G-Protein-Coupled Receptors in CNS: A Potential Therapeutic Target for Intervention in Neurodegenerative Disorders and Associated Cognitive Deficits

 ,
, 
 ,
, 

Abstract
1. Introduction
2. GPCRs’ Relevance in Cognition and Neurodegenerative Disorders
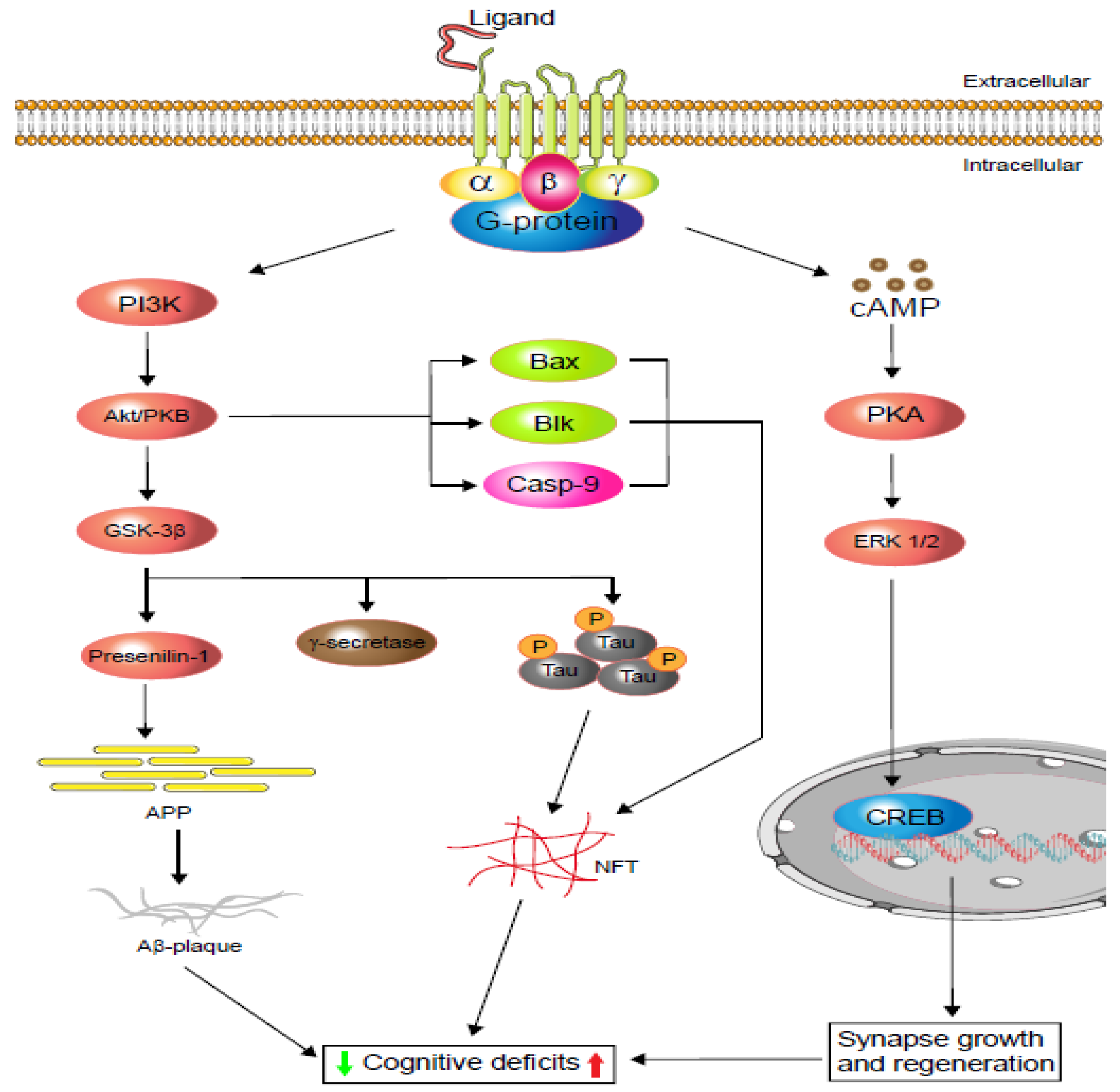
2.1. Alzheimer’s Disease
2.2. Vascular Dementia
2.3. Frontotemporal Dementia
2.4. Parkinson’s Disease
2.5. Multiple Sclerosis
2.6. Huntington’s Disease
3. Therapeutic Targets for Neurodegenerative Disorders Based on Different GPCR Classes
3.1. 5-HT Receptors
3.2. Dopamine Receptors
3.3. Cannabinoid Receptors
3.4. Cholinergic Receptors
3.5. Metabotropic Glutamate Receptors
3.6. Orphan GPCRs
4. An Emerging Paradigm in the Development of Therapeutics for Neurodegenerative Disorders
4.1. Allosteric Modulators of GPCRs in the Treatment of Neurodegeneration
4.2. Neuropeptides as a Target to Treat Neurodegenerations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HT1AR | 5-hydroxy tryptamine receptor 1A |
| 5-HT2AR | 5-hydroxy tryptamine receptor 2A |
| A2AR | adenosine 2a receptor |
| CB1R | cannabinoid type 1 receptor |
| CB2R | cannabinoid type 2 receptor |
| CRHR1 | corticotrophin-releasing hormone receptor 1 |
| D1R | dopamine D1 receptor |
| D2R | dopamine D2 receptor |
| GABA | γ-aminobutyric acid |
| GPR3 | G-protein-coupled receptor 3 |
| GPR88 | G-protein-coupled receptor 88 |
| GPR55 | G-protein-coupled receptor 55 |
| GPR52 | G-protein-coupled receptor 52 |
| 6-OHDA | 6-hydroxydopamine |
| mGluR | metabotropic glutamate receptors |
| nAChRs | nicotinic acetylcholin receptors |
| mAChR | muscharinic acetylcholin receptors |
| THC | Δ9-tetrahydrocannabinol |
| TRP | tryptophan |
References
- Sachdev, P.; Kalaria, R.; O’Brien, J.; Skoog, I.; Alladi, S.; Black, S.E.; Blacker, D.; Blazer, D.G.; Chen, C.; Chui, H.; et al. Diagnostic criteria for vascular cognitive disorders: A VASCOG statement. Alzheimer Dis. Assoc. Disord. 2014, 28, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Jakaria, M.; Azam, S.; Cho, D.-Y.; Haque, M.E.; Kim, I.-S.; Choi, D.-K. The Methanol Extract of Allium cepa L. Protects Inflammatory Markers in LPS-Induced BV-2 Microglial Cells and Upregulates the Antiapoptotic Gene and Antioxidant Enzymes in N27-A Cells. Antioxidants 2019, 8, 348. [Google Scholar] [CrossRef] [PubMed]
- Jakaria, M.; Azam, S.; Jo, S.-H.; Kim, I.-S.; Dash, R.; Choi, D.-K. Potential Therapeutic Targets of Quercetin and Its Derivatives: Its Role in the Therapy of Cognitive Impairment. J. Clin. Med. 2019, 8, 1789. [Google Scholar] [CrossRef] [PubMed]
- Jakaria, M.; Kim, J.; Karthivashan, G.; Park, S.-Y.; Ganesan, P.; Choi, D.-K. Emerging signals modulating potential of ginseng and its active compounds focusing on neurodegenerative diseases. J. Ginseng Res. 2019, 43, 163–171. [Google Scholar] [CrossRef]
- Jakaria, M.; Haque, M.E.; Kim, J.; Cho, D.Y.; Kim, I.S.; Choi, D.K. Active ginseng components in cognitive impairment: Therapeutic potential and prospects for delivery and clinical study. Oncotarget 2018, 9, 33601–33620. [Google Scholar] [CrossRef] [PubMed]
- Jakaria, M.; Azam, S.; Haque, M.E.; Jo, S.-H.; Uddin, M.S.; Kim, I.-S.; Choi, D.-K. Taurine and its analogs in neurological disorders: Focus on therapeutic potential and molecular mechanisms. Redox Biol. 2019, 24, 101223. [Google Scholar] [CrossRef]
- Huang, Y.; Todd, N.; Thathiah, A. The role of GPCRs in neurodegenerative diseases: Avenues for therapeutic intervention. Curr. Opin. Pharmacol. 2017, 32, 96–110. [Google Scholar] [CrossRef]
- Warren, J.D.; Rohrer, J.D.; Rossor, M.N. Clinical review. Frontotemporal dementia. BMJ Clin. Res. Ed. 2013, 347, f4827. [Google Scholar] [CrossRef]
- Jicha, G.A.; Nelson, P.T. Management of frontotemporal dementia: Targeting symptom management in such a heterogeneous disease requires a wide range of therapeutic options. Neurodegener. Dis. Manag. 2011, 1, 141–156. [Google Scholar] [CrossRef]
- Gabryelewicz, T.; Masellis, M.; Berdynski, M.; Bilbao, J.M.; Rogaeva, E.; St George-Hyslop, P.; Barczak, A.; Czyzewski, K.; Barcikowska, M.; Wszolek, Z.; et al. Intra-familial clinical heterogeneity due to FTLD-U with TDP-43 proteinopathy caused by a novel deletion in progranulin gene (PGRN). J. Alzheimers Dis. JAD 2010, 22, 11–1133. [Google Scholar] [CrossRef] [PubMed]
- Guerram, M.; Zhang, L.Y.; Jiang, Z.Z. G-protein coupled receptors as therapeutic targets for neurodegenerative and cerebrovascular diseases. Neurochem. Int. 2016, 101, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, P.G.; Ramos, M.L.S.; Amaro, A.J.; Dias, R.A.; Vieira, S.I. Gi/o-Protein Coupled Receptors in the Aging Brain. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef]
- Mombaerts, P.J. Genes and ligands for odorant, vomeronasal and taste receptors. Nat. Rev. Neurosci. 2004, 5, 263–278. [Google Scholar] [CrossRef]
- Heng, B.C.; Aubel, D.; Fussenegger, M. An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol. Adv. 2013, 31, 1676–1694. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A.J. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef]
- Rask-Andersen, M.; Masuram, S.; Schiöth, H.B. The druggable genome: Evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 9–26. [Google Scholar] [CrossRef]
- Laschet, C.; Dupuis, N.; Hanson, J.J. The G protein-coupled receptors deorphanization landscape. Biochem. Pharmacol. 2018, 153, 62–74. [Google Scholar] [CrossRef]
- Barthet, G.; Framery, B.; Gaven, F.; Pellissier, L.; Reiter, E.; Claeysen, S.; Bockaert, J.; Dumuis, A. 5-hydroxytryptamine 4 receptor activation of the extracellular signal-regulated kinase pathway depends on Src activation but not on G protein or beta-arrestin signaling. Mol. Biol. Cell 2007, 18, 1979–1991. [Google Scholar] [CrossRef]
- Maletínská, L.; Popelová, A.; Železná, B.; Bencze, M.; Kuneš, J. The impact of anorexigenic peptides in experimental models of Alzheimer’s disease pathology. J. Endocrinol. 2019, 240, R47. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Colović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The therapeutics of Alzheimer’s disease: Where we stand and where we are heading. Ann. Neurol. 2013, 74, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Farlow, M.R.; Graham, S.M.; Alva, G. Memantine for the treatment of Alzheimer’s disease: Tolerability and safety data from clinical trials. Drug Saf. 2008, 31, 577–585. [Google Scholar] [CrossRef]
- Willard, S.S.; Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 2013, 9, 948–959. [Google Scholar] [CrossRef]
- Hamilton, A.; Esseltine, J.L.; DeVries, R.A.; Cregan, S.P.; Ferguson, S.S. Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol. Brain 2014, 7, 40. [Google Scholar] [CrossRef]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef]
- Lo, A.C.; De Maeyer, J.H.; Vermaercke, B.; Callaerts-Vegh, Z.; Schuurkes, J.A.; D’Hooge, R. SSP-002392, a new 5-HT4 receptor agonist, dose-dependently reverses scopolamine-induced learning and memory impairments in C57Bl/6 mice. Neuropharmacology 2014, 85, 178–189. [Google Scholar] [CrossRef]
- Zhang, G.; Ásgeirsdóttir, H.N.; Cohen, S.J.; Munchow, A.H.; Barrera, M.P.; Stackman, R.W., Jr. Stimulation of serotonin 2A receptors facilitates consolidation and extinction of fear memory in C57BL/6J mice. Neuropharmacology 2013, 64, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. The pathology of “vascular dementia”: A critical update. J. Alzheimers Dis. JAD 2008, 14, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.-I.; Ogawa, N.; Asanuma, M.; Kondo, Y.; Nomura, M.J. Relationship between cholinergic dysfunction and discrimination learning disabilities in Wistar rats following chronic cerebral hypoperfusion. Brain Res. 1996, 729, 55–65. [Google Scholar] [CrossRef]
- Wan, P.; Wang, S.; Zhang, Y.; Lv, J.; Jin, Q.J. Involvement of dopamine D1 receptors of the hippocampal dentate gyrus in spatial learning and memory deficits in a rat model of vascular dementia. Die Pharm. Int. J. Pharm. Sci. 2014, 69, 709–710. [Google Scholar]
- Li, C.-J.; Lu, Y.; Zhou, M.; Zong, X.-G.; Li, C.; Xu, X.-L.; Guo, L.-J.; Lu, Q.J. Activation of GABA B receptors ameliorates cognitive impairment via restoring the balance of HCN1/HCN2 surface expression in the hippocampal CA1 area in rats with chronic cerebral hypoperfusion. Mol. Neurobiol. 2014, 50, 704–720. [Google Scholar] [CrossRef] [PubMed]
- Glikmann-Johnston, Y.; Saling, M.M.; Reutens, D.C.; Stout, J.C.J. Hippocampal 5-HT1A receptor and spatial learning and memory. Front. Pharmacol. 2015, 6, 289. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.S.; Ballard, C.G.; Kalaria, R.N.; Perry, R.; Hortobagyi, T.; Francis, P.T. Increased binding to 5-HT1A and 5-HT2A receptors is associated with large vessel infarction and relative preservation of cognition. Brain 2009, 132, 1858–1865. [Google Scholar] [CrossRef]
- King, M.V.; Marsden, C.A.; Fone, K.C.J. A role for the 5-HT1A, 5-HT4 and 5-HT6 receptors in learning and memory. Trends Pharmacol. Sci. 2008, 29, 482–492. [Google Scholar] [CrossRef]
- Neary, D.; Snowden, J.; Mann, D. Frontotemporal dementia. Lancet Neurol. 2005, 4, 771–780. [Google Scholar] [CrossRef]
- Jesso, S.; Morlog, D.; Ross, S.; Pell, M.D.; Pasternak, S.H.; Mitchell, D.G.; Kertesz, A.; Finger, E.C. The effects of oxytocin on social cognition and behaviour in frontotemporal dementia. Brain J. Neurol. 2011, 134, 2493–2501. [Google Scholar] [CrossRef]
- Baribeau, D.A.; Anagnostou, E. Oxytocin and vasopressin: Linking pituitary neuropeptides and their receptors to social neurocircuits. Front. Neurosci. 2015, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, Z.R.; Young, L.J. Oxytocin, Vasopressin, and the Neurogenetics of Sociality. Science 2008, 322, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Theodosis, D.T.; Koksma, J.-J.; Trailin, A.; Langle, S.L.; Piet, R.; Lodder, J.C.; Timmerman, J.; Mansvelder, H.; Poulain, D.A.; Oliet, S.H.R.; et al. Oxytocin and estrogen promote rapid formation of functional GABA synapses in the adult supraoptic nucleus. Mol. Cell. Neurosci. 2006, 31, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, M.; Anchisi, D.; Pelati, O.; Zuffi, M.; Matarrese, M.; Moresco, R.M.; Fazio, F.; Perani, D. Glucose metabolism and serotonin receptors in the frontotemporal lobe degeneration. Ann. Neurol. 2005, 57, 216–225. [Google Scholar] [CrossRef]
- Bowen, D.; Procter, A.; Mann, D.; Snowden, J.; Esiri, M.; Neary, D.; Francis, P. Imbalance of a serotonergic system in frontotemporal dementia: Implication for pharmacotherapy. Psychopharmacology 2008, 196, 603–610. [Google Scholar] [CrossRef]
- Alagarsamy, S.; Rouse, S.T.; Junge, C.; Hubert, G.; Gutman, D.; Smith, Y.; Conn, P.J. NMDA-induced phosphorylation and regulation of mGluR5. Pharmacol. Biochem. Behav. 2002, 73, 299–306. [Google Scholar] [CrossRef]
- Leuzy, A.; Zimmer, E.R.; Dubois, J.; Pruessner, J.; Cooperman, C.; Soucy, J.-P.; Kostikov, A.; Schirmaccher, E.; Désautels, R.; Gauthier, S.J.B.S.; et al. In vivo characterization of metabotropic glutamate receptor type 5 abnormalities in behavioral variant FTD. Brain Struct. Funct. 2016, 221, 1387–1402. [Google Scholar] [CrossRef]
- Homayoun, H.; Moghaddam, B.J. Bursting of prefrontal cortex neurons in awake rats is regulated by metabotropic glutamate 5 (mGlu5) receptors: Rate-dependent influence and interaction with NMDA receptors. Cereb. Cortex 2005, 16, 93–105. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 13, 318–324. [Google Scholar] [CrossRef]
- More, S.V.; Choi, D.-K. Emerging preclinical pharmacological targets for Parkinson’s disease. Oncotarget 2016, 7, 29835–29863. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Bronnick, K.; Williams-Gray, C.; Weintraub, D.; Marder, K.; Kulisevsky, J.; Burn, D.; Barone, P.; Pagonabarraga, J.; Allcock, L.; et al. Mild cognitive impairment in Parkinson disease: A multicenter pooled analysis. Neurology 2010, 75, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Hely, M.A.; Reid, W.G.; Adena, M.A.; Halliday, G.M.; Morris, J.G. The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. 2008, 23, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Hisahara, S.; Shimohama, S. Dopamine Receptors and Parkinson’s Disease. Int. J. Med. Chem. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Pezze, M.-A.; Dalley, J.W.; Robbins, T.W. Differential roles of dopamine D1 and D2 receptors in the nucleus accumbens in attentional performance on the five-choice serial reaction time task. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2007, 32, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Leverenz, J.B.; Schneider, J.S.; Adler, C.H. The neurobiological basis of cognitive impairment in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 634–650. [Google Scholar] [CrossRef]
- Ballard, C.; Ziabreva, I.; Perry, R.; Larsen, J.P.; O’Brien, J.; McKeith, I.; Perry, E.; Aarsland, D. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology 2006, 67, 1931–1934. [Google Scholar] [CrossRef]
- Shimada, H.; Hirano, S.; Shinotoh, H.; Aotsuka, A.; Sato, K.; Tanaka, N.; Ota, T.; Asahina, M.; Fukushi, K.; Kuwabara, S.; et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 2009, 73, 273–278. [Google Scholar] [CrossRef]
- Varrone, A.; Svenningsson, P.; Marklund, P.; Fatouros-Bergman, H.; Forsberg, A.; Halldin, C.; Nilsson, L.G.; Farde, L. 5-HT1B receptor imaging and cognition: A positron emission tomography study in control subjects and Parkinson’s disease patients. Synapse 2015, 69, 365–374. [Google Scholar] [CrossRef]
- Benedict, R.H.; Cookfair, D.; Gavett, R.; Gunther, M.; Munschauer, F.; Garg, N.; Weinstock-Guttman, B. Validity of the minimal assessment of cognitive function in multiple sclerosis (MACFIMS). J. Int. Neuropsychol. Soc. JINS 2006, 12, 549–558. [Google Scholar] [CrossRef]
- Lubrini, G.; Perianez, J.A.; Rios-Lago, M.; Frank, A. Processing speed in relapsing-remitting multiple sclerosis: The role played by the depressive symptoms. Rev. Neurol. 2012, 55, 585–592. [Google Scholar] [PubMed]
- Arnett, P.A.; Higginson, C.I.; Voss, W.D.; Bender, W.I.; Wurst, J.M.; Tippin, J.M. Depression in multiple sclerosis: Relationship to working memory capacity. Neuropsychology 1999, 13, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, A.; Magalhaes, S.; Richard, J.F.; Audet, B.; Moore, C. The link between multiple sclerosis and depression. Nat. Rev. Neurol. 2014, 10, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Ehde, D.M.; Kraft, G.H.; Chwastiak, L.; Sullivan, M.D.; Gibbons, L.E.; Bombardier, C.H.; Wadhwani, R. Efficacy of paroxetine in treating major depressive disorder in persons with multiple sclerosis. Gen. Hosp. Psychiatry 2008, 30, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Ferrante, R.J.; Swartz, K.J.; Kowall, N.W. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. Off. J. Soc. Neurosci. 1991, 11, 1649–1659. [Google Scholar] [CrossRef]
- Bruno, V.; Ksiazek, I.; Battaglia, G.; Lukic, S.; Leonhardt, T.; Sauer, D.; Gasparini, F.; Kuhn, R.; Nicoletti, F.; Flor, P.J. Selective blockade of metabotropic glutamate receptor subtype 5 is neuroprotective. Neuropharmacology 2000, 39, 2223–2230. [Google Scholar] [CrossRef]
- Schiefer, J.; Sprunken, A.; Puls, C.; Luesse, H.G.; Milkereit, A.; Milkereit, E.; Johann, V.; Kosinski, C.M. The metabotropic glutamate receptor 5 antagonist MPEP and the mGluR2 agonist LY379268 modify disease progression in a transgenic mouse model of Huntington’s disease. Brain Res. 2004, 1019, 246–254. [Google Scholar] [CrossRef]
- Chen, J.Y.; Wang, E.A.; Cepeda, C.; Levine, M.S. Dopamine imbalance in Huntington’s disease: A mechanism for the lack of behavioral flexibility. Front. Neurosci. 2013, 7, 114. [Google Scholar] [CrossRef]
- Charvin, D.; Vanhoutte, P.; Pages, C.; Borrelli, E.; Caboche, J. Unraveling a role for dopamine in Huntington’s disease: The dual role of reactive oxygen species and D2 receptor stimulation. Proc. Natl. Acad. Sci. USA 2005, 102, 12218–12223. [Google Scholar] [CrossRef]
- Charvin, D.; Roze, E.; Perrin, V.; Deyts, C.; Betuing, S.; Pages, C.; Regulier, E.; Luthi-Carter, R.; Brouillet, E.; Deglon, N.; et al. Haloperidol protects striatal neurons from dysfunction induced by mutated huntingtin in vivo. Neurobiol. Dis. 2008, 29, 22–29. [Google Scholar] [CrossRef]
- Meneses, A. 5-HT system and cognition. Neurosci. Biobehav. Rev. 1999, 23, 1111–1125. [Google Scholar] [CrossRef]
- Buhot, M.C.; Martin, S.; Segu, L. Role of serotonin in memory impairment. Ann. Med. 2000, 32, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Codony, X.; Vela, J.M.; Ramírez, M.J. 5-HT6 receptor and cognition. Curr. Opin. Pharmacol. 2011, 11, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Fone, K.C. An update on the role of the 5-hydroxytryptamine6 receptor in cognitive function. Neuropharmacology 2008, 55, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Meneses, A.; Pérez-García, G.; Ponce-Lopez, T.; Castillo, C. 5-HT6 receptor memory and amnesia: Behavioral pharmacology–learning and memory processes. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 96, pp. 27–47. [Google Scholar]
- González-Vera, J.A.; Medina, R.A.; Martín-Fontecha, M.; Gonzalez, A.; de la Fuente, T.; Vázquez-Villa, H.; García-Cárceles, J.; Botta, J.; McCormick, P.J.; Benhamú, B.; et al. A new serotonin 5-HT6 receptor antagonist with procognitive activity—Importance of a halogen bond interaction to stabilize the binding. Sci. Rep. 2017, 7, 41293. [Google Scholar] [CrossRef] [PubMed]
- Benhamú, B.; Martin-Fontecha, M.; Vázquez-Villa, H.; Pardo, L.; Lopez-Rodriguez, M.L.J. Serotonin 5-HT6 receptor antagonists for the treatment of cognitive deficiency in Alzheimer’s disease. J. Med. Chem. 2014, 57, 7160–7181. [Google Scholar] [CrossRef]
- Ramírez, M.J. 5-HT 6 receptors and Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 15. [Google Scholar]
- Marazziti, D.; Rutigliano, G.; Catena-Dell’Osso, M.; Baroni, S.; Dell’Osso, L. The 5-HT6 receptor antagonism approach in Alzheimer’s disease. Drugs Future 2014, 39, 133–140. [Google Scholar] [CrossRef]
- Mohler, E.G.; Baker, P.M.; Gannon, K.S.; Jones, S.S.; Shacham, S.; Sweeney, J.A.; Ragozzino, M.E. The effects of PRX-07034, a novel 5-HT6 antagonist, on cognitive flexibility and working memory in rats. Psychopharmacology 2012, 220, 687–696. [Google Scholar] [CrossRef]
- Ferrero, H.; Solas, M.; Francis, P.T.; Ramirez, M.J. Serotonin 5-HT6 Receptor Antagonists in Alzheimer’s Disease: Therapeutic Rationale and Current Development Status. CNS Drugs 2017, 31, 19–32. [Google Scholar] [CrossRef]
- Andrews, M.; Tousi, B.; Sabbagh, M.N. 5HT6 Antagonists in the Treatment of Alzheimer’s Dementia: Current Progress. Neurol. Ther. 2018, 7, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Halford, J.C.; Harrold, J.A.; Boyland, E.J.; Lawton, C.L.; Blundell, J.E. Serotonergic drugs: Effects on appetite expression and use for the treatment of obesity. Drugs 2007, 67, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Ivachtchenko, A.V.; Ivanenkov, Y.A.; Veselov, M.S.; Okun, I.M. AVN-322 is a Safe Orally Bio-Available Potent and Highly Selective Antagonist of 5-HT6R with Demonstrated Ability to Improve Impaired Memory in Animal Models. Curr. Alzheimer Res. 2017, 14, 268–294. [Google Scholar] [CrossRef] [PubMed]
- Ivachtchenko, A.V.; Lavrovsky, Y.; Okun, I. AVN-101: A Multi-Target Drug Candidate for the Treatment of CNS Disorders. J. Alzheimers Dis. JAD 2016, 53, 583–620. [Google Scholar] [CrossRef]
- Ivachtchenko, A.V.; Lavrovsky, Y.; Ivanenkov, Y.A. AVN-211, Novel and Highly Selective 5-HT6 Receptor Small Molecule Antagonist, for the Treatment of Alzheimer’s Disease. Mol. Pharm. 2016, 13, 945–963. [Google Scholar] [CrossRef]
- Schneider, L.S. Idalopirdine for Alzheimer’s disease: Written in the stars. Lancet Neurol. 2014, 13, 1063–1065. [Google Scholar] [CrossRef]
- Roman, G.C.; Wilkinson, D.G.; Doody, R.S.; Black, S.E.; Salloway, S.P.; Schindler, R.J. Donepezil in vascular dementia: Combined analysis of two large-scale clinical trials. Dement. Geriatr. Cogn. Disord. 2005, 20, 338–344. [Google Scholar] [CrossRef]
- Maher-Edwards, G.; Zvartau-Hind, M.; Hunter, A.J.; Gold, M.; Hopton, G.; Jacobs, G.; Davy, M.; Williams, P. Double-blind, controlled phase II study of a 5-HT6 receptor antagonist, SB-742457, in Alzheimer’s disease. Curr. Alzheimer Res. 2010, 7, 374–385. [Google Scholar] [CrossRef]
- Maher-Edwards, G.; Dixon, R.; Hunter, J.; Gold, M.; Hopton, G.; Jacobs, G.; Hunter, J.; Williams, P. SB-742457 and donepezil in Alzheimer disease: A randomized, placebo-controlled study. Int. J. Geriatr. Psychiatry 2011, 26, 536–544. [Google Scholar] [CrossRef]
- Maher-Edwards, G.; Watson, C.; Ascher, J.; Barnett, C.; Boswell, D.; Davies, J.; Fernandez, M.; Kurz, A.; Zanetti, O.; Safirstein, B.; et al. Two randomized controlled trials of SB742457 in mild-to-moderate Alzheimer’s disease. Alzheimers Dement. 2015, 1, 23–36. [Google Scholar] [CrossRef]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [CrossRef]
- Nirogi, R.; Kandikere, V.; Mudigonda, K.; Bhyrapuneni, G.; Jasti, V. Safety, tolerability and pharmacokinetics of SUVN-502—A 5-HT6 receptor antagonist, first in human phase-1 Single Ascending Dose (SAD) study. Alzheimers Dement. J. Alzheimers Assoc. 2009, 5, P250. [Google Scholar] [CrossRef]
- Nirogi, R.; Ajjala, D.R.; Aleti, R.; Rayapati, L.; Pantangi, H.R.; Boggavarapu, R.K.; Padala, N.S.P. Development and validation of sensitive LC-MS/MS method for the quantification of SUVN-502 and its metabolite and its application for first in human pharmacokinetic study. J. Pharm. Biomed. Anal. 2017, 145, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Nirogi, R.; Shinde, A.; Kambhampati, R.S.; Mohammed, A.R.; Saraf, S.K.; Badange, R.K.; Bandyala, T.R.; Bhatta, V.; Bojja, K.; Reballi, V.J. Discovery and Development of 1-[(2-Bromophenyl) sulfonyl]-5-methoxy-3-[(4-methyl-1-piperazinyl) methyl]-1 H-indole Dimesylate Monohydrate (SUVN-502): A Novel, Potent, Selective and Orally Active Serotonin 6 (5-HT6) Receptor Antagonist for Potential Treatment of Alzheimer’s Disease. J. Med. Chem. 2017, 60, 1843–1859. [Google Scholar] [PubMed]
- Callegari, I.; Mattei, C.; Benassi, F.; Krueger, F.; Grafman, J.; Yaldizli, Ö.; Sassos, D.; Massucco, D.; Scialò, C.; Nobili, F. Agomelatine improves apathy in frontotemporal dementia. Neurodegener. Dis. 2016, 16, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Karaiskos, D.; Pappa, D.; Katirtzoglou, E.; Siarkos, K.; Tzavellas, E.; Papadimitriou, G.; Politis, A. EPA-1197–Agomelatine for treating apathy in alzheimer’s disease. Eur. Psychiatry 2014, 29, 1. [Google Scholar] [CrossRef]
- Avila, A.; Cardona, X.; Martin-Baranera, M.; Leon, L.; Caballol, N.; Millet, P.; Bello, J.J. Agomelatine for depression in Parkinson disease: Additional effect on sleep and motor dysfunction. J. Clin. Psychopharmacol. 2015, 35, 719–723. [Google Scholar] [CrossRef]
- Biskup, C.S.; Sanchez, C.L.; Arrant, A.; Van Swearingen, A.E.; Kuhn, C.; Zepf, F.D. Effects of acute tryptophan depletion on brain serotonin function and concentrations of dopamine and norepinephrine in C57BL/6J and BALB/cJ mice. PLoS ONE 2012, 7, e35916. [Google Scholar] [CrossRef]
- Jans, L.A.; Korte-Bouws, G.A.; Korte, S.M.; Blokland, A. The effects of acute tryptophan depletion on affective behaviour and cognition in Brown Norway and Sprague Dawley rats. J. Psychopharmacol. 2010, 24, 605–614. [Google Scholar] [CrossRef]
- Young, S.N.; Ervin, F.R.; Pihl, R.O.; Finn, P. Biochemical aspects of tryptophan depletion in primates. Psychopharmacology 1989, 98, 508–511. [Google Scholar] [CrossRef]
- Riedel, W.J.; Klaassen, T.; Deutz, N.E.; van Someren, A.; van Praag, H.M. Tryptophan depletion in normal volunteers produces selective impairment in memory consolidation. Psychopharmacology 1999, 141, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, D.; Riedel, W.J.; Sambeth, A. Effects of acute tryptophan depletion on memory, attention and executive functions: A systematic review. Neurosci. Biobehav. Rev. 2009, 33, 926–952. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.L.; Silliman, R.A.; Guadagnoli, E.; Mor, V. The effect of less than definitive care on breast carcinoma recurrence and mortality. Cancer 2000, 89, 1739–1747. [Google Scholar] [CrossRef]
- Mohajeri, M.H.; Wittwer, J.; Vargas, K.; Hogan, E.; Holmes, A.; Rogers, P.J.; Goralczyk, R.; Gibson, E.L. Chronic treatment with a tryptophan-rich protein hydrolysate improves emotional processing, mental energy levels and reaction time in middle-aged women. Br. J. Nutr. 2015, 113, 350–365. [Google Scholar] [CrossRef]
- Rondanelli, M.; Opizzi, A.; Faliva, M.; Mozzoni, M.; Antoniello, N.; Cazzola, R.; Savare, R.; Cerutti, R.; Grossi, E.; Cestaro, B. Effects of a diet integration with an oily emulsion of DHA-phospholipids containing melatonin and tryptophan in elderly patients suffering from mild cognitive impairment. Nutr. Neurosci. 2012, 15, 46–54. [Google Scholar] [CrossRef]
- Pae, C.-U.; Wang, S.-M.; Han, C.; Lee, S.-J.; Patkar, A.A.; Masand, P.S.; Serretti, A. Vortioxetine: A meta-analysis of 12 short-term, randomized, placebo-controlled clinical trials for the treatment of major depressive disorder. J. Psychiatry Neurosci. 2015, 40, 174–186. [Google Scholar] [CrossRef]
- Almeida, S.; Glahn, D.C.; Argyropoulos, S.V.; Frangou, S. Acute citalopram administration may disrupt contextual information processing in healthy males. Eur. Psychiatry J. Assoc. Eur. Psychiatr. 2010, 25, 87–91. [Google Scholar] [CrossRef]
- Yu, Y.; Patch, C.; Weston-Green, K.; Zhou, Y.; Zheng, K.; Huang, X.F. Dietary Galacto-Oligosaccharides and Resistant Starch Protect Against Altered CB1 and 5-HT1A and 2A Receptor Densities in Rat Brain: Implications for Preventing Cognitive and Appetite Dysfunction During a High-Fat Diet. Mol. Nutr. Food Res. 2018, 62, e1800422. [Google Scholar] [CrossRef]
- Carlsson, A. The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacol. Rev. 1959, 11, 490–493. [Google Scholar]
- Girault, J.-A.; Greengard, P. The Neurobiology of Dopamine Signaling. Arch. Neurol. 2004, 61, 641–644. [Google Scholar] [CrossRef]
- Westbrook, A.; Braver, T.S. Dopamine Does Double Duty in Motivating Cognitive Effort. Neuron 2016, 89, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Grace, A.A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 2016, 17, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.; Wang, M.; Paspalas, C.D. Dopamine’s Actions in Primate Prefrontal Cortex: Challenges for Treating Cognitive Disorders. Pharmacol. Rev. 2015, 67, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.; Badgaiyan, R.D.; Dunston, G.M.; Baron, D.; Modestino, E.J.; McLaughlin, T.; Steinberg, B.; Gold, M.S.; Gondre-Lewis, M.C. The DRD2 Taq1A A1 Allele May Magnify the Risk of Alzheimer’s in Aging African-Americans. Mol. Neurobiol. 2018, 55, 5526–5536. [Google Scholar] [CrossRef]
- Karthivashan, G.; Ganesan, P.; Park, S.-Y.; Lee, H.-W.; Choi, D.-K. Lipid-based nanodelivery approaches for dopamine-replacement therapies in Parkinson’s disease: From preclinical to translational studies. Biomaterials 2020, 232, 119704. [Google Scholar] [CrossRef]
- van Wimersma Greidanus, T.B.; Jolles, J.; De Wied, D. Hypothalamic neuropeptides and memory. Acta Neurochir. 1985, 75, 99–105. [Google Scholar] [CrossRef]
- Fallon, S.J.; Muhammed, K.; Drew, D.S.; Ang, Y.-S.; Manohar, S.G.; Husain, M. Dopamine guides competition for cognitive control: Common effects of haloperidol on working memory and response conflict. Cortex 2019, 113, 156–168. [Google Scholar] [CrossRef]
- Kapur, S.; Seeman, P. Does fast dissociation from the dopamine D2 receptor explain the action of atypical antipsychotics? A new hypothesis. Am. J. Psychiatry 2001, 158, 360–369. [Google Scholar] [CrossRef]
- Marshall, C.A.; Brodnik, Z.D.; Mortensen, O.V.; Reith, M.E.A.; Shumsky, J.S.; Waterhouse, B.D.; España, R.A.; Kortagere, S. Selective activation of Dopamine D3 receptors and norepinephrine transporter blockade enhances sustained attention. Neuropharmacology 2019, 148, 178–188. [Google Scholar] [CrossRef]
- Morena, M.; Campolongo, P. The endocannabinoid system: An emotional buffer in the modulation of memory function. Neurobiol. Learn. Mem. 2014, 112, 30–43. [Google Scholar] [CrossRef]
- Campolongo, P.; Ratano, P.; Manduca, A.; Scattoni, M.L.; Palmery, M.; Trezza, V.; Cuomo, V. The endocannabinoid transport inhibitor AM404 differentially modulates recognition memory in rats depending on environmental aversiveness. Front. Behav. Neurosci. 2012, 6. [Google Scholar] [CrossRef] [PubMed]
- Campolongo, P.; Roozendaal, B.; Trezza, V.; Hauer, D.; Schelling, G.; McGaugh, J.L.; Cuomo, V. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc. Natl. Acad. Sci. USA 2009, 106, 4888–4893. [Google Scholar] [CrossRef] [PubMed]
- Ferland, J.-M.N.; Carr, M.R.; Lee, A.M.; Hoogeland, M.E.; Winstanley, C.A.; Pattij, T. Examination of the effects of cannabinoid ligands on decision making in a rat gambling task. Pharmacol. Biochem. Behav. 2018, 170, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Irimia, C.; Polis, I.Y.; Stouffer, D.; Parsons, L.H. Persistent effects of chronic Δ9-THC exposure on motor impulsivity in rats. Psychopharmacology 2015, 232, 3033–3043. [Google Scholar] [CrossRef] [PubMed]
- Milton, N.G. Anandamide and noladin ether prevent neurotoxicity of the human amyloid-beta peptide. Neurosci. Lett. 2002, 332, 127–130. [Google Scholar] [CrossRef]
- Mazzola, C.; Micale, V.; Drago, F. Amnesia induced by beta-amyloid fragments is counteracted by cannabinoid CB1 receptor blockade. Eur. J. Pharmacol. 2003, 477, 219–225. [Google Scholar] [CrossRef]
- Manuel, I.; Lombardero, L.; LaFerla, F.M.; Gimenez-Llort, L.; Rodriguez-Puertas, R. Activity of muscarinic, galanin and cannabinoid receptors in the prodromal and advanced stages in the triple transgenic mice model of Alzheimer’s disease. Neuroscience 2016, 329, 284–293. [Google Scholar] [CrossRef]
- Fakhfouri, G.; Ahmadiani, A.; Rahimian, R.; Grolla, A.A.; Moradi, F.; Haeri, A. WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-gamma pathway. Neuropharmacology 2012, 63, 653–666. [Google Scholar] [CrossRef]
- Tolon, R.M.; Nunez, E.; Pazos, M.R.; Benito, C.; Castillo, A.I.; Martinez-Orgado, J.A.; Romero, J. The activation of cannabinoid CB2 receptors stimulates in situ and in vitro beta-amyloid removal by human macrophages. Brain Res. 2009, 1283, 148–154. [Google Scholar] [CrossRef]
- Wu, J.; Bie, B.; Yang, H.; Xu, J.J.; Brown, D.L.; Naguib, M. Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol. Aging 2013, 34, 791–804. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, J.; Romero, J.; Velasco, G.; Tolon, R.M.; Ramos, J.A.; Guzman, M. Cannabinoid CB2 receptor: A new target for controlling neural cell survival? Trends Pharmacol. Sci. 2007, 28, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Sagredo, O.; Gonzalez, S.; Aroyo, I.; Pazos, M.R.; Benito, C.; Lastres-Becker, I.; Romero, J.P.; Tolon, R.M.; Mechoulam, R.; Brouillet, E.; et al. Cannabinoid CB2 receptor agonists protect the striatum against malonate toxicity: Relevance for Huntington’s disease. Glia 2009, 57, 1154–1167. [Google Scholar] [CrossRef] [PubMed]
- Palazuelos, J.; Aguado, T.; Pazos, M.R.; Julien, B.; Carrasco, C.; Resel, E.; Sagredo, O.; Benito, C.; Romero, J.; Azcoitia, I.; et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain J. Neurol. 2009, 132, 3152–3164. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Bizat, N.; Boyer, F.; Hantraye, P.; Brouillet, E.; Fernandez-Ruiz, J. Effects of cannabinoids in the rat model of Huntington’s disease generated by an intrastriatal injection of malonate. Neuroreport 2003, 14, 813–816. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Denovan-Wright, E.M. Biased Type 1 Cannabinoid Receptor Signaling Influences Neuronal Viability in a Cell Culture Model of Huntington Disease. Mol. Pharmacol. 2016, 89, 364–375. [Google Scholar] [CrossRef]
- Diaz-Alonso, J.; Paraiso-Luna, J.; Navarrete, C.; Del Rio, C.; Cantarero, I.; Palomares, B.; Aguareles, J.; Fernandez-Ruiz, J.; Bellido, M.L.; Pollastro, F.; et al. VCE-003.2, a novel cannabigerol derivative, enhances neuronal progenitor cell survival and alleviates symptomatology in murine models of Huntington’s disease. Sci. Rep. 2016, 6, 29789. [Google Scholar] [CrossRef]
- Rodriguez-Cueto, C.; Santos-Garcia, I.; Garcia-Toscano, L.; Espejo-Porras, F.; Bellido, M.; Fernandez-Ruiz, J.; Munoz, E.; de Lago, E. Neuroprotective effects of the cannabigerol quinone derivative VCE-003.2 in SOD1(G93A) transgenic mice, an experimental model of amyotrophic lateral sclerosis. Biochem. Pharmacol. 2018, 157, 217–226. [Google Scholar] [CrossRef]
- Colangelo, C.; Shichkova, P.; Keller, D.; Markram, H.; Ramaswamy, S. Cellular, Synaptic and Network Effects of Acetylcholine in the Neocortex. Front. Neural Circuits 2019, 13. [Google Scholar] [CrossRef]
- Wevers, A. Localisation of pre- and postsynaptic cholinergic markers in the human brain. Behav. Brain Res. 2011, 221, 341–355. [Google Scholar] [CrossRef]
- Dani, J.A. Overview of nicotinic receptors and their roles in the central nervous system. Biol. Psychiatry 2001, 49, 166–174. [Google Scholar] [CrossRef]
- Quik, M.; Bordia, T.; O’Leary, K. Nicotinic receptors as CNS targets for Parkinson’s disease. Biochem. Pharmacol. 2007, 74, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Quik, M.; Wonnacott, S. α6β2* and α4β2* Nicotinic Acetylcholine Receptors as Drug Targets for Parkinson’s Disease. J. Pharmacol. Rev. 2011, 63, 938–966. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.B.; Bussert, T.G.; Kreitzer, A.C.; Seal, R.P. Striatal Cholinergic Neurotransmission Requires VGLUT3. J. Neurosci. 2014, 34, 8772–8777. [Google Scholar] [CrossRef] [PubMed]
- English, D.F.; Ibanez-Sandoval, O.; Stark, E.; Tecuapetla, F.; Buzsáki, G.; Deisseroth, K.; Tepper, J.M.; Koos, T. GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat. Neurosci. 2011, 15, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.E.; Patel, J.C.; Cragg, S.J. Dopamine release in the basal ganglia. Neuroscience 2011, 198, 112–137. [Google Scholar] [CrossRef]
- Zhou, F.-M.; Liang, Y.; Dani, J.A. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat. Neurosci. 2001, 4, 1224–1229. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, J.; Wu, J.; Zhu, C.; Hui, Y.; Han, Y.; Huang, Z.; Ellsworth, K.; Fan, W. α7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J. Neuroinflammation 2012, 9, 98. [Google Scholar] [CrossRef]
- Foucault-Fruchard, L.; Doméné, A.; Page, G.; Windsor, M.; Emond, P.; Rodrigues, N.; Dollé, F.; Damont, A.; Buron, F.; Routier, S.; et al. Neuroprotective effect of the alpha 7 nicotinic receptor agonist PHA 543613 in an in vivo excitotoxic adult rat model. Neuroscience 2017, 356, 52–63. [Google Scholar] [CrossRef]
- Medeiros, R.; Castello, N.A.; Cheng, D.; Kitazawa, M.; Baglietto-Vargas, D.; Green, K.N.; Esbenshade, T.A.; Bitner, R.S.; Decker, M.W.; LaFerla, F.M. alpha7 Nicotinic receptor agonist enhances cognition in aged 3xTg-AD mice with robust plaques and tangles. Am. J. Pathol. 2014, 184, 520–529. [Google Scholar] [CrossRef]
- Vicens, P.; Heredia, L.; Torrente, M.; Domingo, J.L. Behavioural effects of PNU-282987 and stress in an animal model of Alzheimer’s disease. Psychogeriatr. Off. J. Jpn. Psychogeriatr. Soc. 2017, 17, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Van Kampen, M.; Selbach, K.; Schneider, R.; Schiegel, E.; Boess, F.; Schreiber, R. AR-R 17779 improves social recognition in rats by activation of nicotinic alpha7 receptors. Psychopharmacology 2004, 172, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Boess, F.G.; De Vry, J.; Erb, C.; Flessner, T.; Hendrix, M.; Luithle, J.; Methfessel, C.; Riedl, B.; Schnizler, K.; van der Staay, F.J.; et al. The novel alpha7 nicotinic acetylcholine receptor agonist N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-7-[2-(methoxy)phenyl]-1-benzofuran-2-carboxa mide improves working and recognition memory in rodents. J. Pharmacol. Exp. Ther. 2007, 321, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Lahmy, V.; Long, R.; Morin, D.; Villard, V.; Maurice, T. Mitochondrial protection by the mixed muscarinic/sigma1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Abeta25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model. Front. Cell. Neurosci. 2014, 8, 463. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Bockaert, J.; Collingridge, G.L.; Conn, P.J.; Ferraguti, F.; Schoepp, D.D.; Wroblewski, J.T.; Pin, J.P. Metabotropic glutamate receptors: From the workbench to the bedside. Neuropharmacology 2011, 60, 1017–1041. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef]
- Iyer, A.M.; van Scheppingen, J.; Milenkovic, I.; Anink, J.J.; Lim, D.; Genazzani, A.A.; Adle-Biassette, H.; Kovacs, G.G.; Aronica, E. Metabotropic glutamate receptor 5 in Down’s syndrome hippocampus during development: Increased expression in astrocytes. Curr. Alzheimer Res. 2014, 11, 694–705. [Google Scholar] [CrossRef]
- Spampinato, S.F.; Copani, A.; Nicoletti, F.; Sortino, M.A.; Caraci, F. Metabotropic Glutamate Receptors in Glial Cells: A New Potential Target for Neuroprotection? Front. Mol. Neurosci. 2018, 11, 414. [Google Scholar] [CrossRef]
- Chen, X.; Lin, R.; Chang, L.; Xu, S.; Wei, X.; Zhang, J.; Wang, C.; Anwyl, R.; Wang, Q. Enhancement of long-term depression by soluble amyloid β protein in rat hippocampus is mediated by metabotropic glutamate receptor and involves activation of p38MAPK, STEP and caspase-3. Neuroscience 2013, 253, 435–443. [Google Scholar] [CrossRef]
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious Effects of Amyloid β Oligomers Acting as an Extracellular Scaffold for mGluR5. Neuron 2010, 66, 739–754. [Google Scholar] [CrossRef]
- Caraci, F.; Molinaro, G.; Battaglia, G.; Giuffrida, M.L.; Riozzi, B.; Traficante, A.; Bruno, V.; Cannella, M.; Merlo, S.; Wang, X.; et al. Targeting Group II Metabotropic Glutamate (mGlu) Receptors for the Treatment of Psychosis Associated with Alzheimer’s Disease: Selective Activation of mGlu2 Receptors Amplifies β-Amyloid Toxicity in Cultured Neurons, Whereas Dual Activation of mGlu2 and mGlu3 Receptors Is Neuroprotective. Mol. Pharmacol. 2011, 79, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-L.; Wang, Y.; Li, D.-L.; Luo, J.; Liu, M.-Y. Orphan G protein-coupled receptors (GPCRs): Biological functions and potential drug targets. Acta Pharmacol. Sin. 2012, 33, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Grottick, A.; Schuelert, N.; Rosenbrock, H.; von Heimendahl, M.; Arban, R.; Hobson, S.J.B.P. 669. GPR52 Agonists Represent a Novel Approach to Treat Cognitive Deficits Associated with Schizophrenia. Boil. Psychiatry 2017, 81, S271. [Google Scholar] [CrossRef]
- Haque, M.E.; Kim, I.-S.; Jakaria, M.; Akther, M.; Choi, D.-K. Importance of GPCR-Mediated Microglial Activation in Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Skwarek-Maruszewska, A.; Horré, K.; Vandewyer, E.; Wolfs, L.; Snellinx, A.; Saito, T.; Radaelli, E.; Corthout, N.; Colombelli, J. Loss of GPR3 reduces the amyloid plaque burden and improves memory in Alzheimer’s disease mouse models. Sci. Transl. Med. 2015, 7, 309ra164. [Google Scholar] [CrossRef] [PubMed]
- Khrimian, L.; Obri, A.; Ramos-Brossier, M.; Rousseaud, A.; Moriceau, S.; Nicot, A.-S.; Mera, P.; Kosmidis, S.; Karnavas, T.; Saudou, F.J. Gpr158 mediates osteocalcin’s regulation of cognition. J. Exp. Med. 2017, 214, 2859–2873. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, A.T.; Maroteaux, G.; Robe, A.; Venteo, L.; Nasseef, M.T.; van Kempen, L.C.; Mechawar, N.; Turecki, G.; Darcq, E.; Kieffer, B.L.J.C.b. Expression map of 78 brain-expressed mouse orphan GPCRs provides a translational resource for neuropsychiatric research. Commun. Biol. 2018, 1, 102. [Google Scholar] [CrossRef]
- Thathiah, A.; Spittaels, K.; Hoffmann, M.; Staes, M.; Cohen, A.; Horre, K.; Vanbrabant, M.; Coun, F.; Baekelandt, V.; Delacourte, A.; et al. The orphan G protein-coupled receptor 3 modulates amyloid-beta peptide generation in neurons. Science 2009, 323, 946–951. [Google Scholar] [CrossRef]
- Sutton, L.P.; Orlandi, C.; Song, C.; Oh, W.C.; Muntean, B.S.; Xie, K.; Filippini, A.; Xie, X.; Satterfield, R.; Yaeger, J.D. Orphan receptor GPR158 controls stress-induced depression. eLife 2018, 7, e33273. [Google Scholar] [CrossRef]
- Komatsu, H.; Maruyama, M.; Yao, S.; Shinohara, T.; Sakuma, K.; Imaichi, S.; Chikatsu, T.; Kuniyeda, K.; Siu, F.K.; Peng, L.S. Anatomical transcriptome of G protein-coupled receptors leads to the identification of a novel therapeutic candidate GPR52 for psychiatric disorders. PLoS ONE 2014, 9, e90134. [Google Scholar] [CrossRef]
- Valverde, O.; Celerier, E.; Baranyi, M.; Vanderhaeghen, P.; Maldonado, R.; Sperlagh, B.; Vassart, G.; Ledent, C. GPR3 receptor, a novel actor in the emotional-like responses. PLoS ONE 2009, 4, e4704. [Google Scholar] [CrossRef] [PubMed]
- Hurst, K.; Badgley, C.; Ellsworth, T.; Bell, S.; Friend, L.; Prince, B.; Welch, J.; Cowan, Z.; Williamson, R.; Lyon, C. A putative lysophosphatidylinositol receptor GPR55 modulates hippocampal synaptic plasticity. Hippocampus 2017, 27, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Saito, T.; Takasaki, J.; Kamohara, M.; Sugimoto, T.; Kobayashi, M.; Tadokoro, M.; Matsumoto, S.-I.; Ohishi, T.; Furuichi, K.J.B.; et al. An evolutionarily conserved G-protein coupled receptor family, SREB, expressed in the central nervous system. Biochem. Biophys. Res. Commun. 2000, 272, 576–582. [Google Scholar] [CrossRef]
- Yanai, T.; Kurosawa, A.; Nikaido, Y.; Nakajima, N.; Saito, T.; Osada, H.; Konno, A.; Hirai, H.; Takeda, S. Identification and molecular docking studies for novel inverse agonists of SREB, super conserved receptor expressed in brain. Genes Cells 2016, 21, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Hellebrand, S.; Wittenberger, T.; Schaller, H.C.; Hermans-Borgmeyer, I. Gpr85, a novel member of the G-protein coupled receptor family, prominently expressed in the developing mouse cerebral cortex. Gene Expr. Patterns 2001, 1, 13–16. [Google Scholar] [CrossRef]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef]
- Lutjens, R.; Rocher, J.P. Recent advances in drug discovery of GPCR allosteric modulators for neurodegenerative disorders. Curr. Opin. Pharmacol. 2017, 32, 91–95. [Google Scholar] [CrossRef]
- Notartomaso, S.; Zappulla, C.; Biagioni, F.; Cannella, M.; Bucci, D.; Mascio, G.; Scarselli, P.; Fazio, F.; Weisz, F.; Lionetto, L. Pharmacological enhancement of mGlu1 metabotropic glutamate receptors causes a prolonged symptomatic benefit in a mouse model of spinocerebellar ataxia type 1. Mol. Brain 2013, 6, 48. [Google Scholar] [CrossRef]
- Tison, F.; Keywood, C.; Wakefield, M.; Durif, F.; Corvol, J.C.; Eggert, K.; Lew, M.; Isaacson, S.; Bezard, E.; Poli, S.M.J.M.D. A phase 2a trial of the novel mglur5-negative allosteric modulator dipraglurant for levodopa-induced dyskinesia in Parkinson’s disease. Mov. Disord. 2016, 31, 1373–1380. [Google Scholar] [CrossRef]
- Foster, D.J.; Choi, D.L.; Conn, P.J.; Rook, J.M. Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer’s disease and schizophrenia. Neuropsychiatr. Dis. Treat. 2014, 10, 183–191. [Google Scholar]
- Jakubík, J.; El-Fakahany, E.E. Allosteric modulation of muscarinic acetylcholine receptors. Pharmacological 2010, 3, 2838–2860. [Google Scholar] [CrossRef] [PubMed]
- Davoren, J.E.; Lee, C.-W.; Garnsey, M.; Brodney, M.A.; Cordes, J.; Dlugolenski, K.; Edgerton, J.R.; Harris, A.R.; Helal, C.J.; Jenkinson, S.; et al. Discovery of the Potent and Selective M1 PAM-Agonist N-[(3R,4S)-3-Hydroxytetrahydro-2H-pyran-4-yl]-5-methyl-4-[4-(1,3-thiazol-4-yl)benzyl]pyridine-2-carboxamide (PF-06767832): Evaluation of Efficacy and Cholinergic Side Effects. J. Med. Chem. 2016, 59, 6313–6328. [Google Scholar] [CrossRef] [PubMed]
- Melancon, B.J.; Tarr, J.C.; Panarese, J.D.; Wood, M.R.; Lindsley, C.W. Allosteric modulation of the M1 muscarinic acetylcholine receptor: Improving cognition and a potential treatment for schizophrenia and Alzheimer’s disease. Drug Discov. Today 2013, 18, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.R.; Noetzel, M.J.; Melancon, B.J.; Poslusney, M.S.; Nance, K.D.; Hurtado, M.A.; Luscombe, V.B.; Weiner, R.L.; Rodriguez, A.L.; Lamsal, A. Discovery of VU0467485/AZ13713945: An M4 PAM evaluated as a preclinical candidate for the treatment of schizophrenia. ACS Med. Chem. Lett. 2016, 8, 233–238. [Google Scholar] [CrossRef]
- Thal, D.M.; Sun, B.; Feng, D.; Nawaratne, V.; Leach, K.; Felder, C.C.; Bures, M.G.; Evans, D.A.; Weis, W.I.; Bachhawat, P.J.N. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 2016, 531, 335–340. [Google Scholar] [CrossRef]
- Rizzi, L.; Roriz-Cruz, M. Sirtuin 1 and Alzheimer’s disease: An up-to-date review. Neuropeptides 2018, 71, 54–60. [Google Scholar] [CrossRef]
- Azam, S.; Jakaria, M.; Kim, I.S.; Kim, J.; Haque, M.E.; Choi, D.K. Regulation of Toll-Like Receptor (TLR) Signaling Pathway by Polyphenols in the Treatment of Age-Linked Neurodegenerative Diseases: Focus on TLR4 Signaling. Front. Immunol. 2019, 10, 1000. [Google Scholar] [CrossRef]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Cilostazol suppresses beta-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-beta. J. Neurosci. Res. 2014, 92, 1581–1590. [Google Scholar] [CrossRef]
- Min, S.-W.; Sohn, P.D.; Li, Y.; Devidze, N.; Johnson, J.R.; Krogan, N.J.; Masliah, E.; Mok, S.-A.; Gestwicki, J.E.; Gan, L. SIRT1 deacetylates tau and reduces pathogenic tau spread in a mouse model of tauopathy. J. Neurosci. 2018, 38, 3680–3688. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Z.-X.; Zheng, L.-P.; Wang, L.; Liu, Y.-F.; Yin, W.-Y.; Chen, Y.-Y.; Wang, X.-S.; Hou, S.-T.; Chen, J.-F. The adenosine A2A receptor antagonist SCH58261 reduces macrophage/microglia activation and protects against experimental autoimmune encephalomyelitis in mice. Neurochem. Int. 2019, 129, 104490. [Google Scholar] [CrossRef]
- Li, C.; Wu, X.; Liu, S.; Zhao, Y.; Zhu, J.; Liu, K. Roles of Neuropeptide Y in Neurodegenerative and Neuroimmune Diseases. Front. Mol. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Santos-Carvalho, A.; Elvas, F.; Álvaro, A.R.; Ambrósio, A.F.; Cavadas, C. Neuropeptide Y receptors activation protects rat retinal neural cells against necrotic and apoptotic cell death induced by glutamate. Cell Death Dis. 2013, 4, e636. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Potkar, R.; Metcalf, J.; Thrin, I.; Adame, A.; Rockenstein, E.; Masliah, E.J. Systemic central nervous system (CNS)-targeted delivery of neuropeptide Y (NPY) reduces neurodegeneration and increases neural precursor cell proliferation in a mouse model of Alzheimer disease. J. Biol. Chem. 2016, 291, 1905–1920. [Google Scholar] [CrossRef] [PubMed]
- Thiriet, N.; Agasse, F.; Nicoleau, C.; Guégan, C.; Vallette, F.; Cadet, J.L.; Jaber, M.; Malva, J.O.; Coronas, V.J. NPY promotes chemokinesis and neurogenesis in the rat subventricular zone. J. Neurochem. 2011, 116, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Pain, S.; Vergote, J.; Gulhan, Z.; Bodard, S.; Chalon, S.; Gaillard, A. Inflammatory process in Parkinson disease: Neuroprotection by Neuropeptide Y. Fundam. Clin. Pharmacol. 2019, 33, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Carniglia, L.; Ramírez, D.; Durand, D.; Saba, J.; Turati, J.; Caruso, C.; Scimonelli, T.N.; Lasaga, M. Neuropeptides and Microglial Activation in Inflammation, Pain, and Neurodegenerative Diseases. J. Mediat. Inflamm. 2017, 2017, 23. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.G. Neurodevelopmental actions of leptin. Brain Res. 2010, 1350, 2–9. [Google Scholar] [CrossRef]
- Jakaria, M.; Haque, M.E.; Cho, D.Y.; Azam, S.; Kim, I.S.; Choi, D.K. Molecular Insights into NR4A2(Nurr1): An Emerging Target for Neuroprotective Therapy Against Neuroinflammation and Neuronal Cell Death. Mol. Neurobiol. 2019, 56, 5799–5814. [Google Scholar] [CrossRef]
- Uddin, M.S.; Al Mamun, A.; Kabir, M.T.; Jakaria, M.; Mathew, B.; Barreto, G.E.; Ashraf, G.M. Nootropic and anti-Alzheimer’s actions of medicinal plants: Molecular insight into therapeutic potential to alleviate Alzheimer’s neuropathology. Mol. Neurobiol. 2019, 56, 4925–4944. [Google Scholar] [CrossRef]
- Haque, M.E.; Akther, M.; Jakaria, M.; Kim, I.S.; Azam, S.; Choi, D.K. Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov. Disord. 2019. [Google Scholar] [CrossRef]
- Jakaria, M.; Park, S.Y.; Haque, M.E.; Karthivashan, G.; Kim, I.S.; Ganesan, P.; Choi, D.K. Neurotoxic Agent-Induced Injury in Neurodegenerative Disease Model: Focus on Involvement of Glutamate Receptors. Front. Mol. Neurosci. 2018, 11, 307. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Agents | Targets | Receptor Family | Indications |
|---|---|---|---|
| Erenumab | CALCRL | Calcitonin | Migraine |
| Ubrogepant | CALCRL | Calcitonin | Migraine |
| Eptinezumab | CGRP | Calcitonin | Migraine |
| Cannabidivarin | GPR119 | GPR119 | MS and epilepsy |
| Cannabidiol | GPR55 | GPR55 | MS and epilepsy |
| VSN16R | GPR18 | GPR18 | MS and epilepsy |
| Δ-9-Tetrahydrocannabinol-cannabidiol (THC-CBD) | CB1/2 receptor | Cannabinoid | Spasticity in MS |
| Fingolimod | Sphingosine 1-phosphate (S1P) | Sphingosine 1-phosphate (S1P) receptor | MS |
| Xanomeline | M1/M4 | Cholinergic | AD |
| AF267B | M1 and M3 | mAChR | Reduces amyloid and tau pathologies in AD |
| Leuprolide | AChEI | Cholinergic | Synergizes AChEI activities |
| Vortioxetine | 5-HT3, 5-HT7, 5-HT1D | Serotonin | Major Depressive Disorder |
| Haloperidol | D2R | Dopamine | Working memory in PD |
| SK609 | D3R | Dopamine | Locomotor activity in PD |
| VCE-003.2 | CB2R | Cannabinoid | HD |
| DMXBA (GTS-21) and ABT-107 | α7nAChR | Cholinergic | PD |
| PHA 543613 | α7nAChR | Cholinergic | Early-stage HD |
| LY341495 | Group I/II mGluRs | mGluRs | Improves synaptic plasticity in AD |
| Galantamine | AChEI | Cholinergic | VaD, AD, PD, and Lewy bodies with dementia |
| Rivastigmine | AChEI | Cholinergic | VaD, AD, PD, and Lewy bodies with dementia |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azam, S.; Haque, M.E.; Jakaria, M.; Jo, S.-H.; Kim, I.-S.; Choi, D.-K. G-Protein-Coupled Receptors in CNS: A Potential Therapeutic Target for Intervention in Neurodegenerative Disorders and Associated Cognitive Deficits. Cells 2020, 9, 506. https://doi.org/10.3390/cells9020506
Azam S, Haque ME, Jakaria M, Jo S-H, Kim I-S, Choi D-K. G-Protein-Coupled Receptors in CNS: A Potential Therapeutic Target for Intervention in Neurodegenerative Disorders and Associated Cognitive Deficits. Cells. 2020; 9(2):506. https://doi.org/10.3390/cells9020506
Chicago/Turabian StyleAzam, Shofiul, Md. Ezazul Haque, Md. Jakaria, Song-Hee Jo, In-Su Kim, and Dong-Kug Choi. 2020. "G-Protein-Coupled Receptors in CNS: A Potential Therapeutic Target for Intervention in Neurodegenerative Disorders and Associated Cognitive Deficits" Cells 9, no. 2: 506. https://doi.org/10.3390/cells9020506
APA StyleAzam, S., Haque, M. E., Jakaria, M., Jo, S.-H., Kim, I.-S., & Choi, D.-K. (2020). G-Protein-Coupled Receptors in CNS: A Potential Therapeutic Target for Intervention in Neurodegenerative Disorders and Associated Cognitive Deficits. Cells, 9(2), 506. https://doi.org/10.3390/cells9020506