Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer




Abstract
1. Introduction
2. Immunotherapy Against Cancer
2.1. Cancer Vaccines
2.2. Monoclonal Antibodies
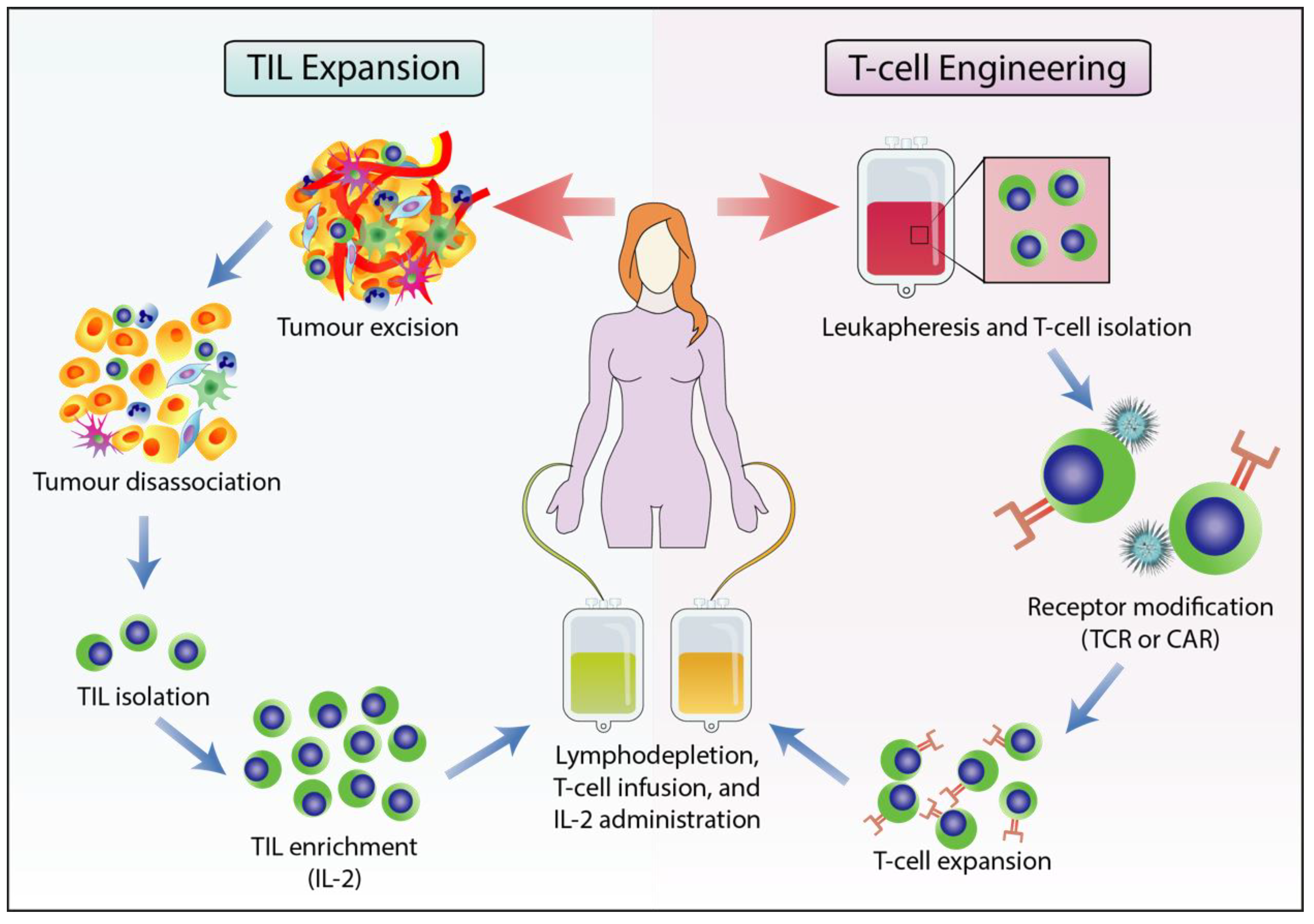
2.3. Adoptive T cell Therapy
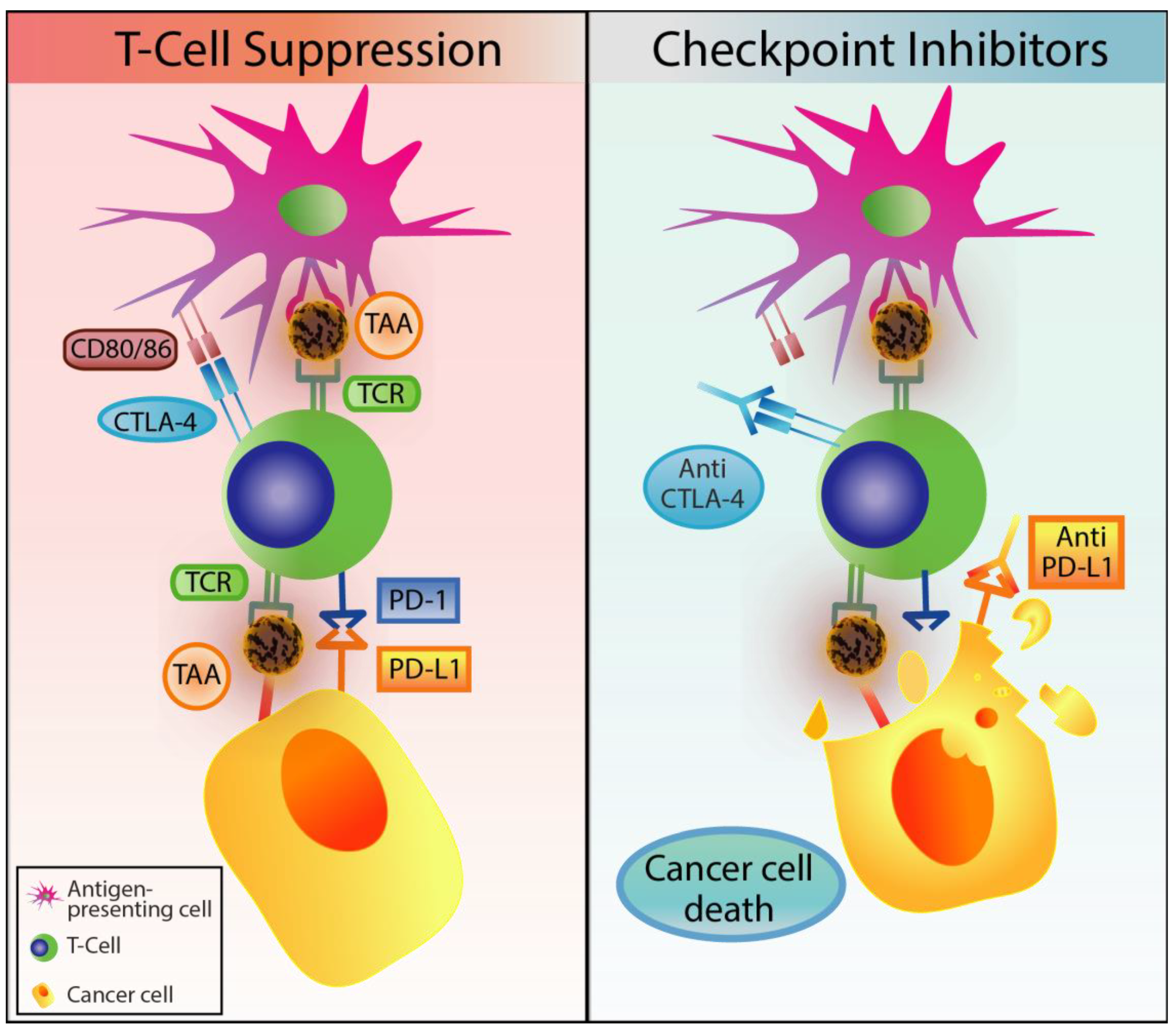
2.4. Immune Checkpoint Inhibitors
3. Classification and Function of Myeloid-Derived Suppressor Cells
3.1. Classification of MDSC
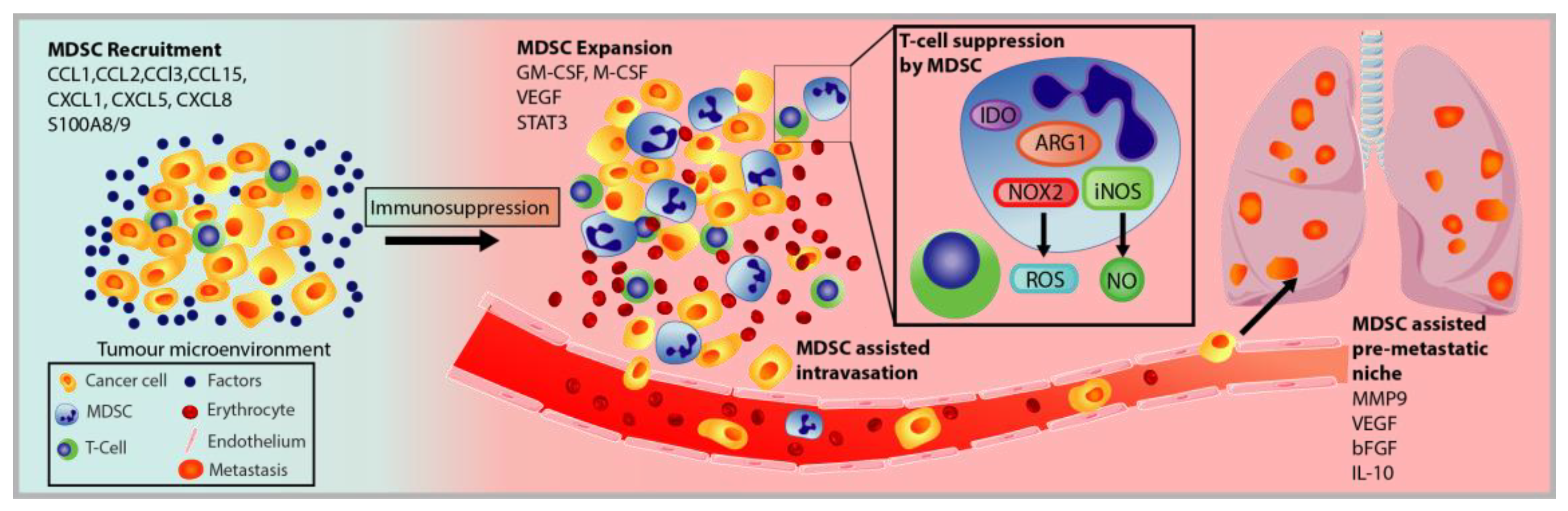
3.2. MDSC Recruitment and Pro-Tumorigenic Activation
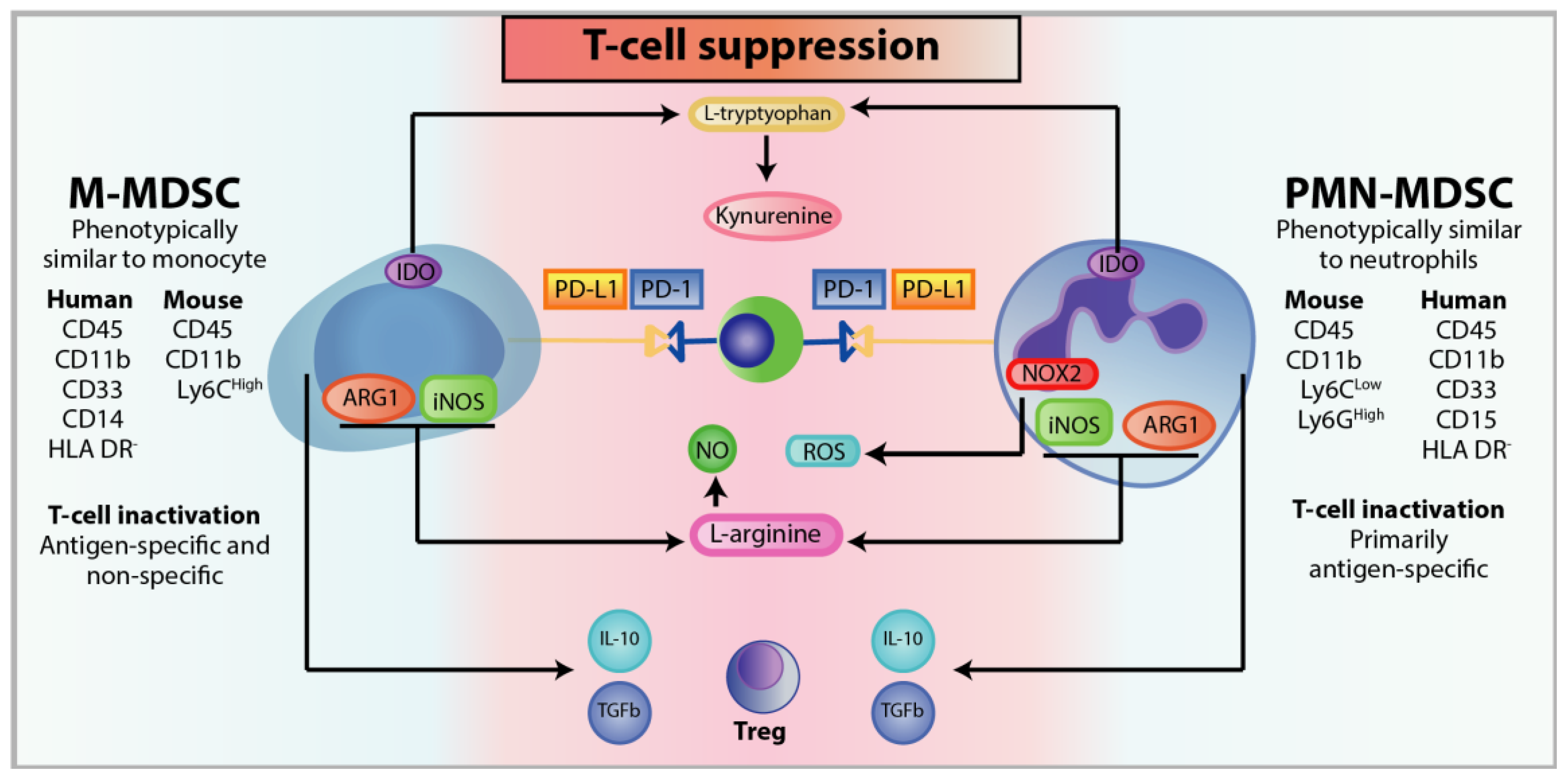
3.3. Immunosuppression of MDSC
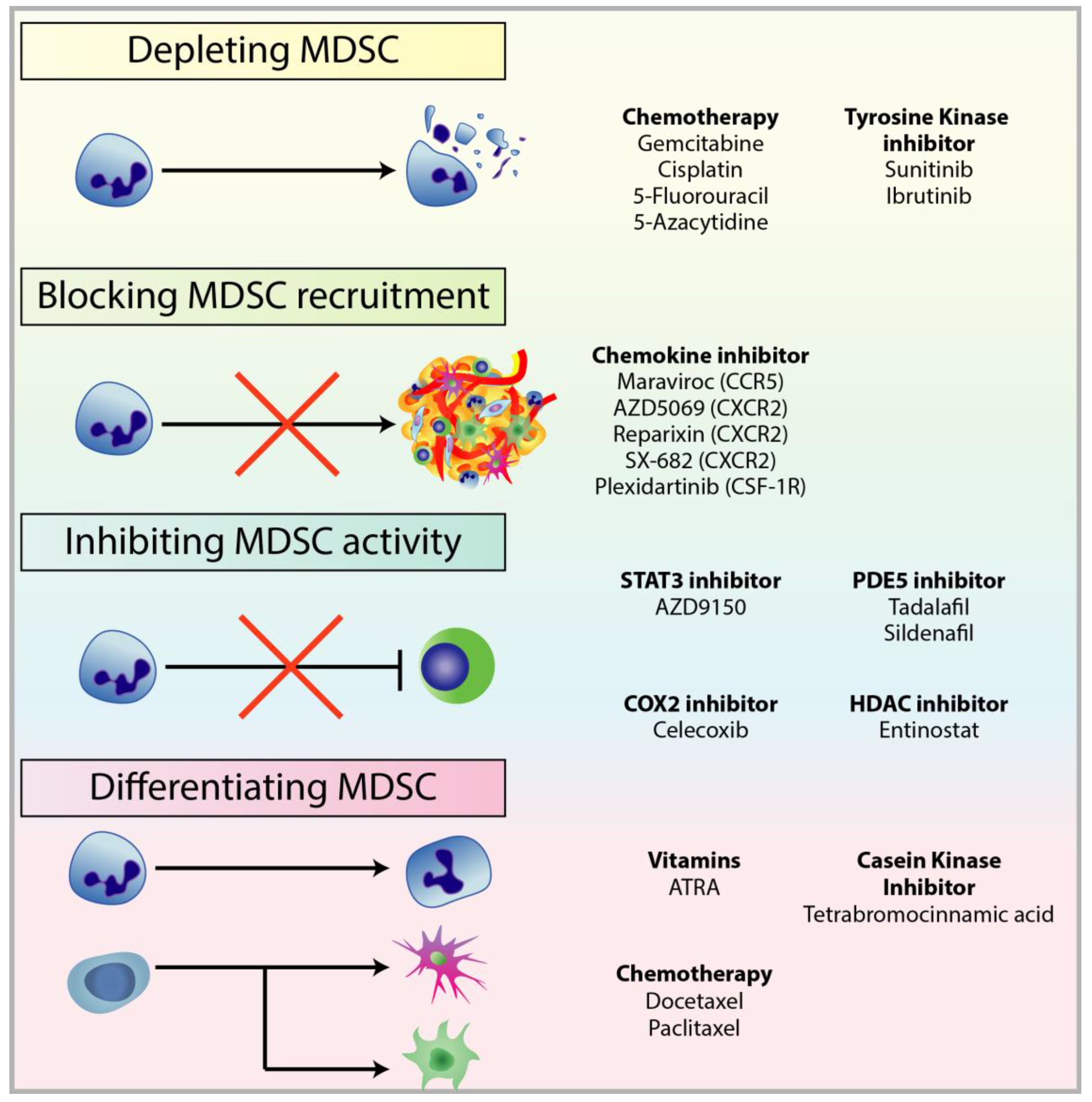
4. Targeting MDSCs in Cancer
4.1. Depleting MDSC Populations
4.2. Blockade of MDSC Migration
4.3. Attenuating MDSC Immunosuppressive Functions
4.4. Inducing MDSC Differentiation
5. Combining MDSC-Targeted Treatments with Immunotherapy
5.1. Checkpoint Inhibitors Combined with MDSC Depletion
5.2. Immunotherapy Combined with Obstructing MDSC Trafficking Therapy
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC class I antigens, immune surveillance, and tumor immune escape. J. Cell Physiol. 2003, 195, 346–355. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef]
- Burkholder, B. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta 2014, 1845, 182–201. [Google Scholar] [CrossRef]
- Salgado, R. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef]
- Chow, M.T.; Moller, A.; Smyth, M.J. Inflammation and immune surveillance in cancer. Semin Cancer Biol. 2012, 22, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Mora, F. Single-Cell Transcriptomics in Cancer Immunobiology: The Future of Precision Oncology. Front. Immunol. 2018, 9, 2582. [Google Scholar] [CrossRef] [PubMed]
- Lollini, P.L. The Promise of Preventive Cancer Vaccines. Vaccines (Basel) 2015, 3, 467–489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef]
- Lim, S.G. Prevention of hepatocellular carcinoma in hepatitis B virus infection. J. Gastroenterol. Hepatol. 2009, 24, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, A.; Monk, B.J. Vaccines against human papillomavirus and cervical cancer: Promises and challenges. Oncologist 2005, 10, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, N. Human Tumor Antigens and Cancer Immunotherapy. Biomed. Res. Int. 2015, 2015, 948501. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, S.; Pritchard, A.L. Identifying neoantigens for use in immunotherapy. Mamm. Genome 2018, 29, 714–730. [Google Scholar] [CrossRef] [PubMed]
- Vermaelen, K. Vaccine Strategies to Improve Anti-cancer Cellular Immune Responses. Front. Immunol. 2019, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Mougel, A.; Terme, M.; Tanchot, C. Therapeutic Cancer Vaccine and Combinations With Antiangiogenic Therapies and Immune Checkpoint Blockade. Front. Immunol. 2019, 10, 467. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Karlitepe, A.; Ozalp, O.; Avci, C.B. New approaches for cancer immunotherapy. Tumour Biol. 2015, 36, 4075–4078. [Google Scholar] [CrossRef]
- Dahlen, E.; Veitonmaki, N.; Norlen, P. Bispecific antibodies in cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2018, 6, 3–17. [Google Scholar] [CrossRef]
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar]
- McKeage, K.; Perry, C.M. Trastuzumab: A review of its use in the treatment of metastatic breast cancer overexpressing HER2. Drugs 2002, 62, 209–243. [Google Scholar] [CrossRef]
- Met, O. Principles of adoptive T cell therapy in cancer. Semin Immunopathol. 2019, 41, 49–58. [Google Scholar] [CrossRef]
- Dudley, M.E. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Besser, M.J. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: Intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin. Cancer Res. 2013, 19, 4792–4800. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J. Expansion of tumor-infiltrating lymphocytes and their potential for application as adoptive cell transfer therapy in human breast cancer. Oncotarget 2017, 8, 113345–113359. [Google Scholar] [CrossRef]
- Houot, R. T-cell-based Immunotherapy: Adoptive Cell Transfer and Checkpoint Inhibition. Cancer Immunol. Res. 2015, 3, 1115–1122. [Google Scholar] [CrossRef]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J. Adoptive cellular therapies: The current landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef]
- June, C.H.; Riddell, S.R.; Schumacher, T.N. Adoptive cellular therapy: A race to the finish line. Sci. Transl. Med. 2015, 7, 280ps7. [Google Scholar] [CrossRef]
- Maude, S.L.; Shpall, E.J.; Grupp, S.A. Chimeric antigen receptor T-cell therapy for ALL. Hematol. Am. Soc. Hematol. Educ. Program. 2014, 2014, 559–564. [Google Scholar] [CrossRef]
- Pehlivan, K.C.; Duncan, B.B.; Lee, D.W. CAR-T Cell Therapy for Acute Lymphoblastic Leukemia: Transforming the Treatment of Relapsed and Refractory Disease. Curr. Hematol. Malig. Rep. 2018, 13, 396–406. [Google Scholar] [CrossRef]
- Gattinoni, L. Adoptive T cell transfer: Imagining the next generation of cancer immunotherapies. Semin. Immunol. 2016, 28, 1–2. [Google Scholar] [CrossRef]
- Li, J. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef]
- Chu, F.; Cao, J.; Neelalpu, S.S. Versatile CAR T-cells for cancer immunotherapy. Contemp. Oncol. (Pozn) 2018, 22, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S. The 2018 Nobel Prize in medicine goes to cancer immunotherapy (editorial for BMC cancer). BMC Cancer 2018, 18, 1086. [Google Scholar] [CrossRef] [PubMed]
- Sambi, M.; Bagheri, L.; Szewczuk, M.R. Current Challenges in Cancer Immunotherapy: Multimodal Approaches to Improve Efficacy and Patient Response Rates. J. Oncol. 2019, 2019, 4508794. [Google Scholar] [CrossRef]
- Escors, D. Tumour immunogenicity, antigen presentation and immunological barriers in cancer immunotherapy. New J. Sci. 2014, 2014. [Google Scholar] [CrossRef]
- Lipson, E.J. Antagonists of PD-1 and PD-L1 in Cancer Treatment. Semin. Oncol. 2015, 42, 587–600. [Google Scholar] [CrossRef]
- Nanda, R. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Rugo, H.S. Safety and Antitumor Activity of Pembrolizumab in Patients with Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 2804–2811. [Google Scholar] [CrossRef]
- Yang, L. Recognizing and managing on toxicities in cancer immunotherapy. Tumour Biol. 2017, 39, 1010428317694542. [Google Scholar] [CrossRef] [PubMed]
- Nikanjam, M.; Patel, H.; Kurzrock, R. Dosing immunotherapy combinations: Analysis of 3,526 patients for toxicity and response patterns. Oncoimmunology 2017, 6, e1338997. [Google Scholar] [CrossRef] [PubMed]
- Hendry, S.A. The Role of the Tumor Vasculature in the Host Immune Response: Implications for Therapeutic Strategies Targeting the Tumor Microenvironment. Front. Immunol. 2016, 7, 621. [Google Scholar] [CrossRef] [PubMed]
- Tuccitto, A. Immunosuppressive circuits in tumor microenvironment and their influence on cancer treatment efficacy. Virchows Arch. 2019, 474, 407–420. [Google Scholar] [CrossRef]
- Yang, L. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef]
- Motz, G.T. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Disis, M.L. Immune regulation of cancer. J. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef]
- Frydrychowicz, M. The Dual Role of Treg in Cancer. Scand. J. Immunol. 2017, 86, 436–443. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Nishikawa, H. Roles of regulatory T cells in cancer immunity. Int. Immunol. 2016, 28, 401–409. [Google Scholar] [CrossRef]
- Weber, R. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Boettcher, S.; Manz, M.G. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood 2012, 119, 2991–3002. [Google Scholar] [CrossRef] [PubMed]
- de Haas, N. Improving cancer immunotherapy by targeting the STATe of MDSCs. Oncoimmunology 2016, 5, e1196312. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Umansky, V. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines (Basel) 2016, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Meirow, Y.; Kanterman, J.; Baniyash, M. Paving the Road to Tumor Development and Spreading: Myeloid-Derived Suppressor Cells are Ruling the Fate. Front. Immunol. 2015, 6, 523. [Google Scholar] [CrossRef]
- Sawanobori, Y. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood 2008, 111, 5457–5466. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Invest. 2007, 117, 1155–1166. [Google Scholar] [CrossRef]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.H. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010, 70, 6139–6149. [Google Scholar] [CrossRef] [PubMed]
- Gao, D. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 2012, 72, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Sceneay, J. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012, 72, 3906–3911. [Google Scholar] [CrossRef]
- Zhang, S. The Role of Myeloid-Derived Suppressor Cells in Patients with Solid Tumors: A Meta-Analysis. PLoS ONE 2016, 11, e0164514. [Google Scholar] [CrossRef]
- Law, A.M. The innate and adaptive infiltrating immune systems as targets for breast cancer immunotherapy. Endocr. Relat. Cancer 2017, 24, R123–R144. [Google Scholar] [CrossRef]
- Solito, S. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N Y Acad. Sci. 2014, 1319, 47–65. [Google Scholar] [CrossRef]
- Vetsika, E.K. A circulating subpopulation of monocytic myeloid-derived suppressor cells as an independent prognostic/predictive factor in untreated non-small lung cancer patients. J. Immunol. Res. 2014, 2014, 659294. [Google Scholar] [CrossRef]
- Hoechst, B. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef]
- Obermajer, N. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F. S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology 2012, 136, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Trikha, P.; Carson, W.E., 3rd. Signaling pathways involved in MDSC regulation. Biochim. Biophys. Acta 2014, 1846, 55–65. [Google Scholar] [PubMed]
- Lesokhin, A.M. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef]
- Liang, H. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 2017, 8, 1736. [Google Scholar] [CrossRef] [PubMed]
- Blattner, C. CCR5(+) Myeloid-Derived Suppressor Cells Are Enriched and Activated in Melanoma Lesions. Cancer Res. 2018, 78, 157–167. [Google Scholar] [CrossRef]
- Umansky, V. CCR5 in recruitment and activation of myeloid-derived suppressor cells in melanoma. Cancer Immunol. Immunother. 2017, 66, 1015–1023. [Google Scholar] [CrossRef]
- Metelitsa, L.S. Anti-tumor potential of type-I NKT cells against CD1d-positive and CD1d-negative tumors in humans. Clin. Immunol. 2011, 140, 119–129. [Google Scholar] [CrossRef]
- Youn, J.I.; Gabrilovich, D.I. The biology of myeloid-derived suppressor cells: The blessing and the curse of morphological and functional heterogeneity. Eur. J. Immunol. 2010, 40, 2969–2975. [Google Scholar] [CrossRef]
- Youn, J.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Karakasheva, T.A. CD38-Expressing Myeloid-Derived Suppressor Cells Promote Tumor Growth in a Murine Model of Esophageal Cancer. Cancer Res. 2015, 75, 4074–4085. [Google Scholar] [CrossRef] [PubMed]
- Ryzhov, S. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J. Immunol. 2011, 187, 6120–6129. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V. Extracellular adenosine metabolism in immune cells in melanoma. Cancer Immunol. Immunother. 2014, 63, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Functional characterization of myeloid-derived suppressor cell subpopulations during the development of experimental arthritis. Eur. J. Immunol. 2015, 45, 464–473. [Google Scholar] [CrossRef]
- Ding, Y. CD40 controls CXCR5-induced recruitment of myeloid-derived suppressor cells to gastric cancer. Oncotarget 2015, 6, 38901–38911. [Google Scholar] [CrossRef]
- Pan, P.Y. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010, 70, 99–108. [Google Scholar] [CrossRef]
- Shen, J. Downregulation of CD40 expression contributes to the accumulation of myeloid-derived suppressor cells in gastric tumors. Oncol Lett. 2014, 8, 775–780. [Google Scholar] [CrossRef]
- Cho, W.K. Immunomodulatory effect of captopril and local irradiation on myeloid-derived suppressor cells. Radiat. Oncol. J. 2016, 34, 223–229. [Google Scholar] [CrossRef]
- Gallego-Ortega, D. ELF5 Drives Lung Metastasis in Luminal Breast Cancer through Recruitment of Gr1+ CD11b+ Myeloid-Derived Suppressor Cells. PLoS Biol. 2015, 13, e1002330. [Google Scholar] [CrossRef]
- Haile, L.A. CD49d is a new marker for distinct myeloid-derived suppressor cell subpopulations in mice. J. Immunol. 2010, 185, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: More mechanisms for inhibiting antitumor immunity. Cancer Immunol. Immunother. 2010, 59, 1593–1600. [Google Scholar] [CrossRef] [PubMed]
- Lacotte, S. Impact of myeloid-derived suppressor cell on Kupffer cells from mouse livers with hepatocellular carcinoma. Oncoimmunology 2016, 5, e1234565. [Google Scholar] [CrossRef] [PubMed]
- Marigo, I. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010, 32, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Liechtenstein, T. A highly efficient tumor-infiltrating MDSC differentiation system for discovery of anti-neoplastic targets, which circumvents the need for tumor establishment in mice. Oncotarget 2014, 5, 7843–7857. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.Y.; Wilkes, S.; Yang, H. CD71 is selectively and ubiquitously expressed at high levels in erythroid precursors of all maturation stages: A comparative immunochemical study with glycophorin A and hemoglobin A. Am. J. Surg. Pathol. 2011, 35, 723–732. [Google Scholar] [CrossRef]
- Movahedi, K. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef]
- Bunt, S.K. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J. Leukoc. Biol. 2009, 85, 996–1004. [Google Scholar] [CrossRef]
- Sansom, D.M. CD28, CTLA-4 and their ligands: Who does what and to whom? Immunology 2000, 101, 169–177. [Google Scholar] [CrossRef]
- Yang, R. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res. 2006, 66, 6807–6815. [Google Scholar] [CrossRef]
- Yang, Y. LPS converts Gr-1(+)CD115(+) myeloid-derived suppressor cells from M2 to M1 via P38 MAPK. Exp. Cell Res. 2013, 319, 1774–1783. [Google Scholar] [CrossRef]
- Youn, J.I. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 2012, 91, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Albeituni, S.H.; Ding, C.; Yan, J. Hampering immune suppressors: Therapeutic targeting of myeloid-derived suppressor cells in cancer. Cancer J. 2013, 19, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Tang, R. M-CSF (monocyte colony stimulating factor) and M-CSF receptor expression by breast tumour cells: M-CSF mediated recruitment of tumour infiltrating monocytes? J. Cell Biochem. 1992, 50, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Hu, X. Transmembrane TNF-alpha promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J. Immunol. 2014, 192, 1320–1331. [Google Scholar] [CrossRef]
- Mandruzzato, S. IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J. Immunol. 2009, 182, 6562–6568. [Google Scholar] [CrossRef]
- Sinha, P. Tumor-induced myeloid-derived suppressor cell function is independent of IFN-gamma and IL-4Ralpha. Eur. J. Immunol. 2012, 42, 2052–2059. [Google Scholar] [CrossRef]
- Gallina, G. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J. Clin. Invest. 2006, 116, 2777–2790. [Google Scholar] [CrossRef]
- Roth, F. Aptamer-mediated blockade of IL4Ralpha triggers apoptosis of MDSCs and limits tumor progression. Cancer Res. 2012, 72, 1373–1383. [Google Scholar] [CrossRef]
- Schouppe, E. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur. J. Immunol. 2013, 43, 2930–2942. [Google Scholar] [CrossRef]
- Goldmann, O.; Beineke, A.; Medina, E. Identification of a Novel Subset of Myeloid-Derived Suppressor Cells During Chronic Staphylococcal Infection That Resembles Immature Eosinophils. J. Infect. Dis. 2017, 216, 1444–1451. [Google Scholar] [CrossRef]
- Georgoudaki, A.M. CD244 is expressed on dendritic cells and regulates their functions. Immunol. Cell Biol. 2015, 93, 581–590. [Google Scholar] [CrossRef]
- Lu, C. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1247135. [Google Scholar] [CrossRef]
- Lee, C.R. Myeloid-Derived Suppressor Cells Are Controlled by Regulatory T Cells via TGF-beta during Murine Colitis. Cell Rep. 2016, 17, 3219–3232. [Google Scholar] [CrossRef]
- Chiu, D.K. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.M. Phenotypic and functional delineation of murine CX(3)CR1 monocyte-derived cells in ovarian cancer. Neoplasia 2009, 11, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W. Subsets of myeloid-derived suppressor cells in hepatocellular carcinoma express chemokines and chemokine receptors differentially. Int. Immunopharmacol. 2015, 26, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013, 24, 631–644. [Google Scholar] [CrossRef]
- O’Connor, M.A. Subpopulations of M-MDSCs from mice infected by an immunodeficiency-causing retrovirus and their differential suppression of T- vs B-cell responses. Virology 2015, 485, 263–273. [Google Scholar] [CrossRef]
- Ortiz, M.L. Myeloid-derived suppressor cells in the development of lung cancer. Cancer Immunol. Res. 2014, 2, 50–58. [Google Scholar] [CrossRef]
- Wang, T. Galectin-3 contributes to cisplatin-induced myeloid derived suppressor cells (MDSCs) recruitment in Lewis lung cancer-bearing mice. Mol. Biol. Rep. 2014, 41, 4069–4076. [Google Scholar] [CrossRef] [PubMed]
- Pyzer, A.R. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int. J. Cancer 2016, 139, 1915–1926. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S. Antigen-specific CD4(+) T cells regulate function of myeloid-derived suppressor cells in cancer via retrograde MHC class II signaling. Cancer Res. 2012, 72, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y. Characterization of the myeloid-derived suppressor cell subset regulated by NK cells in malignant lymphoma. Oncoimmunology 2015, 4, e995541. [Google Scholar] [CrossRef]
- Zoglmeier, C. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin. Cancer Res. 2011, 17, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, N. Expression of Vascular Endothelial Growth Factor in Ovarian Cancer Inhibits Tumor Immunity through the Accumulation of Myeloid-Derived Suppressor Cells. Clin. Cancer Res. 2017, 23, 587–599. [Google Scholar] [CrossRef]
- Kumar, V. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Huang, B. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007, 252, 86–92. [Google Scholar] [CrossRef]
- Obermajer, N. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011, 71, 7463–7470. [Google Scholar] [CrossRef]
- Bronte, V. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Zhang, J. CD13(hi) Neutrophil-like myeloid-derived suppressor cells exert immune suppression through Arginase 1 expression in pancreatic ductal adenocarcinoma. Oncoimmunology 2017, 6, e1258504. [Google Scholar] [CrossRef] [PubMed]
- Resheq, Y.J. Impaired Transmigration of Myeloid-Derived Suppressor Cells across Human Sinusoidal Endothelium Is Associated with Decreased Expression of CD13. J. Immunol. 2017, 199, 1672–1681. [Google Scholar] [CrossRef] [PubMed]
- Haziot, A.; Tsuberi, B.Z.; Goyert, S.M. Neutrophil CD14: Biochemical properties and role in the secretion of tumor necrosis factor-alpha in response to lipopolysaccharide. J. Immunol. 1993, 150, 5556–5565. [Google Scholar] [PubMed]
- Damuzzo, V. Complexity and challenges in defining myeloid-derived suppressor cells. Cytometry B Clin. Cytom. 2015, 88, 77–91. [Google Scholar] [CrossRef]
- Romano, A. Circulating myeloid-derived suppressor cells correlate with clinical outcome in Hodgkin Lymphoma patients treated up-front with a risk-adapted strategy. Br. J. Haematol. 2015, 168, 689–700. [Google Scholar] [CrossRef]
- Karakasheva, T.A. CD38+ M-MDSC expansion characterizes a subset of advanced colorectal cancer patients. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Li, L. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef]
- Li, J. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef]
- Rodriguez, P.C. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009, 69, 1553–1560. [Google Scholar] [CrossRef]
- Shao, X. Distinct alterations of CD68(+)CD163(+) M2-like macrophages and myeloid-derived suppressor cells in newly diagnosed primary immune thrombocytopenia with or without CR after high-dose dexamethasone treatment. J. Transl. Med. 2018, 16, 48. [Google Scholar] [CrossRef] [PubMed]
- Bergenfelz, C. Systemic Monocytic-MDSCs Are Generated from Monocytes and Correlate with Disease Progression in Breast Cancer Patients. PLoS ONE 2015, 10, e0127028. [Google Scholar] [CrossRef] [PubMed]
- Greten, T.F.; Manns, M.P.; Korangy, F. Myeloid derived suppressor cells in human diseases. Int. Immunopharmacol. 2011, 11, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Poschke, I. Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010, 70, 4335–4345. [Google Scholar] [CrossRef]
- Ju, X. The Analysis of CD83 Expression on Human Immune Cells Identifies a Unique CD83+-Activated T Cell Population. J. Immunol. 2016, 197, 4613–4625. [Google Scholar] [CrossRef]
- Zhang, B. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS ONE 2013, 8, e57114. [Google Scholar] [CrossRef]
- OuYang, L.Y. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J. Transl. Med. 2015, 13, 47. [Google Scholar] [CrossRef]
- Kohanbash, G. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res. 2013, 73, 6413–6423. [Google Scholar] [CrossRef]
- Seo, E.H. Association of Chemokines and Chemokine Receptor Expression with Monocytic-Myeloid-Derived Suppressor Cells during Tumor Progression. Immune Netw 2018, 18, e23. [Google Scholar] [CrossRef]
- Highfill, S.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl Med. 2014, 6, 237ra67. [Google Scholar] [CrossRef]
- Kusmartsev, S. Oxidative stress regulates expression of VEGFR1 in myeloid cells: Link to tumor-induced immune suppression in renal cell carcinoma. J. Immunol. 2008, 181, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Yu, J. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef]
- Qian, B.Z. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Chun, E. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015, 12, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Reichel, C.A. C-C motif chemokine CCL3 and canonical neutrophil attractants promote neutrophil extravasation through common and distinct mechanisms. Blood 2012, 120, 880–890. [Google Scholar] [CrossRef]
- Condamine, T. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [CrossRef]
- Steele, C.W. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef]
- Nefedova, Y. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J. Immunol. 2004, 172, 464–474. [Google Scholar] [CrossRef]
- Arai, K. S100A8 and S100A9 overexpression is associated with poor pathological parameters in invasive ductal carcinoma of the breast. Curr. Cancer Drug Targets 2008, 8, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J. Immunol. 2013, 190, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef]
- Corzo, C.A. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Liu, G. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res. 2014, 74, 727–737. [Google Scholar] [CrossRef]
- Noman, M.Z. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Huang, B. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef]
- Srivastava, M.K. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef]
- Fletcher, M. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 2015, 75, 275–283. [Google Scholar] [CrossRef]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Bingisser, R.M. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J. Immunol. 1998, 160, 5729–5734. [Google Scholar] [PubMed]
- Rivoltini, L. Immunity to cancer: Attack and escape in T lymphocyte-tumor cell interaction. Immunol. Rev. 2002, 188, 97–113. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed]
- Harari, O.; Liao, J.K. Inhibition of MHC II gene transcription by nitric oxide and antioxidants. Curr. Pharm. Des. 2004, 10, 893–898. [Google Scholar] [CrossRef]
- Nagaraj, S. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef]
- Lu, T. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J. Clin. Invest. 2011, 121, 4015–4029. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Gabrilovich, D.I. Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J. Leukoc. Biol. 2003, 74, 186–196. [Google Scholar] [CrossRef]
- Kusmartsev, S. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef]
- Molon, B. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef]
- Beury, D.W. Myeloid-Derived Suppressor Cell Survival and Function Are Regulated by the Transcription Factor Nrf2. J. Immunol. 2016, 196, 3470–3478. [Google Scholar] [CrossRef]
- Della Chiesa, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef] [PubMed]
- Song, H. L-kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int. Immunopharmacol. 2011, 11, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Fleming, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Fallarino, F. T cell apoptosis by kynurenines. Adv. Exp. Med. Biol. 2003, 527, 183–190. [Google Scholar]
- Curti, A. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877. [Google Scholar] [CrossRef]
- Cheng, P. S100A9-induced overexpression of PD-1/PD-L1 contributes to ineffective hematopoiesis in myelodysplastic syndromes. Leukemia 2019, 33, 2034–2046. [Google Scholar] [CrossRef]
- Hart, K.M. IL-10 immunomodulation of myeloid cells regulates a murine model of ovarian cancer. Front. Immunol. 2011, 2, 29. [Google Scholar] [CrossRef]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Invest. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Messmer, M.N. Tumor-induced myeloid dysfunction and its implications for cancer immunotherapy. Cancer Immunol. Immunother. 2015, 64, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dolcetti, L. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur. J. Immunol. 2010, 40, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, A.G. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol. Med. 2011, 17, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Maenhout, S.K. Enhanced suppressive capacity of tumor-infiltrating myeloid-derived suppressor cells compared with their peripheral counterparts. Int. J. Cancer 2014, 134, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur. J. Immunol. 2010, 40, 3317–3320. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014, 63, 247–257. [Google Scholar] [CrossRef]
- Tarhini, A.A. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS ONE 2014, 9, e87705. [Google Scholar] [CrossRef]
- Weber, J. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol. Res. 2016, 4, 345–353. [Google Scholar] [CrossRef]
- Groth, C. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef]
- Suzuki, E. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef]
- Sevko, A. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J. Immunol. 2013, 190, 2464–2471. [Google Scholar] [CrossRef]
- Eriksson, E. Gemcitabine reduces MDSCs, tregs and TGFbeta-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 282. [Google Scholar] [CrossRef]
- Vincent, J. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [PubMed]
- Mikyskova, R. Cyclophosphamide-induced myeloid-derived suppressor cell population is immunosuppressive but not identical to myeloid-derived suppressor cells induced by growing TC-1 tumors. J. Immunother. 2012, 35, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y. Contrasting effects of cyclophosphamide on anti-CTL-associated protein 4 blockade therapy in two mouse tumor models. Cancer Sci. 2017, 108, 1974–1984. [Google Scholar] [CrossRef]
- Ko, J.S. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef]
- Kodera, Y. Sunitinib inhibits lymphatic endothelial cell functions and lymph node metastasis in a breast cancer model through inhibition of vascular endothelial growth factor receptor 3. Breast Cancer Res. 2011, 13, R66. [Google Scholar] [CrossRef] [PubMed]
- Qin, H. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat. Med. 2014, 20, 676–681. [Google Scholar] [CrossRef]
- Robinson, S.C. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003, 63, 8360–8365. [Google Scholar]
- Velasco-Velazquez, M. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012, 72, 3839–3850. [Google Scholar] [CrossRef]
- Toh, B. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H. CXCR2(+) MDSCs promote breast cancer progression by inducing EMT and activated T cell exhaustion. Oncotarget 2017, 8, 114554–114567. [Google Scholar] [CrossRef] [PubMed]
- Di Mitri, D. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef]
- Sun, L. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Holmgaard, R.B. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine 2016, 6, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed]
- Richardsen, E. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. AntiCancer Res. 2015, 35, 865–874. [Google Scholar] [PubMed]
- Sluijter, M. Inhibition of CSF-1R supports T-cell mediated melanoma therapy. PLoS ONE 2014, 9, e104230. [Google Scholar] [CrossRef]
- Mok, S. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014, 74, 153–161. [Google Scholar] [CrossRef]
- Kumar, V. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668. [Google Scholar] [CrossRef]
- Elliott, L.A. Human Tumor-Infiltrating Myeloid Cells: Phenotypic and Functional Diversity. Front. Immunol. 2017, 8, 86. [Google Scholar] [CrossRef]
- Sinha, P. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef]
- Eruslanov, E. Pivotal Advance: Tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE(2) catabolism in myeloid cells. J. Leukoc. Biol. 2010, 88, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, A.C. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin. Cancer Res. 2007, 13, 721s–726s. [Google Scholar] [CrossRef] [PubMed]
- Veltman, J.D. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. EBioMedicine 2010, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Lin, S. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am. J. Cancer Res. 2017, 7, 41–52. [Google Scholar]
- Tai, L.H. Phosphodiesterase-5 inhibition reduces postoperative metastatic disease by targeting surgery-induced myeloid derived suppressor cell-dependent inhibition of Natural Killer cell cytotoxicity. Oncoimmunology 2018, 7, e1431082. [Google Scholar] [CrossRef]
- Califano, J.A. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 30–38. [Google Scholar] [CrossRef]
- Hassel, J.C. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology 2017, 6, e1326440. [Google Scholar] [PubMed]
- Weed, D.T. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef]
- Wang, Y.Y. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088. [Google Scholar] [PubMed]
- Hiramoto, K. Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev. Res. (Phila) 2014, 7, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin. Cancer Res. 2010, 16, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- De Santo, C. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc. Natl. Acad. Sci. USA 2005, 102, 4185–4190. [Google Scholar] [CrossRef]
- Reilley, M.J. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef]
- Kusmartsev, S. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2008, 14, 8270–8278. [Google Scholar] [CrossRef]
- Mirza, N. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006, 66, 9299–9307. [Google Scholar] [CrossRef]
- Iclozan, C. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol. Immunother. 2013, 62, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, Y. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007, 67, 11021–11028. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P. Effects of notch signaling on regulation of myeloid cell differentiation in cancer. Cancer Res. 2014, 74, 141–152. [Google Scholar] [CrossRef]
- Orillion, A. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef]
- Christmas, B.J. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577. [Google Scholar] [CrossRef] [PubMed]
- Mikyskova, R. DNA demethylating agent 5-azacytidine inhibits myeloid-derived suppressor cells induced by tumor growth and cyclophosphamide treatment. J. Leukoc. Biol. 2014, 95, 743–753. [Google Scholar] [CrossRef]
- Kodumudi, K.N. A novel chemoimmunomodulating property of docetaxel: Suppression of myeloid-derived suppressor cells in tumor bearers. Clin. Cancer Res. 2010, 16, 4583–4594. [Google Scholar] [CrossRef]
- Michels, T. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J. Immunotoxicol. 2012, 9, 292–300. [Google Scholar] [CrossRef]
- Kim, K. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Marker | M-MDSC | PMN-MDSC | Notes | References |
|---|---|---|---|---|
| CCR2 | +(high) | + | Involved in MDSC recruitment and expansion. Upregulated in MDSCs for multiple cancer types | [75,76] |
| CCR5 | + | + | Involved in MDSC expansion and activation. Upregulated in MDSCs in melanoma. | [77,78] |
| CD1b | + | − | High expression of CD1b used by NKT to target MDSC for anti-tumour immunity. | [79] |
| CD11b (Mac-1) | + | + | Transmembrane glycoprotein for leukocyte adhesion and migration. Commonly used in combination as CD11b+, Gr-1+, Ly6C+ or Ly6G+ for identifying MDSC. | [80] |
| CD11c | − | − | Marker used to differentiate dendritic cells | [81] |
| CD38 | + | + | Associated in early myeloid differentiation, activation, and migration. High expression may be associated with immature MDSC and stronger T-cell suppression | [82] |
| CD39 | + | + | Surface ectonucleotidase that is paired with CD73 and involved in the adenosine-pathway to inhibit T-cell and NK-cell activity. Upregulated in Lewis lung carcinoma and melanoma. | [83,84] |
| CD40 | + | + | Immune stimulatory receptor that suppresses T-cell activation, tumour specific T-reg expansion by MDSC, CXCR5-induced expansion of MDSC, and MDSC accumulation by facilitating apoptosis resistance. Upregulated in MDSC for collagen-induced arthritis, colitis, and gastric cancer | [85,86,87,88] |
| CD43 | Unknown | + | Involved in neutrophil recruitment. Upregulated in PMN-MDSC in mammary carcinoma model. | [89] |
| CD45 | + | + | Leukocyte common antigen used early in FACS gating to discriminate between tumour cells and immune cells. | [90] |
| CD49d (VLA4) | + | − | Specific marker for M-MDSC. CD49d+ MDSC were primarily monocytic and potent suppressors of antigen-specific T-cell responses. | [91] |
| CD54 (ICAM-1) | + (high) | + (low) | Immunostimulatory molecule that binds to CD11b. | [92,93] |
| CD62L (L-selectin) | + | + | Homing molecule that can be used to discriminate between DC and MDSC. | [94,95] |
| CD71 (transferrin receptor) | + | - | Marker for early erythroid precursors and proliferation. Upregulated in subcutaneous lymphoma model. | [96,97] |
| CD73 | + | + (high) | Surface ectonucleotidase that is paired with CD39 and involved in the adenosine-pathway. Inhibits T-cell and NK-cell activity and expansion of MDSC. Highly expressed in PMN-MDSC. Upregulated in Lewis lung carcinoma and melanoma. | [83,84] |
| CD80 (B7.1) | + (low) | +/− (low) | Ligand of CTLA-4 to inhibit T-cell activity. Upregulated in MDSC by chronic inflammation in subcutaneous lymphoma, breast, and ovarian cancer | [81,97,98,99,100] |
| CD86 | + | + | Ligand of CTLA-4 to inhibit T-cell activity. Upregulated in MDSC by chronic inflammation in breast cancer and collagen-induced arthritis. | [85,98,99] |
| CD98 | Unknown | + | Prognostic biomarker in different cancers and functions in cysteine transportation. May also be associated with prolonging lifespan of MDSC through mTOR signalling. Upregulated in PMN-MDSC in mammary carcinoma model. | [89,92] |
| CD115 (M-CSFR) | +/− | +/− | Recruits tumour-infiltrating monocytes. Upregulated in MDSC in multiple cancer types. | [101,102,103,104] |
| CD120b (TNFR2) | +(low) | +(low) | Involved in accumulation and activation of MDSC within the tumour. | [105] |
| CD124 (IL-4 receptor α) | +/− | +/− | May be implicated in T-cell suppression by MDSC and MDSC survival. Upregulated in MDSC in multiple cancer types. | [106,107,108,109] |
| CD162 (PSGL-1) | + | + | Affects T-cell adhesion and entry to sites of inflammation. | [110] |
| CD170 Syglec-F (eosinophil marker) | − | − | Eosinophilic marker used to identify new subset of Eo-MDSC in chronic Staphylococcus aureus infection. | [111] |
| CD244 | − | +/− | Cell surface receptor expressed on NK cells, DC cells and T-cells. Upregulated in MDSC in multiple cancer types. | [102,103,112] |
| CD279 (PD-L1) | + | + | Inhibitory ligand that suppresses T-cell activation. Upregulated in MDSC in colitis and multiple cancer types. | [81,113,114] |
| CX3CR1 | + | + | Involved in MDSC recruitment and expansion. Can be recruited by CCL26 that are secreted by hypoxic cancer cells. Expression levels can change based on tumour progression. | [115,116] |
| CXCR1 | + | + | Involved in MDSC recruitment and expansion. Upregulated in MDSC in multiple cancer types. | [56,117] |
| CXCR2 | + | + | Involved in MDSC recruitment and expansion. Upregulated in MDSC in multiple cancer types. | [56,117,118] |
| CXCR4 | + | + | Involved in MDSC recruitment and expansion. Upregulated in MDSC in multiple cancer types. | [119] |
| F4/80 | +/− | − | Marker used to differentiate macrophages and M-MDSC. | [97,120] |
| Gr-1 | + (low) | + (high) | Recognises epitope in both Ly6C and Ly6G | [80] |
| Ly6C | + (high) | + (low) | Differentiation antigen expressed in M-MDSC, macrophages, and dendritic cell precursors. | [81] |
| Ly6G | − | + (high) | Differentiation antigen expressed in PMN-MDSC, neutrophils, monocytes, and granulocytes. | [81] |
| Mac-2 (galectin-3) | + (high) | + (low) | Recruits MDSC via GM-CSF pathway and induces apoptosis in T-cell via TIM-3. | [121,122] |
| MHC Class I | + | + | Important role in antigen processing and presentation for the activation of adaptive immunity. Expressed in both subsets of MDSC. | [123] |
| MHC Class II | +/− (low) | +/− (low) | MHC Class II expression varies based on disease context and mouse model used. Usually low expression or similar to tumour-free mice. | [124,125] |
| Sca-1, Ly6A/E | + | + | Marker for hematopoietic stem cells. Expression can be highly variable. | [97,126] |
| VEGFR | + | + | Receptor for VEGF, which stimulates angiogenesis and recruits MDSC. MDSC-expressing VEGFR possesses stronger immunosuppressive activities compared to other MDSCs in ovarian cancer. | [127] |
| Human Marker | M-MDSC | PMN-MDSC | Notes | References |
|---|---|---|---|---|
| CCR2 | + (high) | + | Involved in MDSC recruitment and expansion. Upregulated in MDSC in multiple cancer types, such as breast, ovarian, gastric, and melanoma. | [128,129] |
| CXCR4 | + | + | Involved in MDSC recruitment and expansion. Upregulated in MDSC in ovarian cancer patients. | [130] |
| CD11b | + | + | Transmembrane glycoprotein for leukocyte adhesion and migration. Used as a myeloid marker similar to CD33. | [131] |
| CD13 | + (low) | + (high) | Myeloid marker involved in cell motility. | [132,133] |
| CD14 | + (high) | − | Differentiation antigen expressed in M-MDSC, macrophages, and dendritic cell precursors. | [134] |
| CD15 | − | + | Differentiation antigen expressed in PMN-MDSC, neutrophils, monocytes, and granulocytes | [81] |
| CD16 (FcyR) | + (high) | +/− (low) | Discriminating antigen to exclude PMN-MDSC. Can be used to separate immature MDSC (CD16−) from PMN-MDSC (CD16+) in whole blood. | [135] |
| CD33 | + (high) | + (low) | Myeloid marker that is more highly expressed in M-MDSC and dimly expressed in PMN-MDSC | [131] |
| CD34 | + (high) | + (low) | Marker for hematopoietic progenitor cells used to discriminate immature MDSC. | [70,123,136] |
| CD38 | + | + | Associated with poor prognosis. Advanced stages in cancer patients have been found to have expansion of CD38+ MDSC in head and neck, and colorectal cancer. | [82,137] |
| CD39 | + | + | Surface ectonucleotidase that is paired with CD73 and are involved in the adenosine-pathway. Inhibits T-cell and NK-cell activity and exerts tumour cell protection against chemotherapy; for example, rapamycin. Upregulated in ovarian cancer and NSCLC. | [138,139] |
| CD45 | + | + | Leukocyte common antigen used early in FACS gating to discriminate between tumour cells and immune cells. | [102,103,112] |
| CD62L (L-selectin) | + | + | Homing molecule involved in MDSC circulation. Lower expression in MDSC compared to neutrophils. Found in renal cell carcinoma patients. | [140] |
| CD66b | - | + | Differentiation marker expressed in PMN-MDSC. | [131] |
| CD68 | + | − | Macrophage specific marker used to discriminate between TAM and M-MDSC | [131,141] |
| CD73 | + | + | Surface ectonucleotidase that is paired with CD73 and is involved in the adenosine-pathway. Inhibits T-cell and NK-cell activity and exerts tumour cell protection against chemotherapy; for example, rapamycin. Upregulated in ovarian cancer and NSCLC. | [138,139,142] |
| CD80 | +/− | − | Activation marker and ligand of CTLA-4 to inhibit T-cell activity. Expression can vary/no expression. Upregulated in advanced melanoma patients and breast cancer patients. | [80,143,144] |
| CD83 | +/− | − | Marker used for mature dendritic cells. Can also be expressed in B and T lymphocytes. Has functions in immune cell activation and suppression | [143,144,145] |
| CD86 | +/− | − | Activation marker and ligand of CTLA-4 to inhibit T-cell activity. Upregulated in breast cancer patients. | [142,144] |
| CD115 (M-CSFR) | +/− | +/− | Recruits tumour-infiltrating monocytes. Found in MDSC subset similar to precursor myeloid cells. | [146] |
| CD117 (cKIT) | +/− | + | Granulocyte-monocyte progenitor marker. Upregulated in colorectal cancer. | [146,147] |
| CD124 (IL-4 receptor α) | + | + | May be implicated in T-cell suppression by MDSC and MDSC survival. Expression can greatly vary depending on disease type. | [106,135,146,148] |
| CD163 | + | − | Macrophage specific marker used to discriminate between TAM and M-MDSC | [131,141] |
| CXCR1 | + | + | Involved in MDSC recruitment and expansion. Upregulated in MDSC in multiple cancer types. | [149,150] |
| CXCR2 | + | + | Involved in MDSC recruitment and expansion. Upregulated in MDSC in multiple cancer types. | [149,150] |
| HLA-DR | − | − | Important role in antigen processing and presentation. | [67,81] |
| Lin | +/− (low) | +/− (low) | MDSC are generally negative or have very low expression for mature cell lineage markers. | [67] |
| VEGFR | + (low) | + (low) | Receptor for VEGF, which stimulates angiogenesis and recruit MDSC. Upregulated in patients with renal cell carcinoma. | [151] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. https://doi.org/10.3390/cells9030561
Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells. 2020; 9(3):561. https://doi.org/10.3390/cells9030561
Chicago/Turabian StyleLaw, Andrew M. K., Fatima Valdes-Mora, and David Gallego-Ortega. 2020. "Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer" Cells 9, no. 3: 561. https://doi.org/10.3390/cells9030561
APA StyleLaw, A. M. K., Valdes-Mora, F., & Gallego-Ortega, D. (2020). Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells, 9(3), 561. https://doi.org/10.3390/cells9030561