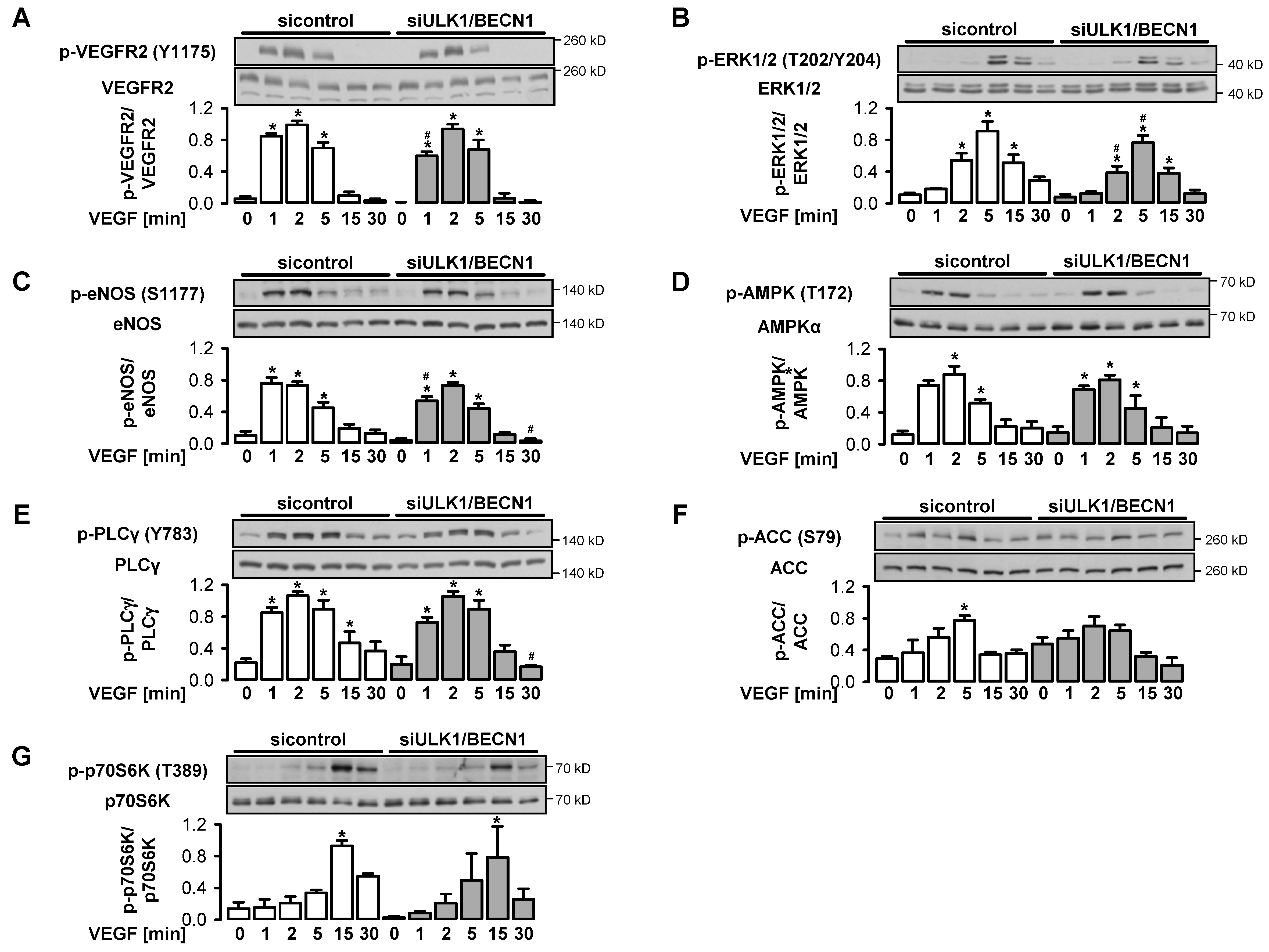
3.3. VEGF-Induced Initiation of Autophagy depends on AMPKα1
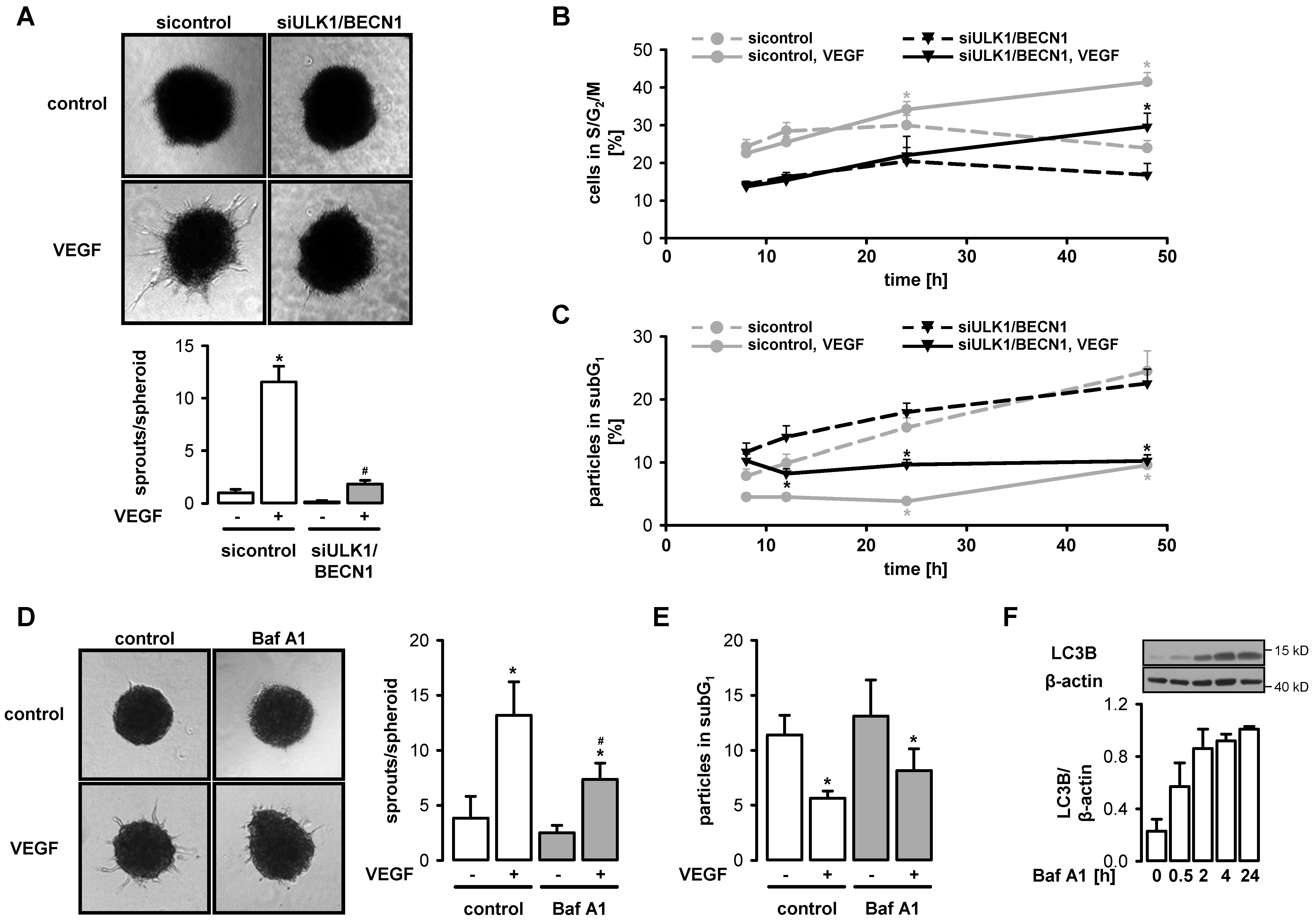
3.4. VEGF-Induced Angiogenesis Requires Functional Autophagy
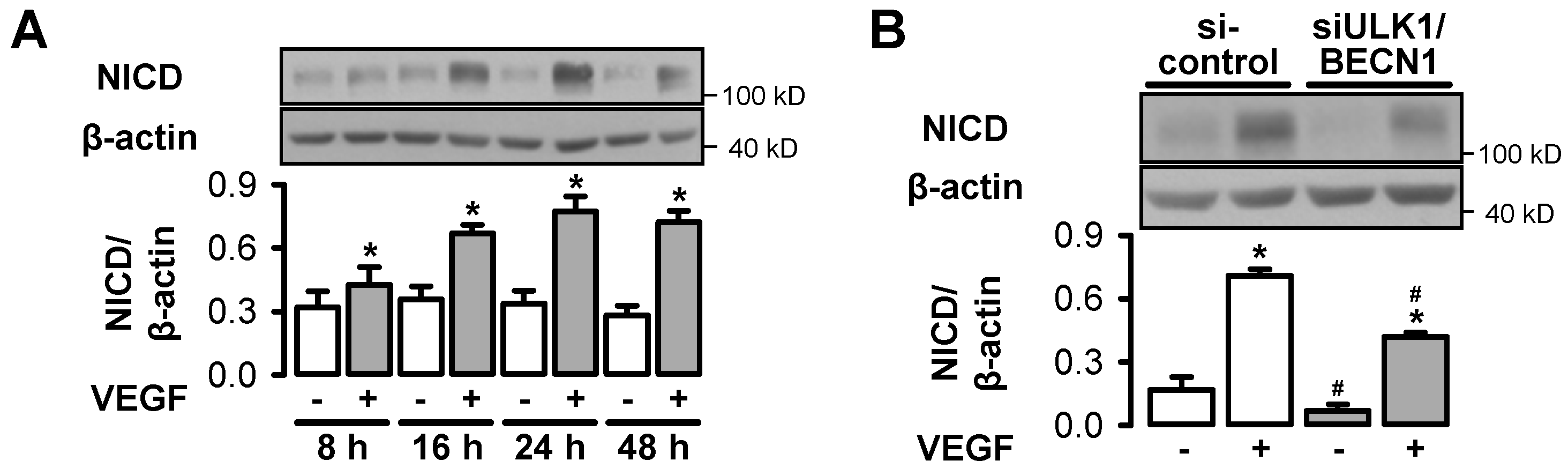
3.5. Autophagy Interferes with VEGF-Induced Notch Signaling
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. New Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Bach, M.; Larance, M.; James, D.E.; Ramm, G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 2011, 440, 283–291. [Google Scholar] [CrossRef]
- Mack, H.I.; Zheng, B.; Asara, J.M.; Thomas, S.M. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012, 8, 1197–1214. [Google Scholar] [CrossRef]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef] [PubMed]
- Nwadike, C.; Williamson, L.E.; Gallagher, L.E.; Guan, J.L.; Chan, E.Y.W. AMPK Inhibits ULK1-Dependent Autophagosome Formation and Lysosomal Acidification via Distinct Mechanisms. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Papinski, D.; Kraft, C. Regulation of Autophagy By Signaling Through the Atg1/ULK1 Complex. J. Mol. Biol. 2016, 428, 1725–1741. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Lystad, A.H.; Simonsen, A. Mechanisms and Pathophysiological Roles of the ATG8 Conjugation Machinery. Cells 2019, 8, 973. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef]
- Bharath, L.P.; Mueller, R.; Li, Y.; Ruan, T.; Kunz, D.; Goodrich, R.; Mills, T.; Deeter, L.; Sargsyan, A.; Anandh Babu, P.V.; et al. Impairment of autophagy in endothelial cells prevents shear-stress-induced increases in nitric oxide bioavailability. Can. J. Physiol. Pharmacol. 2014, 92, 605–612. [Google Scholar] [CrossRef]
- Liu, J.; Bi, X.; Chen, T.; Zhang, Q.; Wang, S.X.; Chiu, J.J.; Liu, G.S.; Zhang, Y.; Bu, P.; Jiang, F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015, 6, e1827. [Google Scholar] [CrossRef]
- Chen, F.; Chen, B.; Xiao, F.Q.; Wu, Y.T.; Wang, R.H.; Sun, Z.W.; Fu, G.S.; Mou, Y.; Tao, W.; Hu, X.S.; et al. Autophagy protects against senescence and apoptosis via the RAS-mitochondria in high-glucose-induced endothelial cells. Cell. Physiol. Biochem. 2014, 33, 1058–1074. [Google Scholar] [CrossRef]
- Torisu, K.; Singh, K.K.; Torisu, T.; Lovren, F.; Liu, J.; Pan, Y.; Quan, A.; Ramadan, A.; Al-Omran, M.; Pankova, N.; et al. Intact endothelial autophagy is required to maintain vascular lipid homeostasis. Aging Cell 2016, 15, 187–191. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef]
- Matsuda, J.; Namba, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Yamamoto, T.; Minami, S.; Sakai, S.; Fujimura, R.; Kaimori, J.Y.; et al. Antioxidant role of autophagy in maintaining the integrity of glomerular capillaries. Autophagy 2018, 14, 53–65. [Google Scholar] [CrossRef]
- Kim, H.S.; Montana, V.; Jang, H.J.; Parpura, V.; Kim, J.A. Epigallocatechin gallate (EGCG) stimulates autophagy in vascular endothelial cells: A potential role for reducing lipid accumulation. J. Biol. Chem. 2013, 288, 22693–22705. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Roth, L.; Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W. Defective Autophagy in Atherosclerosis: To Die or to Senesce? Oxidative Med. Cell. Longev. 2018, 2018, 7687083. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Yi, L.; Jin, X.; Liang, X.Y.; Zhou, Y.; Zhang, T.; Xie, Q.; Zhou, X.; Chang, H.; Fu, Y.J.; et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 2013, 9, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Pan, X.Y.; Xu, Y.; Xiao, Y.; An, Y.; Tie, L.; Pan, Y.; Li, X.J. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy 2012, 8, 812–825. [Google Scholar] [CrossRef]
- Muller, C.; Salvayre, R.; Negre-Salvayre, A.; Vindis, C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 2011, 18, 817–828. [Google Scholar] [CrossRef]
- Menghini, R.; Casagrande, V.; Marino, A.; Marchetti, V.; Cardellini, M.; Stoehr, R.; Rizza, S.; Martelli, E.; Greco, S.; Mauriello, A.; et al. MiR-216a: A link between endothelial dysfunction and autophagy. Cell Death Dis. 2014, 5, e1029. [Google Scholar] [CrossRef]
- Xie, Y.; You, S.J.; Zhang, Y.L.; Han, Q.; Cao, Y.J.; Xu, X.S.; Yang, Y.P.; Li, J.; Liu, C.F. Protective role of autophagy in AGE-induced early injury of human vascular endothelial cells. Mol. Med. Rep. 2011, 4, 459–464. [Google Scholar] [CrossRef]
- LaRocca, T.J.; Henson, G.D.; Thorburn, A.; Sindler, A.L.; Pierce, G.L.; Seals, D.R. Translational evidence that impaired autophagy contributes to arterial ageing. J. Physiol. 2012, 590, 3305–3316. [Google Scholar] [CrossRef]
- Mai, S.; Muster, B.; Bereiter-Hahn, J.; Jendrach, M. Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence lifespan. Autophagy 2012, 8, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Vion, A.C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl. Acad. Sci. USA 2017, 114, E8675–E8684. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, X.; Peng, J.; Tang, Y.; Yang, Q.; Liu, L.; Wang, Z.; Jiang, Z.; Xiao, M.; Ni, C.; et al. Autophagy regulates vascular endothelial cell eNOS and ET-1 expression induced by laminar shear stress in an ex vivo perfused system. Ann. Biomed. Eng. 2014, 42, 1978–1988. [Google Scholar] [CrossRef] [PubMed]
- Fetterman, J.L.; Holbrook, M.; Flint, N.; Feng, B.; Breton-Romero, R.; Linder, E.A.; Berk, B.D.; Duess, M.A.; Farb, M.G.; Gokce, N.; et al. Restoration of autophagy in endothelial cells from patients with diabetes mellitus improves nitric oxide signaling. Atherosclerosis 2016, 247, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Cho, J.M.; Park, S.K.; Ruan, T.; Li, Y.; Mueller, R.; Bean, T.; Reese, V.; Richardson, R.S.; Cai, J.; et al. Endothelial Cell Autophagy Maintains Shear Stress-Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Patella, F.; Neilson, L.J.; Athineos, D.; Erami, Z.; Anderson, K.I.; Blyth, K.; Ryan, K.M.; Zanivan, S. In-Depth Proteomics Identifies a Role for Autophagy in Controlling Reactive Oxygen Species Mediated Endothelial Permeability. J. Proteome Res. 2016, 15, 2187–2197. [Google Scholar] [CrossRef]
- Slavin, S.A.; Leonard, A.; Grose, V.; Fazal, F.; Rahman, A. Autophagy inhibitor 3-methyladenine protects against endothelial cell barrier dysfunction in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L388–L396. [Google Scholar] [CrossRef]
- Torisu, T.; Torisu, K.; Lee, I.H.; Liu, J.; Malide, D.; Combs, C.A.; Wu, X.S.; Rovira, I.I.; Fergusson, M.M.; Weigert, R.; et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat. Med. 2013, 19, 1281–1287. [Google Scholar] [CrossRef]
- Du, J.; Teng, R.J.; Guan, T.; Eis, A.; Kaul, S.; Konduri, G.G.; Shi, Y. Role of autophagy in angiogenesis in aortic endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 302, C383–C391. [Google Scholar] [CrossRef]
- Shen, W.; Tian, C.; Chen, H.; Yang, Y.; Zhu, D.; Gao, P.; Liu, J. Oxidative stress mediates chemerin-induced autophagy in endothelial cells. Free Radic. Biol. Med. 2013, 55, 73–82. [Google Scholar] [CrossRef]
- Lin, J.R.; Shen, W.L.; Yan, C.; Gao, P.J. Downregulation of dynamin-related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Sprott, D.; Poitz, D.M.; Korovina, I.; Ziogas, A.; Phieler, J.; Chatzigeorgiou, A.; Mitroulis, I.; Deussen, A.; Chavakis, T.; Klotzsche-von Ameln, A. Endothelial-Specific Deficiency of ATG5 (Autophagy Protein 5) Attenuates Ischemia-Related Angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, U.; Cui, X.; Hong, G.; Namkoong, S.; Karlsson, J.M.; Baty, C.J.; Tzeng, E. High mobility group box 1 promotes endothelial cell angiogenic behavior in vitro and improves muscle perfusion in vivo in response to ischemic injury. J. Vasc. Surg. 2012, 55, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yao, Y.; Hu, Z.; Hu, C.; Song, Q.; Ye, J.; Xu, C.; Wang, A.Z.; Chen, Q.; Wang, Q.K. Angiogenic Factor AGGF1 Activates Autophagy with an Essential Role in Therapeutic Angiogenesis for Heart Disease. PLoS Biol. 2016, 14, e1002529. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, H.P.; Jin, Y.; Choi, A.M.; Ryter, S.W. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy 2011, 7, 829–839. [Google Scholar] [CrossRef]
- Buraschi, S.; Neill, T.; Goyal, A.; Poluzzi, C.; Smythies, J.; Owens, R.T.; Schaefer, L.; Torres, A.; Iozzo, R.V. Decorin causes autophagy in endothelial cells via Peg3. Proc. Natl. Acad. Sci. USA 2013, 110, E2582–E2591. [Google Scholar] [CrossRef]
- Zhang, L.; Jing, H.; Cui, L.; Li, H.; Zhou, B.; Zhou, G.; Dai, F. 3,4-Dimethoxystilbene, a resveratrol derivative with anti-angiogenic effect, induces both macroautophagy and apoptosis in endothelial cells. J. Cell. Biochem. 2013, 114, 697–707. [Google Scholar] [CrossRef]
- Wang, Q.; Liang, B.; Shirwany, N.A.; Zou, M.H. 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS ONE 2011, 6, e17234. [Google Scholar] [CrossRef]
- Goyal, A.; Neill, T.; Owens, R.T.; Schaefer, L.; Iozzo, R.V. Reprint of: Decorin activates AMPK, an energy sensor kinase, to induce autophagy in endothelial cells. Matrix Biol. 2014, 35, 42–50. [Google Scholar] [CrossRef]
- Xiong, Y.; Yepuri, G.; Forbiteh, M.; Yu, Y.; Montani, J.P.; Yang, Z.; Ming, X.F. ARG2 impairs endothelial autophagy through regulation of MTOR and PRKAA/AMPK signaling in advanced atherosclerosis. Autophagy 2014, 10, 2223–2238. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Stahmann, N.; Woods, A.; Spengler, K.; Heslegrave, A.; Bauer, R.; Krause, S.; Viollet, B.; Carling, D.; Heller, R. Activation of AMP-activated protein kinase by vascular endothelial growth factor mediates endothelial angiogenesis independently of nitric-oxide synthase. J. Biol. Chem. 2010, 285, 10638–10652. [Google Scholar] [CrossRef] [PubMed]
- Nagata, D.; Mogi, M.; Walsh, K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J. Biol. Chem. 2003, 278, 31000–31006. [Google Scholar] [CrossRef] [PubMed]
- Reihill, J.A.; Ewart, M.A.; Salt, I.P. The role of AMP-activated protein kinase in the functional effects of vascular endothelial growth factor-A and -B in human aortic endothelial cells. Vasc. Cell 2011, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Zibrova, D.; Vandermoere, F.; Goransson, O.; Peggie, M.; Marino, K.V.; Knierim, A.; Spengler, K.; Weigert, C.; Viollet, B.; Morrice, N.A.; et al. GFAT1 phosphorylation by AMPK promotes VEGF-induced angiogenesis. Biochem. J. 2017, 474, 983–1001. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.P.; Mitchelhill, K.I.; Michell, B.J.; Stapleton, D.; Rodriguez-Crespo, I.; Witters, L.A.; Power, D.A.; Ortiz de Montellano, P.R.; Kemp, B.E. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999, 443, 285–289. [Google Scholar] [CrossRef]
- Mount, P.F.; Hill, R.E.; Fraser, S.A.; Levidiotis, V.; Katsis, F.; Kemp, B.E.; Power, D.A. Acute renal ischemia rapidly activates the energy sensor AMPK but does not increase phosphorylation of eNOS-Ser1177. Am. J. Physiol. Ren. Physiol. 2005, 289, F1103–F1115. [Google Scholar] [CrossRef]
- Morrow, V.A.; Foufelle, F.; Connell, J.M.; Petrie, J.R.; Gould, G.W.; Salt, I.P. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J. Biol. Chem. 2003, 278, 31629–31639. [Google Scholar] [CrossRef]
- Stahmann, N.; Woods, A.; Carling, D.; Heller, R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase beta. Mol. Cell. Biol. 2006, 26, 5933–5945. [Google Scholar] [CrossRef]
- Chen, Z.; Peng, I.C.; Sun, W.; Su, M.I.; Hsu, P.H.; Fu, Y.; Zhu, Y.; DeFea, K.; Pan, S.; Tsai, M.D.; et al. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ. Res. 2009, 104, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Spengler, K.; Grosse, S.; Kryeziu, N.; Knierim, A.; Heller, R. Studying the Role of AMPK in Angiogenesis. Methods Mol. Biol. 2018, 1732, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Nagy, J.A.; Manseau, E.J.; Phung, T.L.; Dvorak, H.F.; Benjamin, L.E. Rapamycin inhibition of the Akt/mTOR pathway blocks select stages of VEGF-A164-driven angiogenesis, in part by blocking S6Kinase. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Wold, M.S.; Lim, J.; Lachance, V.; Deng, Z.; Yue, Z. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol. Neurodegener. 2016, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Alsaadi, R.; Guo, Z.; Kalinina, A.; Carrier, M.; Tremblay, M.E.; Lacoste, B.; Lagace, D.; Russell, R.C. An antibody for analysis of autophagy induction. Nat. Methods 2020, 17, 232–239. [Google Scholar] [CrossRef]
- Pfisterer, S.G.; Mauthe, M.; Codogno, P.; Proikas-Cezanne, T. Ca2+/calmodulin-dependent kinase (CaMK) signaling via CaMKI and AMP-activated protein kinase contributes to the regulation of WIPI-1 at the onset of autophagy. Mol. Pharmacol. 2011, 80, 1066–1075. [Google Scholar] [CrossRef]
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 12637. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Yuan, H.; Fang, F.; Wei, X.; Liu, L.; Li, Q.; Guan, J.L.; Liu, F. Tsc1 Regulates the Balance Between Osteoblast and Adipocyte Differentiation Through Autophagy/Notch1/beta-Catenin Cascade. J. Bone Miner. Res. 2018, 33, 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Wang, J.; Wang, W.; Tian, Y.; XiangWei, W.; Chen, P.; Ma, K.; Zhou, C. Autophagy eliminates cytoplasmic beta-catenin and NICD to promote the cardiac differentiation of P19CL6 cells. Cell. Signal. 2014, 26, 2299–2305. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yu, S.; Zhang, H.; Xu, J. Angiogenesis impairment in diabetes: Role of methylglyoxal-induced receptor for advanced glycation endproducts, autophagy and vascular endothelial growth factor receptor 2. PLoS ONE 2012, 7, e46720. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, Q.; Xie, W.; Wang, Y.; Wang, G. Anti-tumor effects of triptolide on angiogenesis and cell apoptosis in osteosarcoma cells by inducing autophagy via repressing Wnt/beta-Catenin signaling. Biochem. Biophys. Res. Commun. 2018, 496, 443–449. [Google Scholar] [CrossRef]
- Kim, K.W.; Paul, P.; Qiao, J.; Lee, S.; Chung, D.H. Enhanced autophagy blocks angiogenesis via degradation of gastrin-releasing peptide in neuroblastoma cells. Autophagy 2013, 9, 1579–1590. [Google Scholar] [CrossRef]
- Mack, J.J.; Iruela-Arispe, M.L. NOTCH regulation of the endothelial cell phenotype. Curr. Opin. Hematol. 2018, 25, 212–218. [Google Scholar] [CrossRef]
- Wu, X.; Fleming, A.; Ricketts, T.; Pavel, M.; Virgin, H.; Menzies, F.M.; Rubinsztein, D.C. Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat. Commun. 2016, 7, 10533. [Google Scholar] [CrossRef]
- Ahn, J.S.; Ann, E.J.; Kim, M.Y.; Yoon, J.H.; Lee, H.J.; Jo, E.H.; Lee, K.; Lee, J.S.; Park, H.S. Autophagy negatively regulates tumor cell proliferation through phosphorylation dependent degradation of the Notch1 intracellular domain. Oncotarget 2016, 7, 79047–79063. [Google Scholar] [CrossRef]
- Zhang, T.; Guo, L.; Wang, Y.; Yang, Y. Macroautophagy Regulates Nuclear NOTCH1 Activity Through Multiple p62 Binding Sites. Iubmb Life 2018, 70, 985–994. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spengler, K.; Kryeziu, N.; Große, S.; Mosig, A.S.; Heller, R. VEGF Triggers Transient Induction of Autophagy in Endothelial Cells via AMPKα1. Cells 2020, 9, 687. https://doi.org/10.3390/cells9030687
Spengler K, Kryeziu N, Große S, Mosig AS, Heller R. VEGF Triggers Transient Induction of Autophagy in Endothelial Cells via AMPKα1. Cells. 2020; 9(3):687. https://doi.org/10.3390/cells9030687
Chicago/Turabian StyleSpengler, Katrin, Nderim Kryeziu, Silke Große, Alexander S. Mosig, and Regine Heller. 2020. "VEGF Triggers Transient Induction of Autophagy in Endothelial Cells via AMPKα1" Cells 9, no. 3: 687. https://doi.org/10.3390/cells9030687
APA StyleSpengler, K., Kryeziu, N., Große, S., Mosig, A. S., & Heller, R. (2020). VEGF Triggers Transient Induction of Autophagy in Endothelial Cells via AMPKα1. Cells, 9(3), 687. https://doi.org/10.3390/cells9030687