Molecular and Pharmacological Modulation of CALHM1 Promote Neuroprotection against Oxygen and Glucose Deprivation in a Model of Hippocampal Slices


 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Usage and Care
2.2. Isolation and Preparation of Mouse Hippocampal Slices
2.3. Oxygen and Glucose Deprivation in Mouse Hippocampal Slices
2.4. Glutamate Excitotoxicity in Mouse Hippocampal Slices
2.5. Determination of Hippocampal Viability by the MTT Assay
2.6. Quantification of Reactive Oxygen Species Production
2.7. Western Blot Analysis
2.8. Statistical Analysis
3. Results
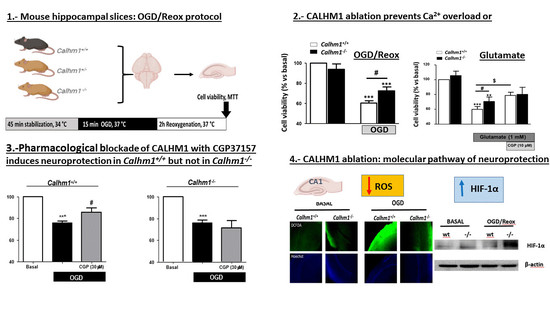
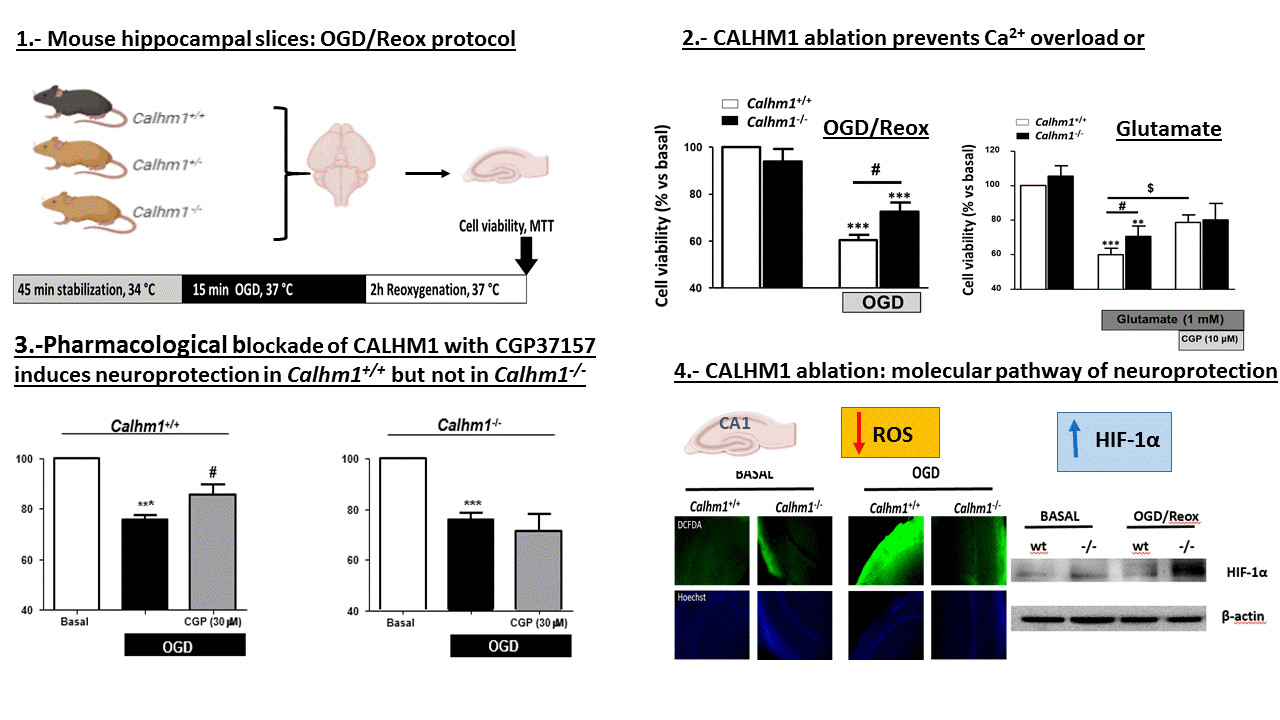
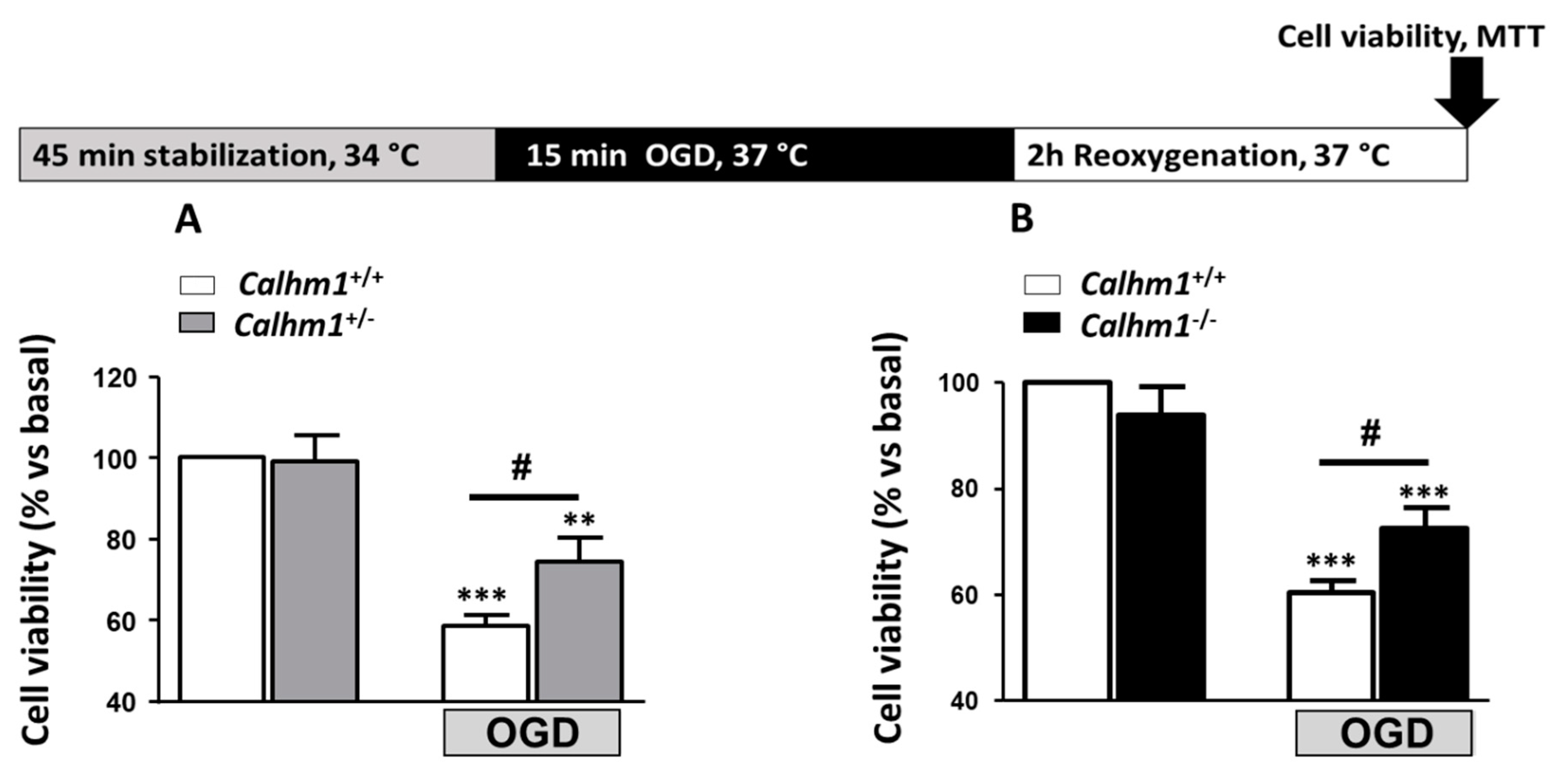
3.1. Partial or Total Molecular Ablation of CALHM1 Results in a Protective Effect in Hippocampal Slices Exposed to OGD/Reox
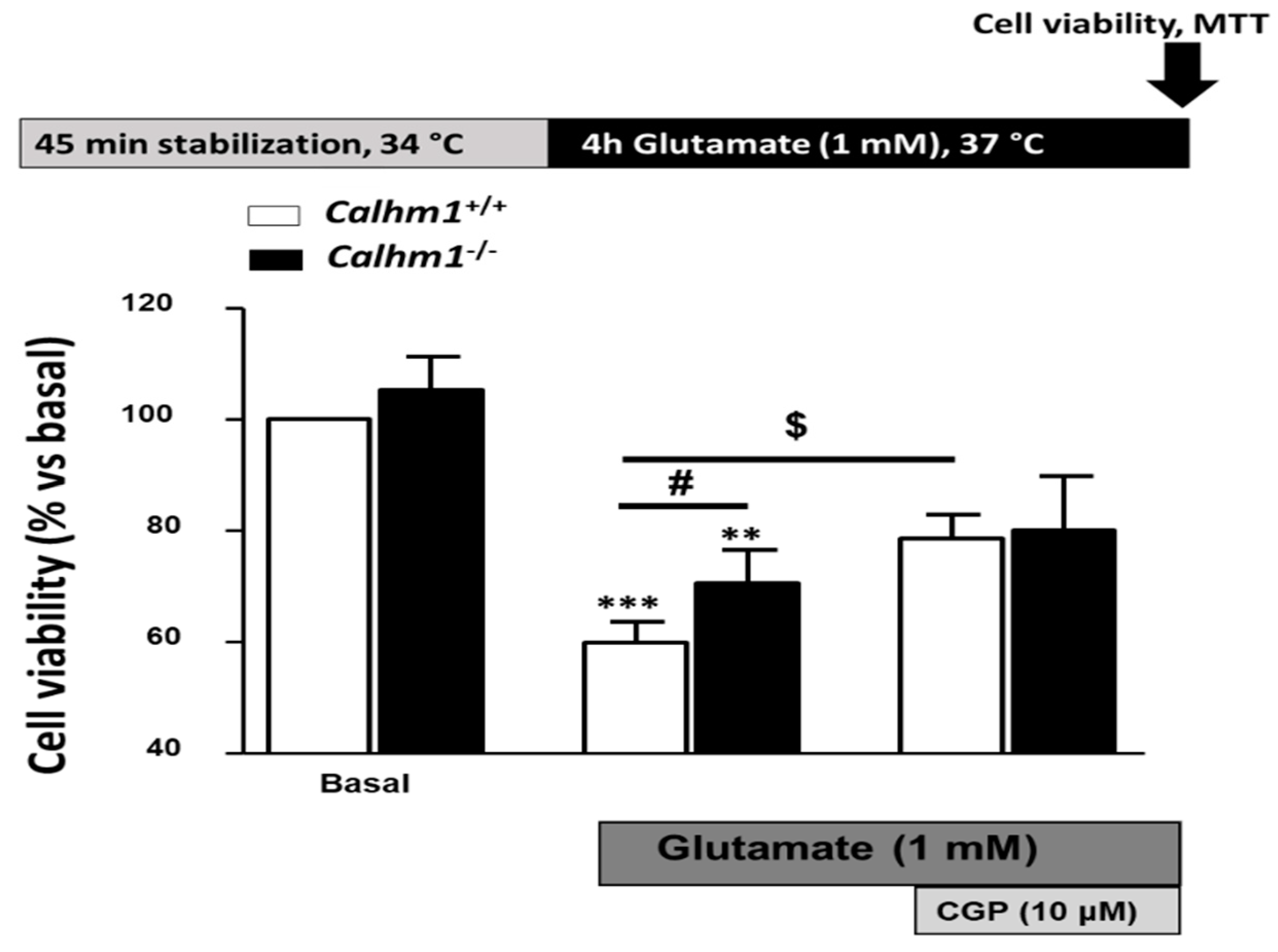
3.2. Genetic Ablation of CALHM1 Promotes Neuroprotection in the Glutamate Excitotoxicity Model
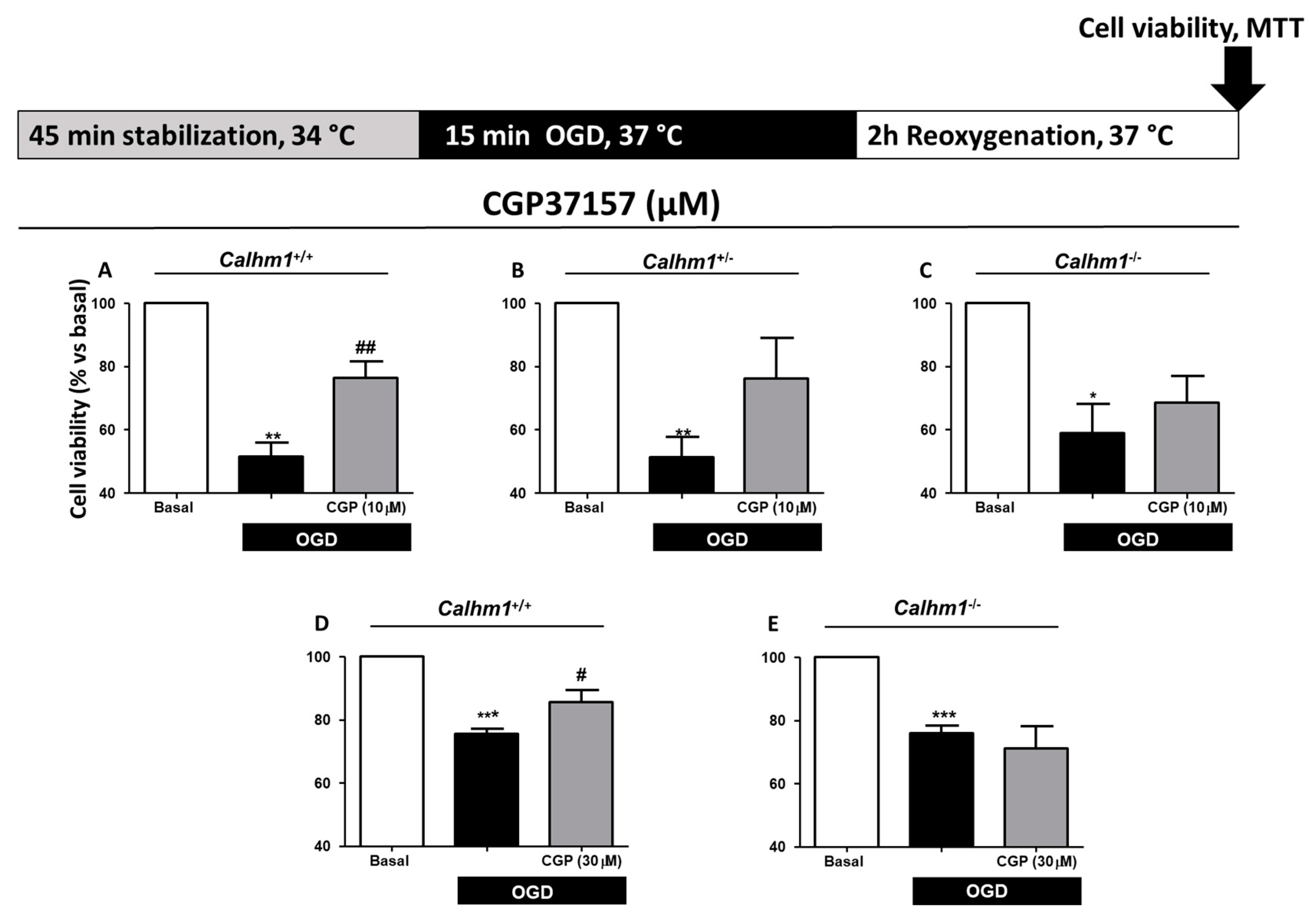
3.3. CGP37157 Requires CALHM1 to Exert Neuroprotection under OGD/Reox Conditions
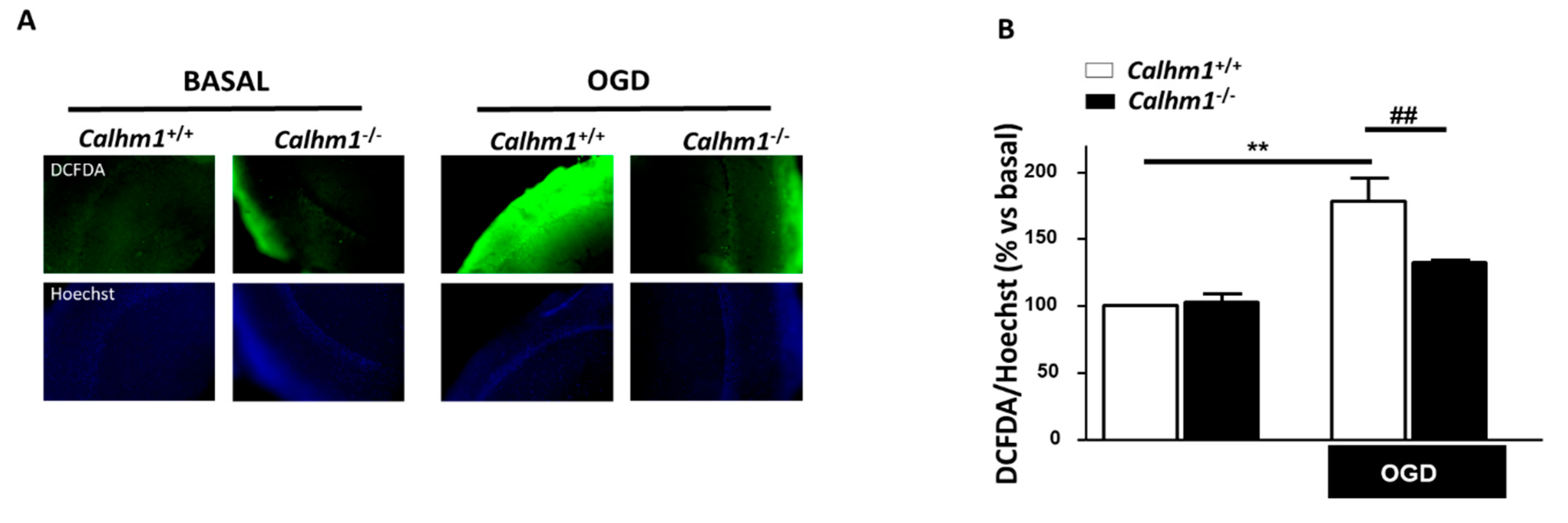
3.4. The Production of Free Radicals Induced by OGD/Reox is Prevented when CALHM1 Is Absent
3.5. HIF-1α is Overexpressed under OGD/Reox Conditions when CALHM1 is Absent
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davenport, R.; Dennis, M. Neurological emergencies: Acute stroke. J. Neurol. Neurosurg. Psychiatry 2000, 68, 277–288. [Google Scholar] [CrossRef]
- Balami, J.S.; Chen, R.L.; Grunwald, I.Q.; Buchan, A.M. Neurological complications of acute ischaemic stroke. Lancet Neurol. 2011, 10, 357–371. [Google Scholar] [CrossRef]
- Van Den Berg, L.A.; Dijkgraaf, M.G.; Berkhemer, O.A.; Fransen, P.S.; Beumer, D.; Lingsma, H.F.; Majoie, C.B.; Dippel, D.W.; van der Lugt, A.; van Oostenbrugge, R.J.; et al. Two-Year Outcome after Endovascular Treatment for Acute Ischemic Stroke. N. Engl. J. Med. 2017, 376, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Staples, M.; Tajiri, N.; Dailey, T.; Mosley, Y.I.; Lau, T.; van Loveren, H.; Kim, S.U.; Yamashima, T.; Yasuhara, T.; Date, I.; et al. In vivo animal stroke models: A rationale for rodent and non-human primate models. Transl. Stroke Res. 2013, 4, 308–321. [Google Scholar] [CrossRef][Green Version]
- Manning, N.W.; Campbell, B.C.; Oxley, T.J.; Chapot, R. Acute ischemic stroke: Time, penumbra, and reperfusion. Stroke 2014, 45, 640–644. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Kiedrowski, L. Critical role of sodium in cytosolic [Ca2+] elevations in cultured hippocampal CA1 neurons during anoxic depolarization. J. Neurochem. 2007, 100, 915–923. [Google Scholar] [CrossRef]
- Saliaris, A.P.; Amado, L.C.; Minhas, K.M.; Schuleri, K.H.; Lehrke, S.; St John, M.; Fitton, T.; Barreiro, C.; Berry, C.; Zheng, M.; et al. A concerted role of Na+ -K+ -Cl- cotransporter and Na+/Ca2+ exchanger in ischemic damage. J. Cereb. Blood Flow Metab. 2008, 28, 737–746. [Google Scholar]
- Adibhatla, R.M.; Hatcher, J.F. Phospholipase A(2), reactive oxygen species, and lipid peroxidation in CNS pathologies. BMB Rep. 2008, 41, 560–567. [Google Scholar] [CrossRef]
- Adibhatla, R.M.; Hatcher, J.F.; Dempsey, R.J. Lipids and lipidomics in brain injury and diseases. AAPS J. 2006, 8, E314–E321. [Google Scholar] [CrossRef]
- Młyniec, K.; Davies, C.L.; de Agüero Sánchez, I.G.; Pytka, K.; Budziszewska, B.; Nowak, G. Essential elements in depression and anxiety. Pharmacol. Rep. 2015, 67, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.D.; Thayer, S.A. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J. Neurosci. 1992, 12, 1882–1895. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.D. Neuroprotection for ischemic stroke: Past, present and future. Neuropharmacology 2008, 55, 363–389. [Google Scholar] [CrossRef] [PubMed]
- Dreses-Werringloer, U.; Lambert, J.C.; Vingtdeux, V.; Zhao, H.; Vais, H.; Siebert, A.; Jain, A.; Koppel, J.; Rovelet-Lecrux, A.; Hannequin, D.; et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Aβ levels, and Alzheimer’s disease risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, J.L.; Michalski, K.; Chou, T.H.; Grant, T.; Rao, S.; Simorowski, N. Structure and assembly of calcium homeostasis modulator proteins. Nat. Struct. Mol. Biol. 2020, 27, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Siebert, A.P.; Ma, Z.; Grevet, J.D.; Demuro, A.; Parker, I.; Foskett, J.K. Structural and functional similarities of calcium homeostasis modulator 1 (CALHM1) ion channel with connexins, pannexins, and innexins. J. Biol. Chem. 2013, 288, 6140–6153. [Google Scholar] [CrossRef]
- Ma, Z.; Siebert, A.P.; Cheung, K.H.; Lee, R.J.; Johnson, B.; Cohen, A.S.; Vingtdeux, V.; Marambaud, P.; Foskett, J.K. Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc. Natl. Acad. Sci. USA 2012, 109, 1963–1971. [Google Scholar] [CrossRef]
- Taruno, A.; Vingtdeux, V.; Ohmoto, M.; Ma, Z.; Dvoryanchikov, G.; Li, A.; Adrien, L.; Zhao, H.; Leung, S.; Abernethy, M.; et al. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature 2013, 495, 223–226. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Chang, E.H.; Frattini, S.A.; Zhao, H.; Chandakkar, P.; Adrien, L.; Strohl, J.J.; Gibson, E.L.; Ohmoto, M.; Matsumoto, I.; et al. CALHM1 deficiency impairs cerebral neuron activity and memory flexibility in mice. Sci. Rep. 2016, 6, 24250. [Google Scholar] [CrossRef]
- Cisneros-Mejorado, A.; Gottlieb, M.; Ruiz, A.; Chara, J.C.; Pérez-Samartín, A.; Marambaud, P.; Matute, C. Blockade and knock-out of CALHM1 channels attenuate ischemic brain damage. J. Cereb. Blood Flow Metab. 2018, 38, 1060–1069. [Google Scholar] [CrossRef]
- Moreno-Ortega, A.J.; Martínez-Sanz, F.J.; Lajarín-Cuesta, R.; de los Rios, C.; Cano-Abad, M.F. Benzothiazepine CGP37157 and its 2’-isopropyl analogue modulate Ca2+ entry through CALHM1. Neuropharmacology 2015, 95, 503–510. [Google Scholar] [CrossRef] [PubMed]
- González-Lafuente, L.; Egea, J.; León, R.; Martínez-Sanz, F.J.; Monjas, L.; Perez, C.; Merino, C.; García-De Diego, A.M.; Rodríguez-Franco, M.I.; García, A.G.; et al. Benzothiazepine CGP37157 and its isosteric 2’-methyl analogue provide neuroprotection and block cell calcium entry. ACS Chem. Neurosci. 2012, 3, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Molz, S.; Decker, H.; Dal-Cim, T.; Cremonez, C.; Cordova, F.M.; Leal, R.B.; Tasca, C.I. Glutamate-induced toxicity in hippocampal slices involves apoptotic features and p38 MAPK signaling. Neurochem. Res. 2008, 33, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Woster, P.M.; Yager, J.D.; Casero, R.A. The role of polyamine catabolism in polyamine analogue-induced programmed cell death. Proc. Natl. Acad. Sci. USA 1997, 94, 11557–11562. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Candal, A.; Argibay, B.; Iglesias-Rey, R.; Vargas, Z.; Vieites-Prado, A.; López-Arias, E.; Rodríguez-Castro, E.; López-Dequidt, I.; Rodríguez-Yáñez, M.; Piñeiro, Y.; et al. Vectorized nanodelivery systems for ischemic stroke: A concept and a need. J. Nanobiotechnol. 2017, 15, 30. [Google Scholar] [CrossRef]
- Lee, G.; Won, H.S.; Lee, Y.M.; Choi, J.W.; Oh, T.I.; Jang, J.H.; Choi, D.K.; Lim, B.O.; Kim, Y.J.; Park, J.W.; et al. Oxidative dimerization of PHD2 is responsible for its inactivation and contributes to metabolic reprogramming via HIF-1α activation. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Cuartero, M.I.; Ballesteros, I.; de la Parra, J.; Harkin, A.L.; Abautret-Daly, A.; Sherwin, E.; Fernández-Salguero, P.; Corbí, Á.L.; Lizasoain, I.; Moro, M.A. L-Kynurenine/Aryl Hydrocarbon Receptor Pathway Mediates Brain Damage After Experimental Stroke. Circulation 2014, 130, 2040–2051. [Google Scholar] [CrossRef]
- Martínez-Sanz, F.J.; Lajarín-Cuesta, R.; Moreno-Ortega, A.J.; González-Lafuente, L.; Fernández-Morales, J.C.; López-Arribas, R. Benzothiazepine CGP37157 Analogues Exert Cytoprotection in Various in Vitro Models of Neurodegeneration. ACS Chem. Neurosci. 2015, 6, 1626–1636. [Google Scholar] [CrossRef]
- Moreno-Ortega, A.J.; Ruiz-Nuño, A.; García, A.G.; Cano-Abad, M.F. Mitochondria sense with different kinetics the calcium entering into HeLa cells through calcium channels CALHM1 and mutated P86L-CALHM1. Biochem. Biophys. Res. Commun. 2010, 391, 722–726. [Google Scholar] [CrossRef]
- Gallego-Sandín, S.; Alonso, M.T.; García-Sancho, J. Calcium homoeostasis modulator 1 (CALHM1) reduces the calcium content of the endoplasmic reticulum (ER) and triggers ER stress. Biochem. J. 2011, 437, 469–475. [Google Scholar] [CrossRef]
- Mammucari, C.; Raffaello, A.; Reane, D.V.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: From molecular mechanism to animal models. Pflugers Arch. Eur. J. Physiol. 2018, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Divolis, G.; Mavroeidi, P.; Mavrofrydi, O.; Papazafiri, P. Differential effects of calcium on PI3K-Akt and HIF-1α survival pathways. Cell Biol. Toxicol. 2016, 32, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I. The interplay between HIF-1 and calcium signalling in cancer. Int. J. Biochem. Cell Biol. 2018, 97, 73–77. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrosa, J.; Paredes, I.; Marambaud, P.; G. López, M.; Cano-Abad, M.F. Molecular and Pharmacological Modulation of CALHM1 Promote Neuroprotection against Oxygen and Glucose Deprivation in a Model of Hippocampal Slices. Cells 2020, 9, 664. https://doi.org/10.3390/cells9030664
Garrosa J, Paredes I, Marambaud P, G. López M, Cano-Abad MF. Molecular and Pharmacological Modulation of CALHM1 Promote Neuroprotection against Oxygen and Glucose Deprivation in a Model of Hippocampal Slices. Cells. 2020; 9(3):664. https://doi.org/10.3390/cells9030664
Chicago/Turabian StyleGarrosa, Javier, Iñigo Paredes, Philippe Marambaud, Manuela G. López, and María F. Cano-Abad. 2020. "Molecular and Pharmacological Modulation of CALHM1 Promote Neuroprotection against Oxygen and Glucose Deprivation in a Model of Hippocampal Slices" Cells 9, no. 3: 664. https://doi.org/10.3390/cells9030664
APA StyleGarrosa, J., Paredes, I., Marambaud, P., G. López, M., & Cano-Abad, M. F. (2020). Molecular and Pharmacological Modulation of CALHM1 Promote Neuroprotection against Oxygen and Glucose Deprivation in a Model of Hippocampal Slices. Cells, 9(3), 664. https://doi.org/10.3390/cells9030664