Transient Receptor Potential Canonical 5-Scramblase Signaling Complex Mediates Neuronal Phosphatidylserine Externalization and Apoptosis

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Solutions
2.2. Cell Culture and Transfection
2.3. Detection of PS Externalization using Annexin V-FITC Assay
2.4. Mouse Model of Cerebral Ischemia-Reperfusion
2.5. Preparation of Mouse Brain Sections
2.6. TUNEL Assay
2.7. Co-Immunoprecipitation and Immunoblots
2.8. Förster Resonance Energy Transfer (FRET) Assay
2.9. PLA
2.10. Statistical Analysis
3. Results
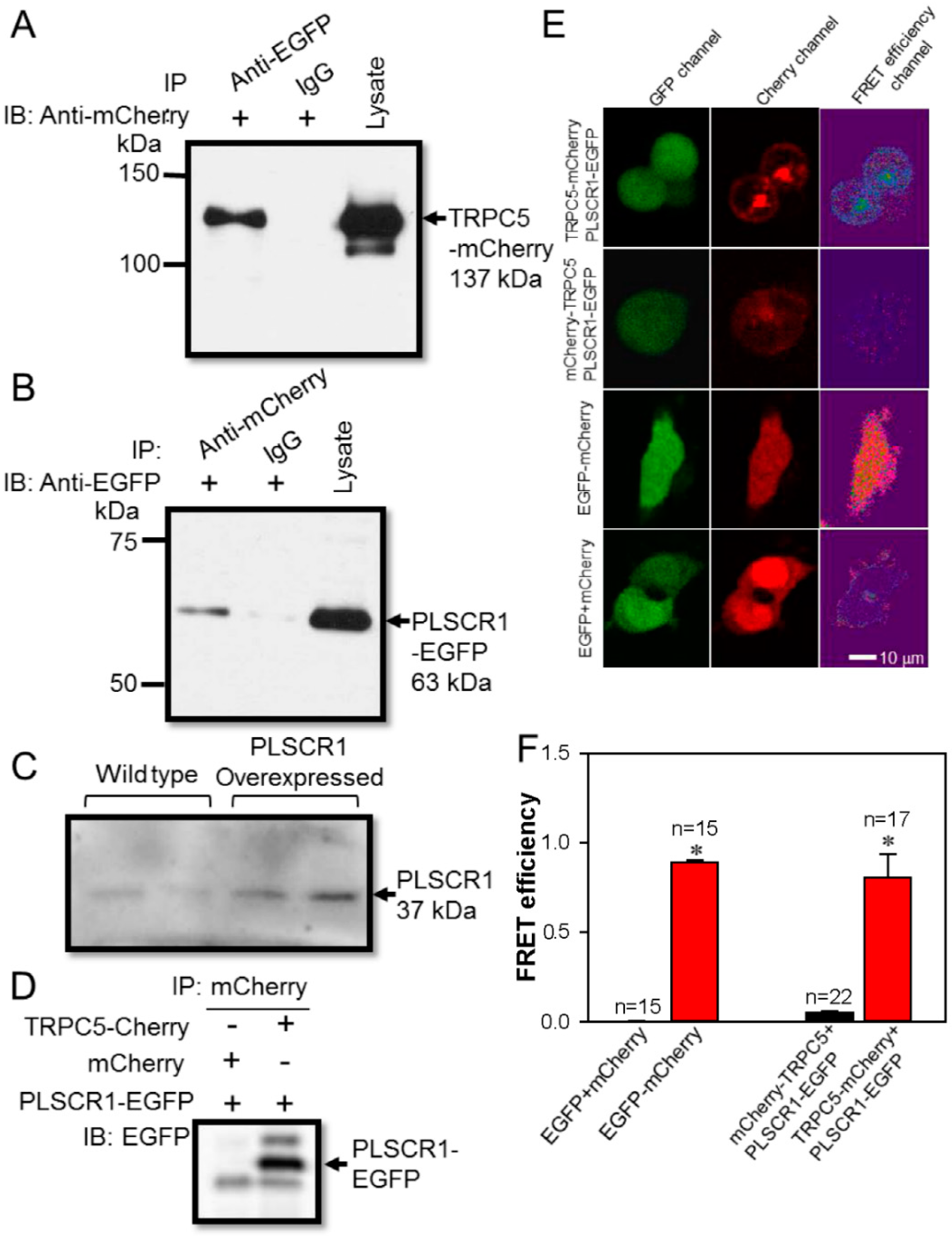
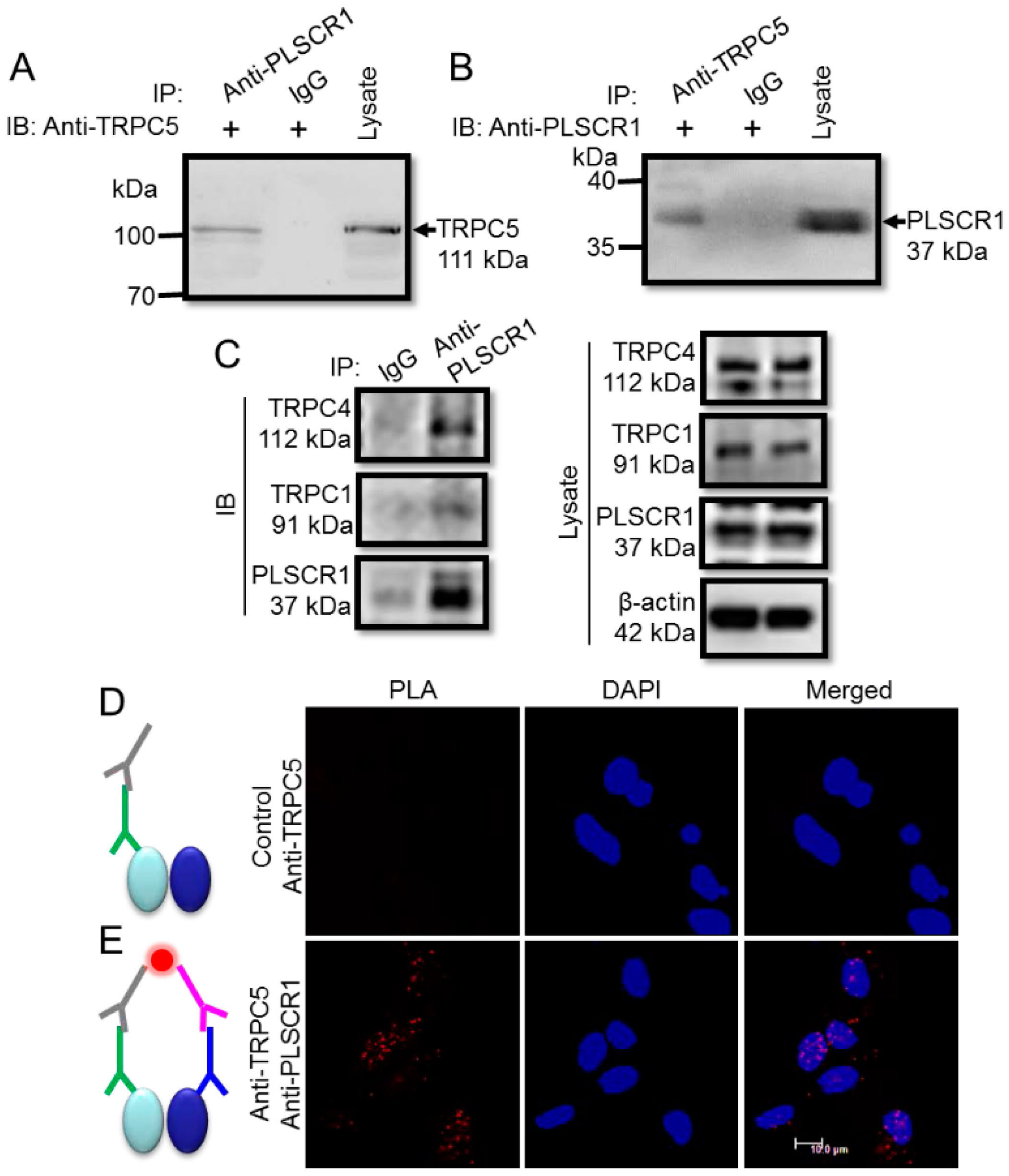
3.1. Spatial Proximity and Association of TRPC5 and PLSCR1 on HEK293 Plasma Membrane
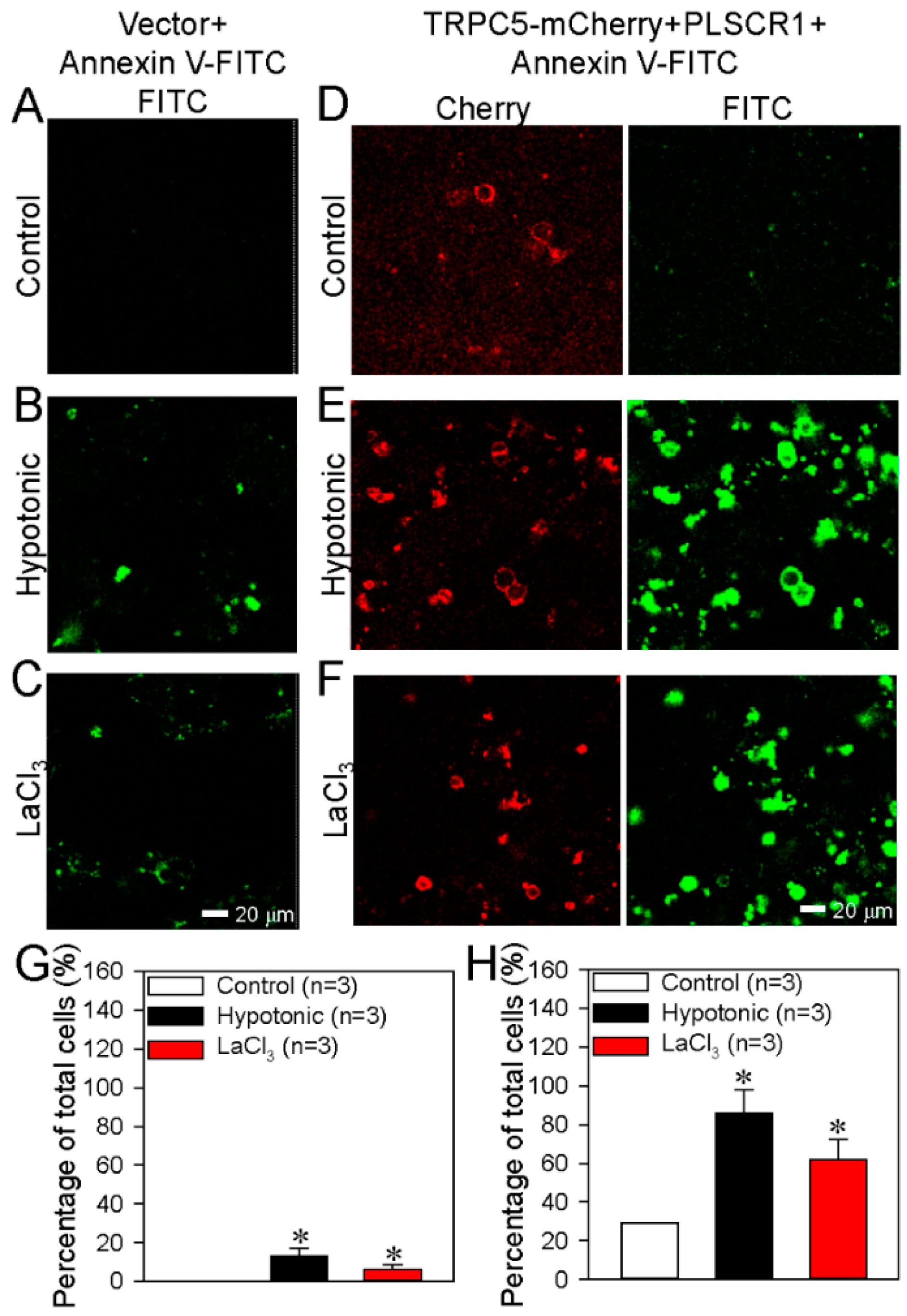
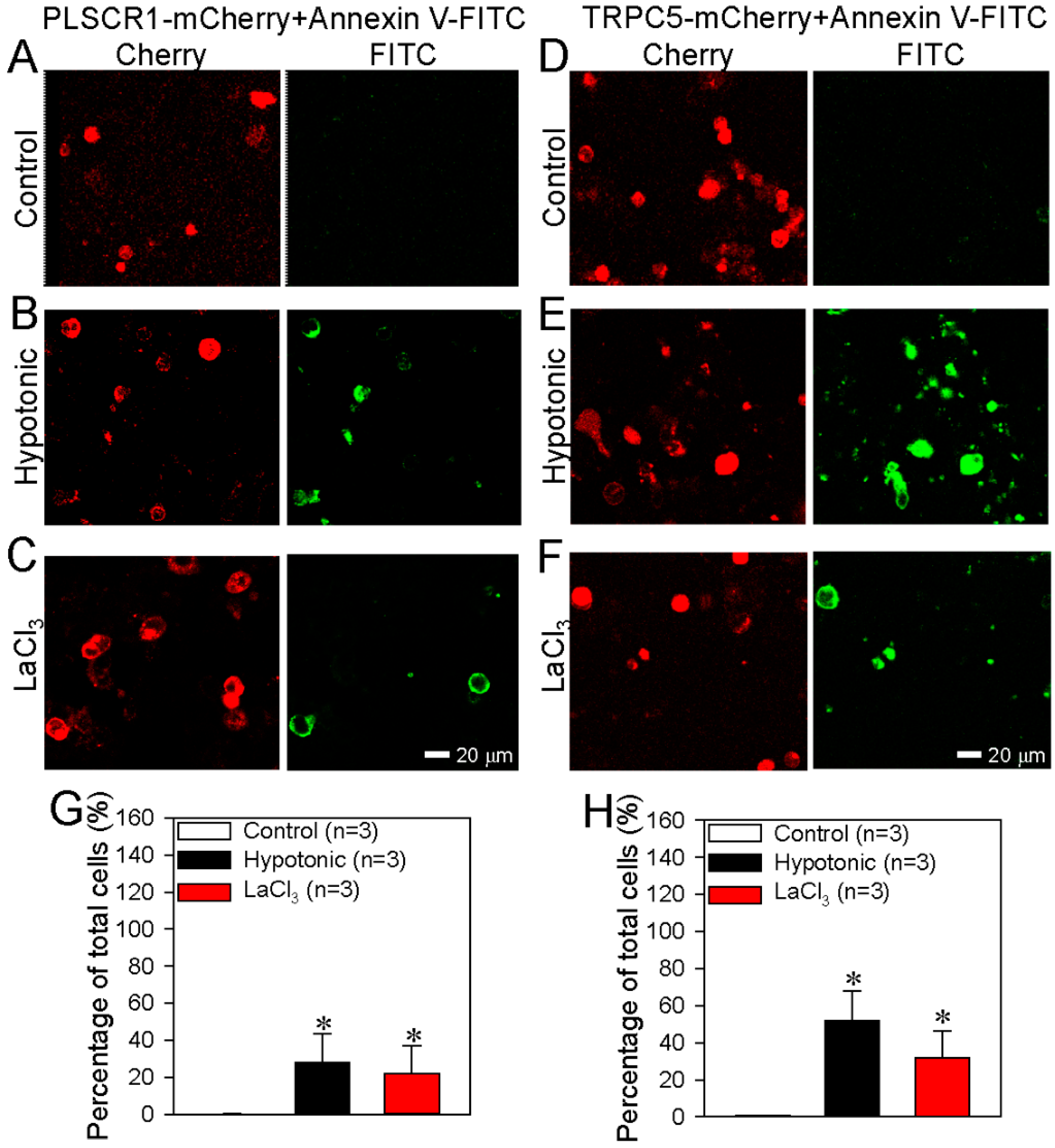
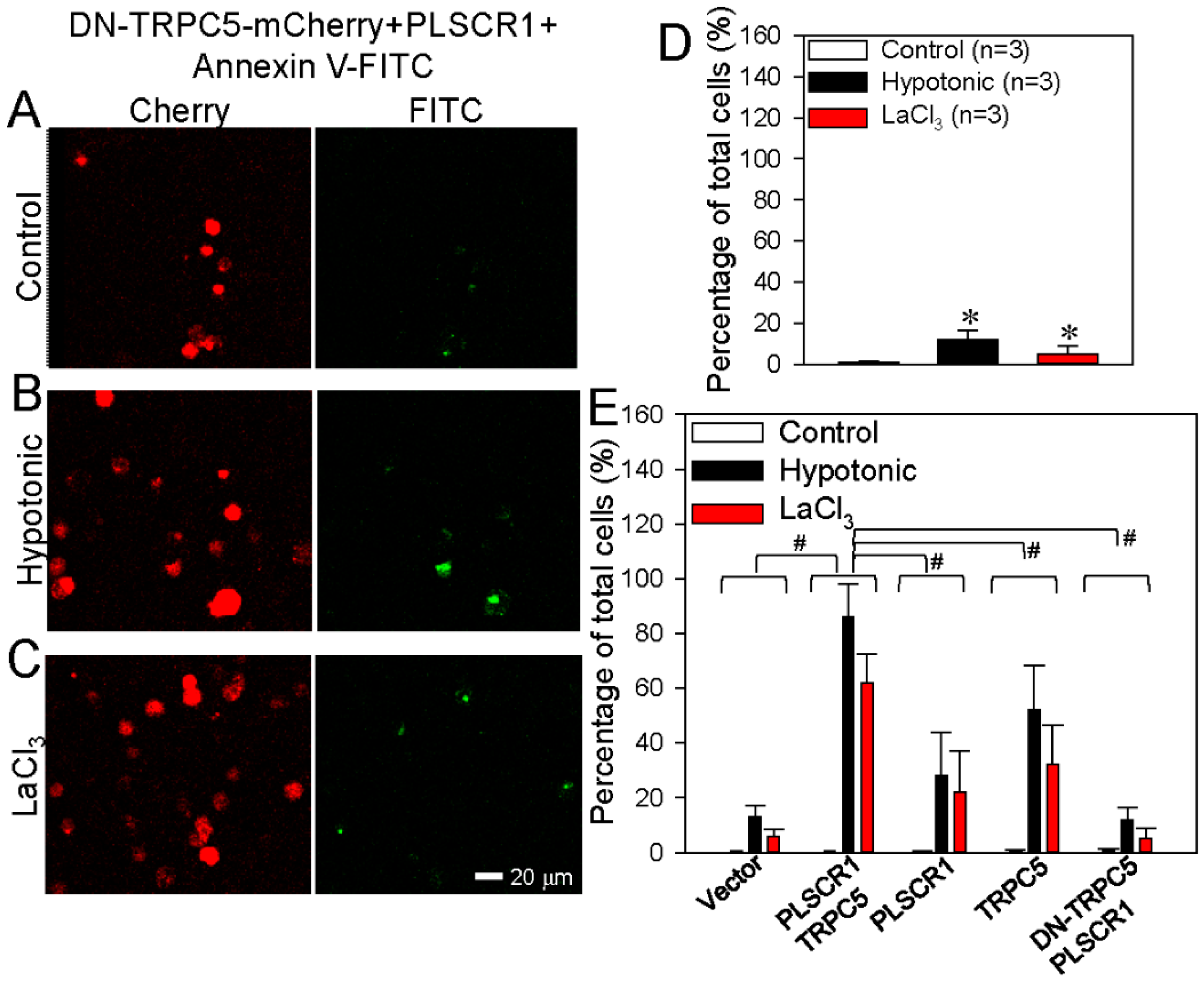
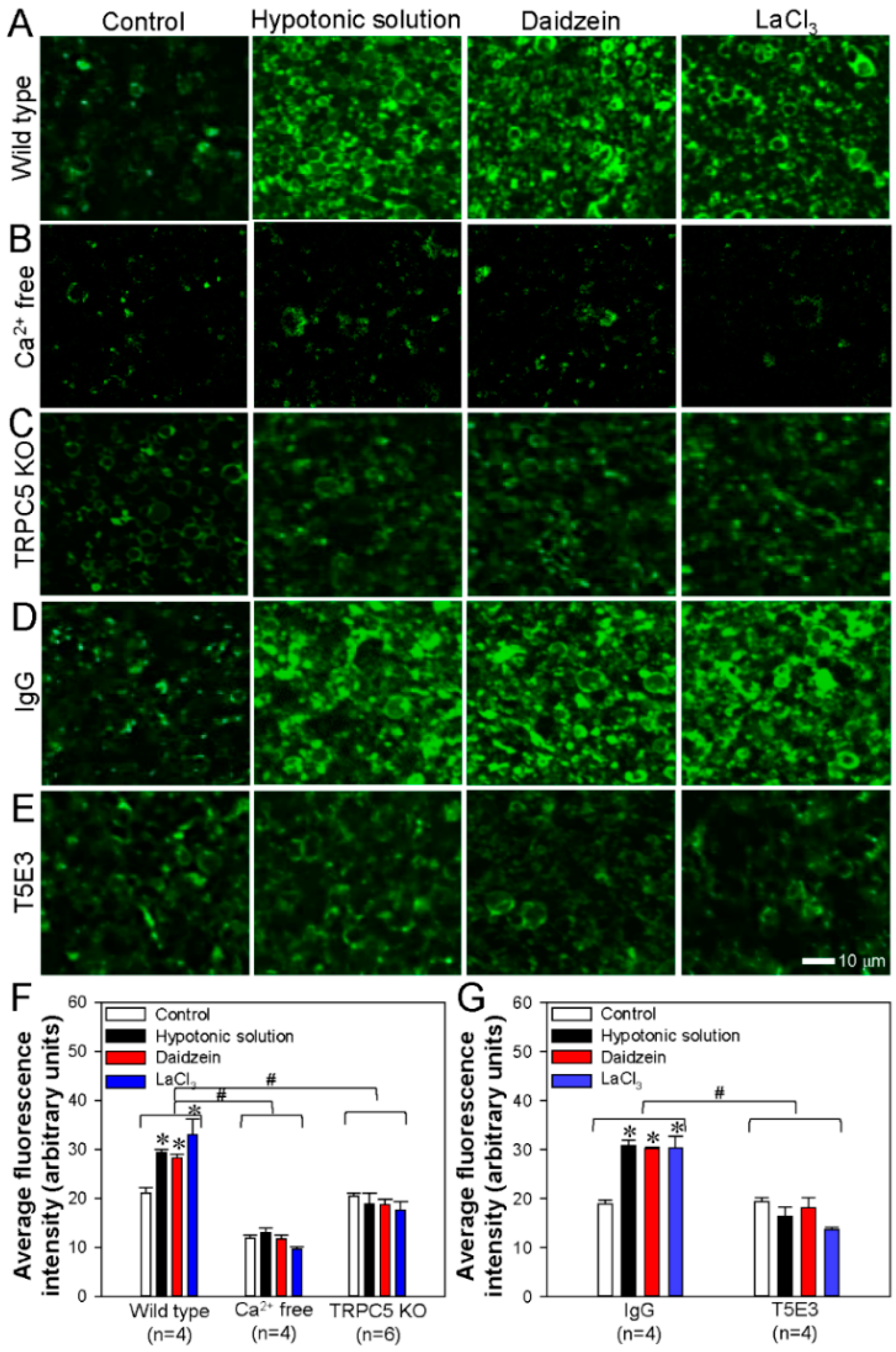
3.2. TRPC5 Promotes PS Externalization in HEK293 Cells
3.3. Physical Association of TRPC5 and PLSCR1 in Native Neurons
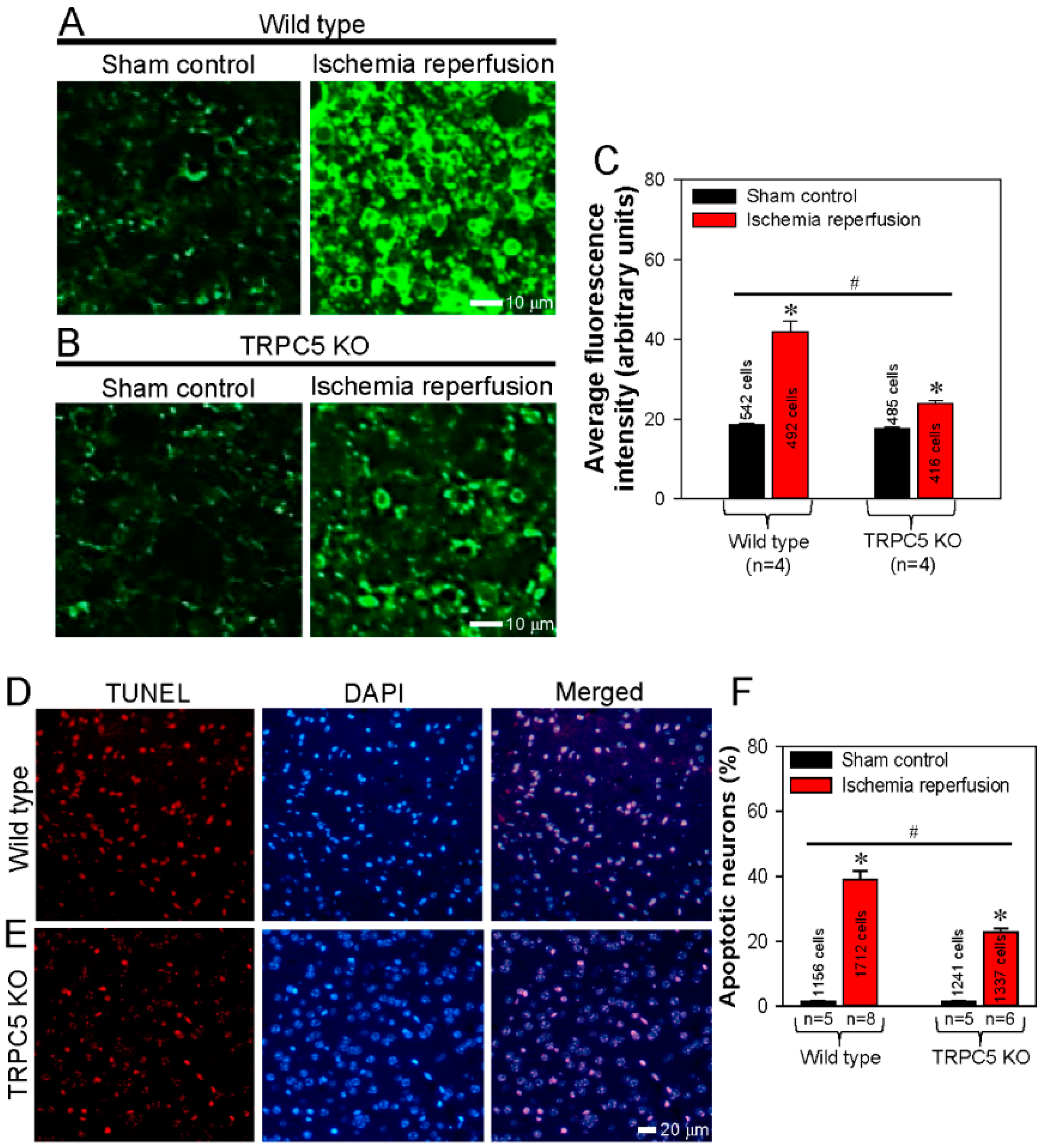
3.4. Activation of TRPC5 Stimulates Neuronal PS Externalization in Cerebral Slides
3.5. Spatial Proximity and Association of TRPC5 and PLSCR1 on HEK293 Plasma Membrane
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Williamson, P.; Schlegel, R.A. Transbilayer Phospholipid Movement and the Clearance of Apoptotic Cells. Biochim. Biophys. Acta 2002, 1585, 53–63. [Google Scholar] [CrossRef]
- Basse, F.; Stout, J.G.; Sims, P.J.; Wiedmer, T. Isolation of an Erythrocyte Membrane Protein That Mediates Ca2+-Dependent Transbilayer Movement of Phospholipid. J. Biol. Chem. 1996, 271, 17205–17210. [Google Scholar] [CrossRef] [PubMed]
- Kodigepalli, M.K.; Bowers, K.; Sharp, A.; Nanjundan, M. Roles and Regulation of Phospholipid Scramblases. FEBS Lett. 2015, 589, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhou, Q.; Wiedmer, T.; Sims, P.J. Level of Expression of Phospholipid Scramblase Regulates Induced Movement of Phosphatidylserine to the Cell Surface. J. Biol. Chem. 1998, 273, 6603–6606. [Google Scholar] [CrossRef]
- Arashiki, N.; Saito, M.; Koshino, I.; Kamata, K.; Hale, J.; Mohandas, N.; Manno, S.; Takakuwa, Y. An Unrecognized Function of Cholesterol: Regulating the Mechanism Controlling Membrane Phospholipid Asymmetry. Biochemistry 2016, 55, 3504–3513. [Google Scholar] [CrossRef]
- Mastorci, K.; Montico, B.; Fae, D.A.; Sigalotti, L.; Ponzoni, M.; Inghirami, G.; Dolcetti, R.; Col, J.D. Phospholipid Scramblase 1 as a Critical Node at the Crossroad between Autophagy and Apoptosis in Mantle Cell Lymphoma. Oncotarget 2016, 7, 41913–41928. [Google Scholar] [CrossRef]
- Chen, H.M.; Ben-Efraim, I.; Mitrousis, G.; Walker-Kopp, N.; Sims, P.J.; Cingolani, G. Phospholipid Scramblase 1 Contains a Nonclassical Nuclear Localization Signal with Unique Binding Site in Importin Alpha. J. Biol. Chem. 2005, 280, 10599–10606. [Google Scholar] [CrossRef]
- Ben-Efraim, I.; Zhou, Q.; Wiedmer, T.; Gerace, L.; Sims, P.J. Phospholipid Scramblase 1 Is Imported into the Nucleus by a Receptor-Mediated Pathway and Interacts with DNA. Biochemistry 2004, 43, 3518–3526. [Google Scholar] [CrossRef]
- Zhu, J.; Sheng, J.; Dong, H.; Kang, L.; Ang, J.; Xu, Z. Phospholipid Scramblase 1 Functionally Interacts with Angiogenin and Regulates Angiogenin-Enhanced Rrna Transcription. Cell Physiol. Biochem. 2013, 32, 1695–1706. [Google Scholar] [CrossRef]
- Rayala, S.; Francis, V.G.; Sivagnanam, U.; Gummadi, S.N. N-Terminal Proline-Rich Domain Is Required for Scrambling Activity of Human Phospholipid Scramblases. J. Biol. Chem. 2014, 289, 13206–13218. [Google Scholar] [CrossRef]
- Francis, G.V.; Padmanabhan, P.; Gummadi, S.N. Snail Interacts with Hplscr1 Promoter and Down Regulates Its Expression in Imr-32. Biochem. Biophys. Res. Commun. 2014, 450, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Sahu, K.S.; Gummadi, S.N.; Manoj, N.; Aradhyam, G.K. Phospholipid Scramblases: An Overview. Arch. Biochem. Biophys. 2007, 462, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Amengual, O.; Atsumi, T.; Oku, K.; Hashimoto, T.; Kataoka, H.; Horita, T.; Yasuda, S.; Ieko, M.; Fukushima, K.; et al. Increased Expression of Phospholipid Scramblase 1 in Monocytes from Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2010, 37, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Amengual, O.; Atsumi, T.; Oku, K.; Suzuki, E.; Horita, T.; Yasuda, S.; Koike, T. Phospholipid Scramblase 1 Expression Is Enhanced in Patients with Antiphospholipid Syndrome. Mod. Rheumatol. 2013, 23, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.K.; Li, X.; Zhao, Q.; Huang, Y.; Li, D.; Peng, Z.G.; Shen, W.Z.; Zhao, J.; Zhou, Q.; Chen, Z.; et al. Protein Kinase Cdelta Mediates Retinoic Acid and Phorbol Myristate Acetate-Induced Phospholipid Scramblase 1 Gene Expression: Its Role in Leukemic Cell Differentiation. Blood 2004, 104, 3731–3738. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Yamashita, T.; Shiozawa, E.; Nagasawa, A.; Okabe-Kado, J.; Nakamaki, T.; Tomoyasu, S.; Kimura, F.; Motoyoshi, K.; Honma, Y.; et al. Mmtra1b/Phospholipid Scramblase 1 Gene Expression Is a New Prognostic Factor for Acute Myelogenous Leukemia. Leuk. Res. 2004, 28, 149–157. [Google Scholar] [CrossRef]
- Kodigepalli, M.K.; Anur, P.; Spellman, P.; Sims, P.J.; Nanjundan, M. Phospholipid Scramblase 1, an Interferon-Regulated Gene Located at 3q23, Is Regulated by Snon/Skil in Ovarian Cancer Cells. Mol. Cancer 2013, 12, 32. [Google Scholar] [CrossRef]
- Fan, W.C.; Chen, C.Y.; Chen, K.T.; Shen, C.R.; Kuo, Y.B.; Chen, Y.S.; Chou, Y.P.; Wei, W.S.; Chan, E.C. Blockade of Phospholipid Scramblase 1 with Its N-Terminal Domain Antibody Reduces Tumorigenesis of Colorectal Carcinomas in Vitro and in Vivo. J. Transl. Med. 2012, 10, 254. [Google Scholar] [CrossRef]
- Kuo, B.Y.; Chan, C.C.; Chang, C.A.; Fan, C.W.; Hung, R.P.; Hung, Y.S.; Chen, K.T.; Yu, J.S.; Chang, Y.S.; Chan, E.C. Identification of Phospholipid Scramblase 1 as a Biomarker and Determination of Its Prognostic Value for Colorectal Cancer. Mol. Med. 2011, 17, 41–47. [Google Scholar] [CrossRef]
- Cui, W.; Li, S.Y.; Du, J.F.; Zhu, Z.M.; An, P. Silencing Phospholipid Scramblase 1 Expression by Rna Interference in Colorectal Cancer and Metastatic Liver Cancer. Hepatobiliary Pancreat. Dis. Int. 2012, 11, 393–400. [Google Scholar] [CrossRef]
- Zwaal, F.R.; Schroit, A.J. Pathophysiologic Implications of Membrane Phospholipid Asymmetry in Blood Cells. Blood 1997, 89, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.D.; Wagner-Britz, L.; Maia, S.; Steffen, P.; Wagner, C.; Kaestner, L.; Bernhardt, I. Regulation of Phosphatidylserine Exposure in Red Blood Cells. Cell Physiol. Biochem. 2011, 28, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Philipp, S.; Hambrecht, J.; Braslavski, L.; Schroth, G.; Freichel, M.; Murakami, M.; Cavalie, A.; Flockerzi, V. A Novel Capacitative Calcium Entry Channel Expressed in Excitable Cells. EMBO J. 1998, 17, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Shimizu, S.; Wakamori, M.; Maeda, A.; Kurosaki, T.; Takada, N.; Imoto, K.; Mori, Y. Molecular Cloning and Functional Characterization of a Novel Receptor-Activated Trp Ca2+ Channel from Mouse Brain. J. Biol. Chem. 1998, 273, 10279–10287. [Google Scholar] [CrossRef]
- Shen, B.; Wong, C.O.; Lau, O.C.; Woo, T.; Bai, S.; Huang, Y.; Yao, X. Plasma Membrane Mechanical Stress Activates Trpc5 Channels. PLoS ONE 2015, 10, e0122227. [Google Scholar] [CrossRef]
- Xu, Z.S.; Muraki, K.; Zeng, F.; Li, J.; Sukumar, P.; Shah, S.; Dedman, A.M.; Flemming, P.K.; McHugh, D.; Naylor, J.; et al. A Sphingosine-1-Phosphate-Activated Calcium Channel Controlling Vascular Smooth Muscle Cell Motility. Circ. Res. 2006, 98, 1381–1389. [Google Scholar] [CrossRef]
- Schaefer, M.; Plant, T.D.; Obukhov, A.G.; Hofmann, T.; Gudermann, T.; Schultz, G. Receptor-Mediated Regulation of the Nonselective Cation Channels Trpc4 and Trpc5. J. Biol. Chem. 2000, 275, 17517–17526. [Google Scholar] [CrossRef]
- Trebak, M.; Lemonnier, L.; DeHaven, W.I.; Wedel, B.J.; Bird, G.S.; Putney, J.W., Jr. Complex Functions of Phosphatidylinositol 4,5-Bisphosphate in Regulation of Trpc5 Cation Channels. Pflugers Arch. 2009, 457, 4757–4769. [Google Scholar] [CrossRef]
- Gomis, A.; Soriano, S.; Belmonte, C.; Viana, F. Hypoosmotic- and Pressure-Induced Membrane Stretch Activate Trpc5 Channels. J. Physiol. 2008, 586, 5633–5649. [Google Scholar] [CrossRef]
- Zimmermann, K.; Lennerz, J.K.; Hein, A.; Link, A.S.; Kaczmarek, J.S.; Delling, M.; Uysal, S.; Pfeifer, J.D.; Riccio, A.; Clapham, D.E. Transient Receptor Potential Cation Channel, Subfamily C, Member 5 (Trpc5) Is a Cold-Transducer in the Peripheral Nervous System. Proc. Natl. Acad. Sci. USA 2011, 108, 18114–18119. [Google Scholar] [CrossRef]
- Greka, A.; Navarro, B.; Oancea, E.; Duggan, A.; Clapham, D.E. Trpc5 Is a Regulator of Hippocampal Neurite Length and Growth Cone Morphology. Nat. Neurosci. 2003, 6, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.; Li, Y.; Moon, J.; Kim, K.S.; Smith, K.S.; Rudolph, U.; Gapon, S.; Yao, G.L.; Tsvetkov, E.; Rodig, S.J.; et al. Essential Role for Trpc5 in Amygdala Function and Fear-Related Behavior. Cell 2009, 137, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Cai, Y.; He, D.; Zou, C.; Zhang, P.; Lo, C.Y.; Xu, Z.; Chan, F.L.; Yu, S.; Chen, Y.; et al. Transient Receptor Potential Channel Trpc5 Is Essential for P-Glycoprotein Induction in Drug-Resistant Cancer Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 16282–16287. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, Z.; Hua, D.; He, D.; Wang, L.; Zhang, P.; Wang, J.; Cai, Y.; Gao, C.; Zhang, X.; et al. Essential Role for Trpc5-Containing Extracellular Vesicles in Breast Cancer with Chemotherapeutic Resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 6389–6394. [Google Scholar] [CrossRef]
- Lau, C.O.; Shen, B.; Wong, C.O.; Tjong, Y.W.; Lo, C.Y.; Wang, H.C.; Huang, Y.; Yung, W.H.; Chen, Y.C.; Fung, M.L.; et al. Trpc5 Channels Participate in Pressure-Sensing in Aortic Baroreceptors. Nat. Commun. 2016, 7, 11947. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, Y.; Zhuo, D.; Lo, C.Y.; Yu, L.; Lau, C.W.; Kwan, Y.W.; Tse, G.; Huang, Y.; Yao, X. Endothelial Cell Transient Receptor Potential Channel C5 (Trpc5) Is Essential for Endothelium-Dependent Contraction in Mouse Carotid Arteries. Biochem. Pharmacol. 2019, 159, 11–24. [Google Scholar] [CrossRef]
- Hong, C.; Seo, H.; Kwak, M.; Jeon, J.; Jang, J.; Jeong, E.M.; Myeong, J.; Hwang, Y.J.; Ha, K.; Kang, M.J.; et al. Increased Trpc5 Glutathionylation Contributes to Striatal Neuron Loss in Huntington’s Disease. Brain 2015, 138, 3030–3047. [Google Scholar] [CrossRef]
- Li, K.; Li, W.; Yin, H.; Cheong, Y.K.; Ren, G.; Yang, Z. Pretreatment-Etidronate Alleviates Cocl2 Induced-Sh-Sy5y Cell Apoptosis Via Decreased Hif-1alpha and Trpc5 Channel Proteins. Neurochem. Res. 2019, 44, 428–440. [Google Scholar] [CrossRef]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. Formation of Novel Trpc Channels by Complex Subunit Interactions in Embryonic Brain. J. Biol. Chem. 2003, 278, 39014–39019. [Google Scholar] [CrossRef]
- Wu, K.; Shen, B.; Jiang, F.; Xia, L.; Fan, T.; Qin, M.; Yang, L.; Guo, J.; Li, Y.; Zhu, M.; et al. Trpp2 Enhances Metastasis by Regulating Epithelial-Mesenchymal Transition in Laryngeal Squamous Cell Carcinoma. Cell Physiol. Biochem. 2016, 39, 2203–2215. [Google Scholar] [CrossRef]
- Longa, Z.E.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible Middle Cerebral Artery Occlusion without Craniectomy in Rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, H.; Xiao, Z.; Li, M.; Xue, P.; Chen, Y.; Zhong, J. Evaluation of Computed Tomography Perfusion Imaging in a Rat Acute Cerebral Ischemia-Reperfusion Model. Turk. Neurosurg. 2016, 26, 246–252. [Google Scholar] [PubMed]
- Chen, M.; Li, J.; Jiang, F.; Fu, J.; Xia, X.; Du, J.; Hu, M.; Huang, J.; Shen, B. Orai1 Forms a Signal Complex with Bkca Channel in Mesenteric Artery Smooth Muscle Cells. Physiol. Rep. 2016, 4, e12682. [Google Scholar] [CrossRef] [PubMed]
- Albertazzi, L.; Arosio, D.; Marchetti, L.; Ricci, F.; Beltram, F. Quantitative Fret Analysis with the Egfp-Mcherry Fluorescent Protein Pair. Photochem. Photobiol. 2009, 85, 287–297. [Google Scholar] [CrossRef]
- Wouters, S.F.; Verveer, P.J.; Bastiaens, P.I. Imaging Biochemistry inside Cells. Trends Cell Biol. 2001, 11, 203–211. [Google Scholar] [CrossRef]
- De Las Heras-Martinez, G.; Andrieu, J.; Larijani, B.; Requejo-Isidro, J. Quantifying Intracellular Equilibrium Dissociation Constants Using Single-Channel Time-Resolved Fret. J. Biophotonics 2018, 11, e201600272. [Google Scholar] [CrossRef]
- Wu, X.; Babnigg, G.; Zagranichnaya, T.; L, M. Villereal. The Role of Endogenous Human Trp4 in Regulating Carbachol-Induced Calcium Oscillations in Hek-293 Cells. J. Biol. Chem. 2002, 277, 13597–13608. [Google Scholar] [CrossRef]
- Wong, O.C.; Huang, Y.; Yao, X. Genistein Potentiates Activity of the Cation Channel Trpc5 Independently of Tyrosine Kinases. Br. J. Pharmacol. 2010, 159, 1486–1496. [Google Scholar] [CrossRef]
- Zwaal, F.R.; Comfurius, P.; Bevers, E.M. Surface Exposure of Phosphatidylserine in Pathological Cells. Cell Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef]
- Williamson, P.; Bevers, E.M.; Smeets, E.F.; Comfurius, P.; Schlegel, R.A.; Zwaal, R.F. Continuous Analysis of the Mechanism of Activated Transbilayer Lipid Movement in Platelets. Biochemistry 1995, 34, 10448–10455. [Google Scholar] [CrossRef]
- Fadok, A.V.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of Phosphatidylserine on the Surface of Apoptotic Lymphocytes Triggers Specific Recognition and Removal by Macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [PubMed]
- Martin, J.S.; Reutelingsperger, C.P.; McGahon, A.J.; Rader, J.A.; van Schie, R.C.; LaFace, D.M.; Green, D.R. Early Redistribution of Plasma Membrane Phosphatidylserine is a General Feature of Apoptosis Regardless of the Initiating Stimulus: Inhibition by Overexpression of Bcl-2 and Abl. J. Exp. Med. 1995, 182, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Fadok, A.V.; de Cathelineau, A.; Daleke, D.L.; Henson, P.M.; Bratton, D.L. Loss of Phospholipid Asymmetry and Surface Exposure of Phosphatidylserine Is Required for Phagocytosis of Apoptotic Cells by Macrophages and Fibroblasts. J. Biol. Chem. 2001, 276, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Fadok, A.V.; Bratton, D.L.; Rose, D.M.; Pearson, A.; Ezekewitz, R.A.; Henson, P.M. A Receptor for Phosphatidylserine-Specific Clearance of Apoptotic Cells. Nature 2000, 405, 85–90. [Google Scholar] [CrossRef]
- Bevers, M.E.; Comfurius, P.; van Rijn, J.L.; Hemker, H.C.; Zwaal, R.F. Generation of Prothrombin-Converting Activity and the Exposure of Phosphatidylserine at the Outer Surface of Platelets. Eur. J. Biochem. 1982, 122, 429–436. [Google Scholar] [CrossRef]
- Bevers, M.E.; Comfurius, P.; Zwaal, R.F. Changes in Membrane Phospholipid Distribution during Platelet Activation. Biochim. Biophys. Acta 1983, 736, 57–66. [Google Scholar] [CrossRef]
- Sims, J.P.; Wiedmer, T. Unraveling the Mysteries of Phospholipid Scrambling. Thromb. Haemost. 2001, 86, 266–275. [Google Scholar]
- Heja, L.; Kardos, J. Ncx Activity Generates Spontaneous Ca(2+) Oscillations in the Astrocytic Leaflet Microdomain. Cell Calcium 2019, 86, 102137. [Google Scholar] [CrossRef]
- Saeki, T.; Suzuki, Y.; Yamamura, H.; Takeshima, H.; Imaizumi, Y. A Junctophilin-Caveolin Interaction Enables Efficient Coupling between Ryanodine Receptors and Bkca Channels in the Ca(2+) Microdomain of Vascular Smooth Muscle. J. Biol. Chem. 2019, 294, 13093–13105. [Google Scholar] [CrossRef]
- Sossey-Alaoui, K.; Lyon, J.A.; Jones, L.; Abidi, F.E.; Hartung, A.J.; Hane, B.; Schwartz, C.E.; Stevenson, R.E.; Srivastava, A.K. Molecular Cloning and Characterization of Trpc5 (Htrp5), the Human Homologue of a Mouse Brain Receptor-Activated Capacitative Ca2+ Entry Channel. Genomics 1999, 60, 330–340. [Google Scholar] [CrossRef]
- Donnan, A.G.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, J.; Li, J.; Xia, L.; Wang, Y.; Zhu, J.; Du, J.; Lu, Y.; Liu, G.; Yao, X.; Shen, B. Transient Receptor Potential Canonical 5-Scramblase Signaling Complex Mediates Neuronal Phosphatidylserine Externalization and Apoptosis. Cells 2020, 9, 547. https://doi.org/10.3390/cells9030547
Guo J, Li J, Xia L, Wang Y, Zhu J, Du J, Lu Y, Liu G, Yao X, Shen B. Transient Receptor Potential Canonical 5-Scramblase Signaling Complex Mediates Neuronal Phosphatidylserine Externalization and Apoptosis. Cells. 2020; 9(3):547. https://doi.org/10.3390/cells9030547
Chicago/Turabian StyleGuo, Jizheng, Jie Li, Lin Xia, Yang Wang, Jinhang Zhu, Juan Du, Yungang Lu, Guodong Liu, Xiaoqiang Yao, and Bing Shen. 2020. "Transient Receptor Potential Canonical 5-Scramblase Signaling Complex Mediates Neuronal Phosphatidylserine Externalization and Apoptosis" Cells 9, no. 3: 547. https://doi.org/10.3390/cells9030547
APA StyleGuo, J., Li, J., Xia, L., Wang, Y., Zhu, J., Du, J., Lu, Y., Liu, G., Yao, X., & Shen, B. (2020). Transient Receptor Potential Canonical 5-Scramblase Signaling Complex Mediates Neuronal Phosphatidylserine Externalization and Apoptosis. Cells, 9(3), 547. https://doi.org/10.3390/cells9030547