Microfluidic Device for On-Chip Immunophenotyping and Cytogenetic Analysis of Rare Biological Cells

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
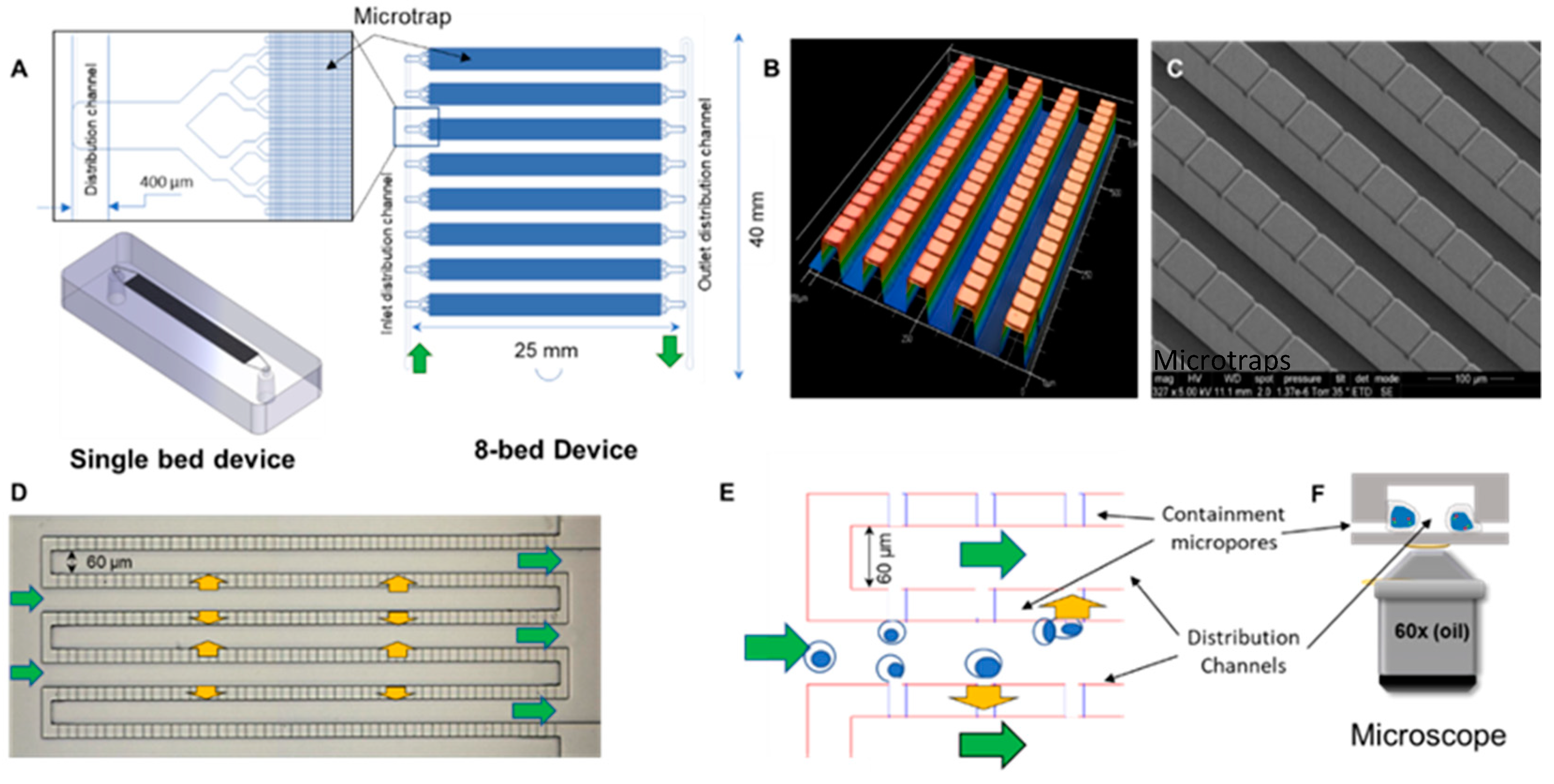
2.1. Design and Fabrication of the Microtrap Device
2.2. Sample loading
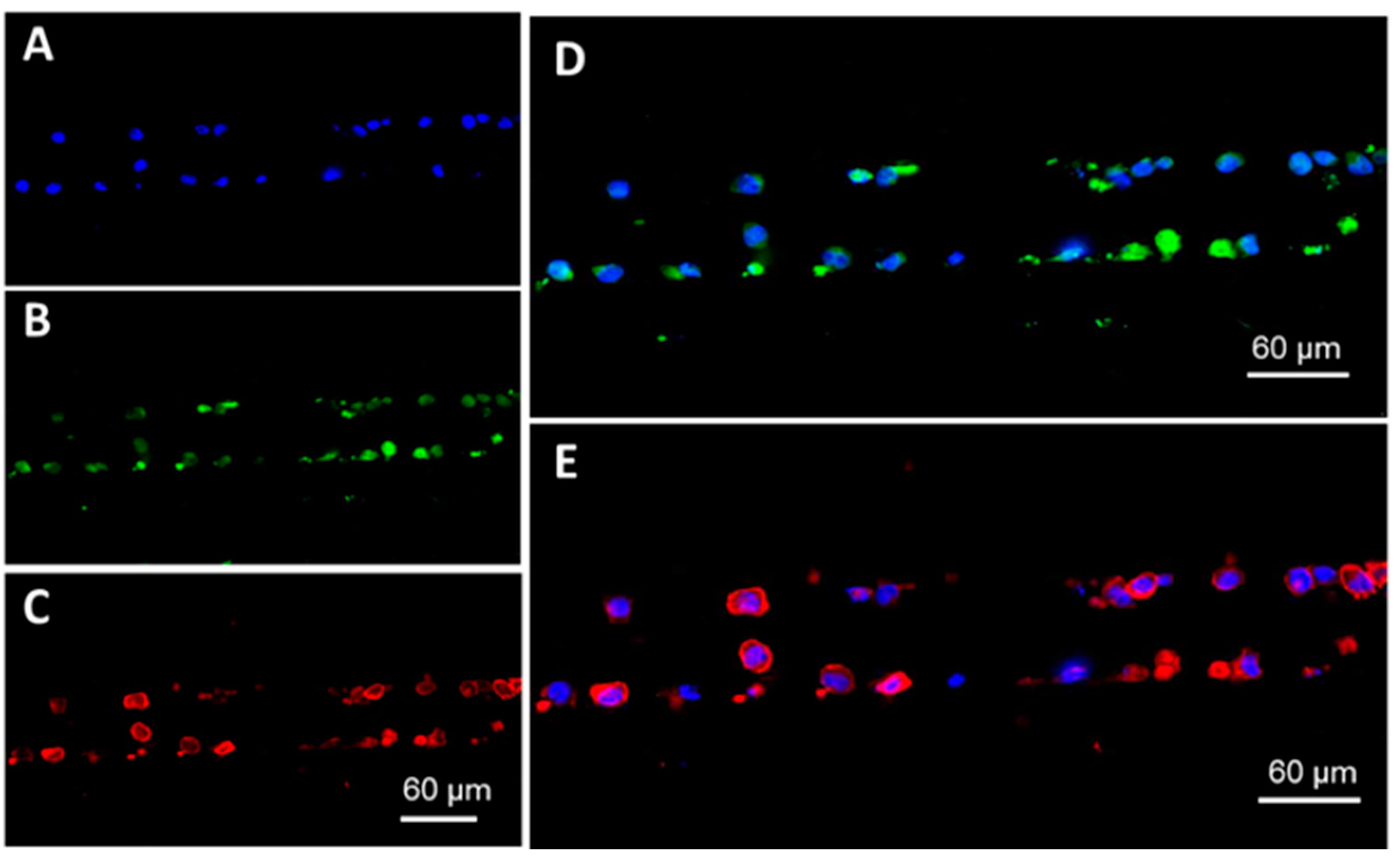
2.3. On-Chip immunostaining
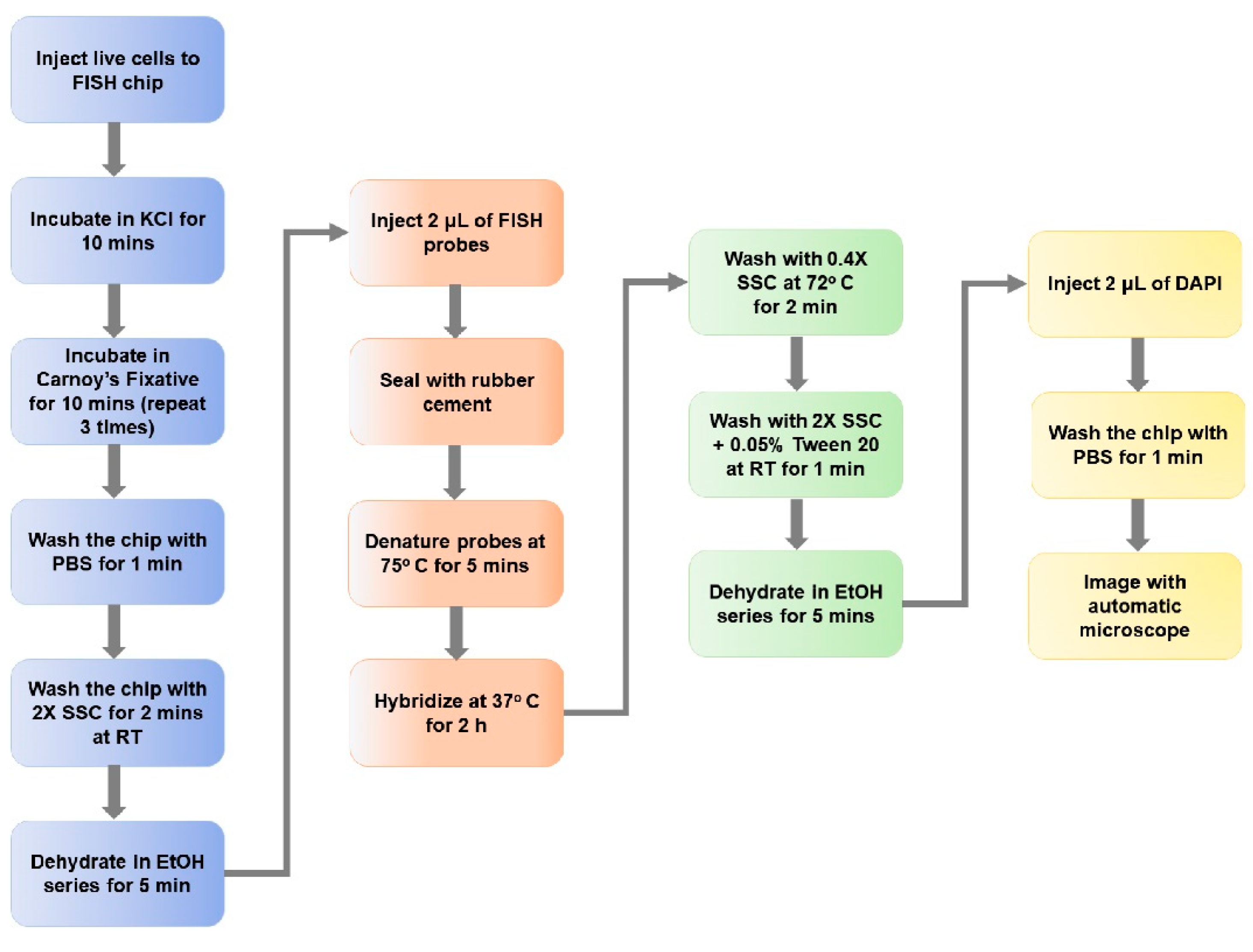
2.4. Sample Preparation for FISH
2.5. On-Chip FISH
2.6. Imaging On-chip and Image Processing
2.7. Patient Sample Processing for FISH
3. Results
3.1. Microtrap Device for Immunophenotyping and Cytogenetic Analysis
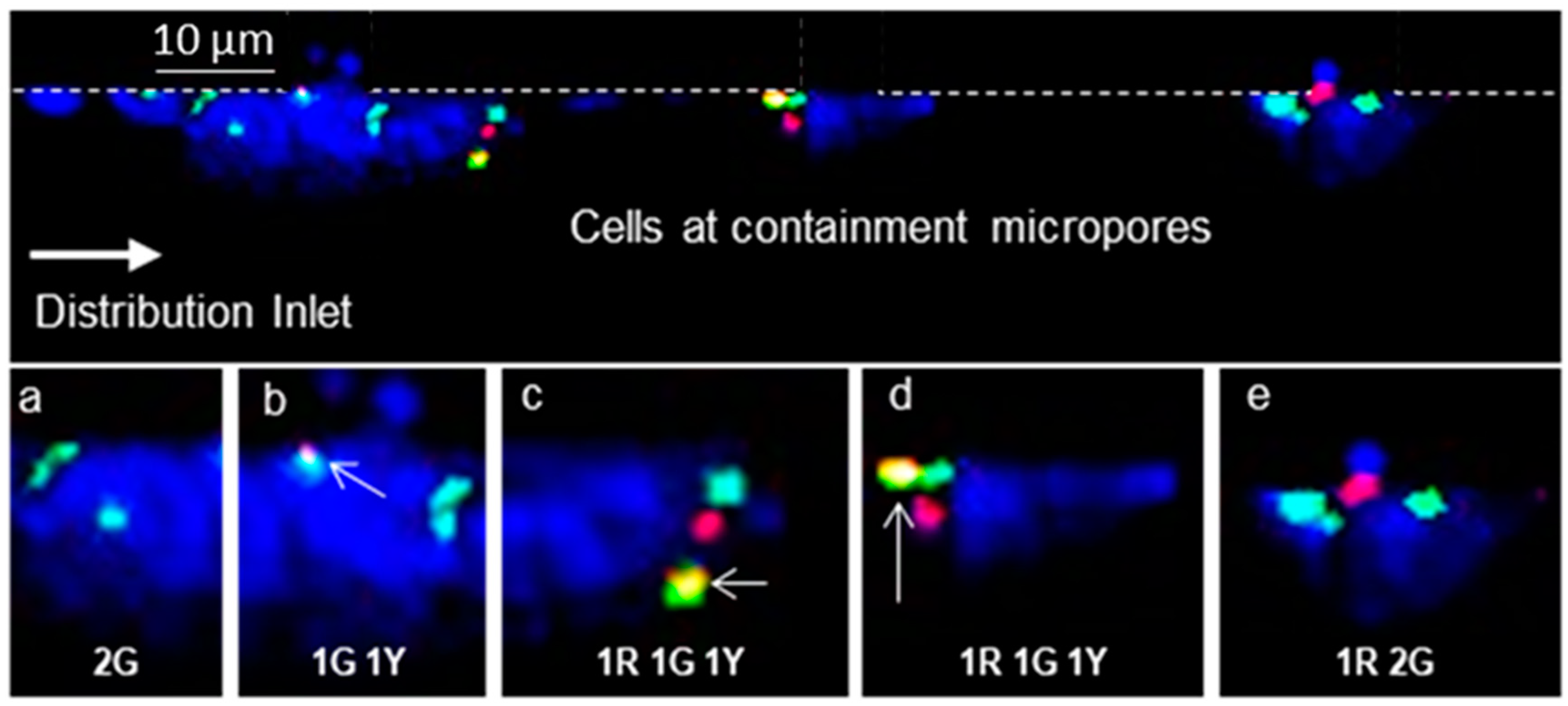
3.2. Microfluidic Containment Device Operation
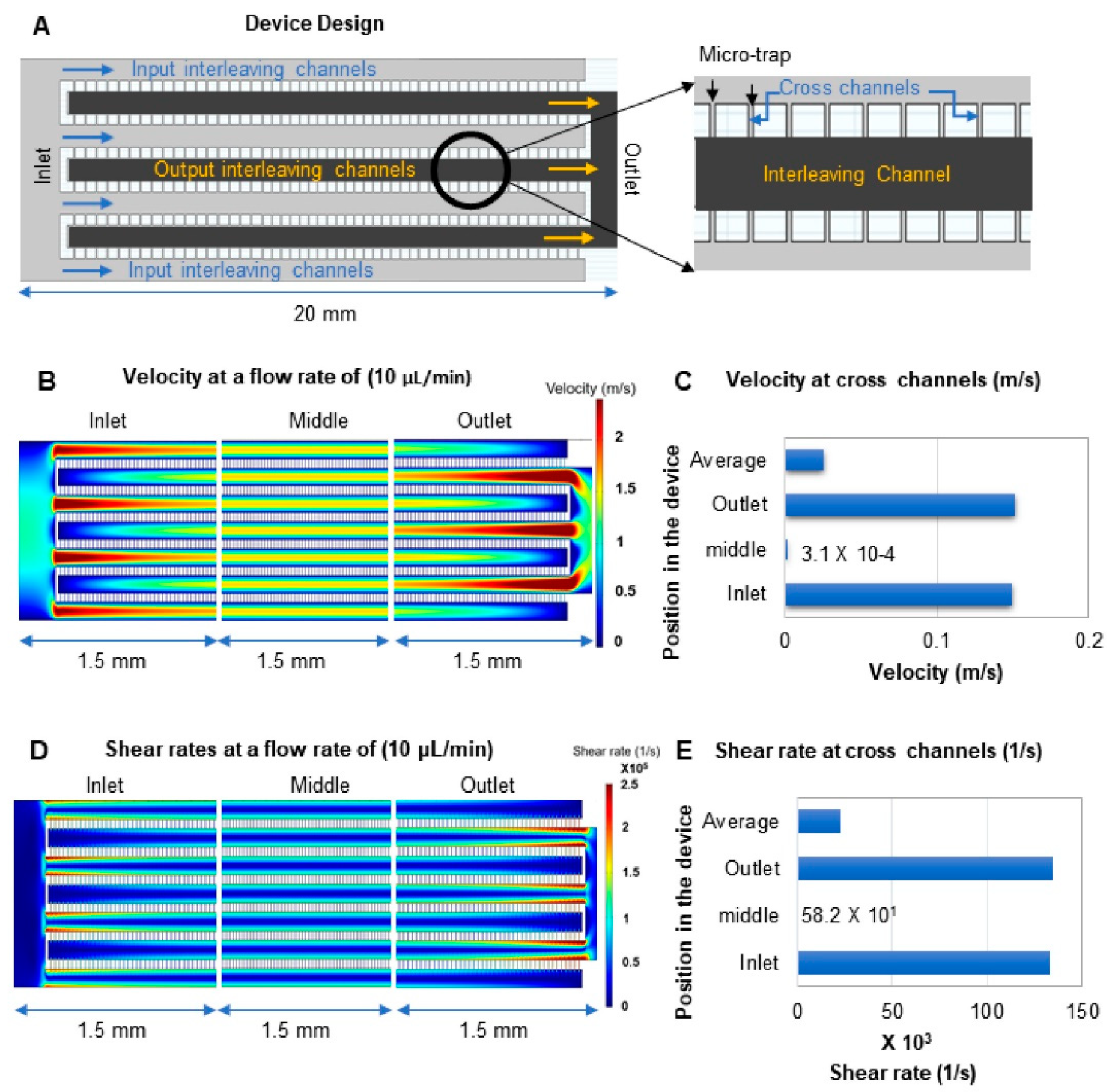
3.3. Device Design and COMSOL Simulations
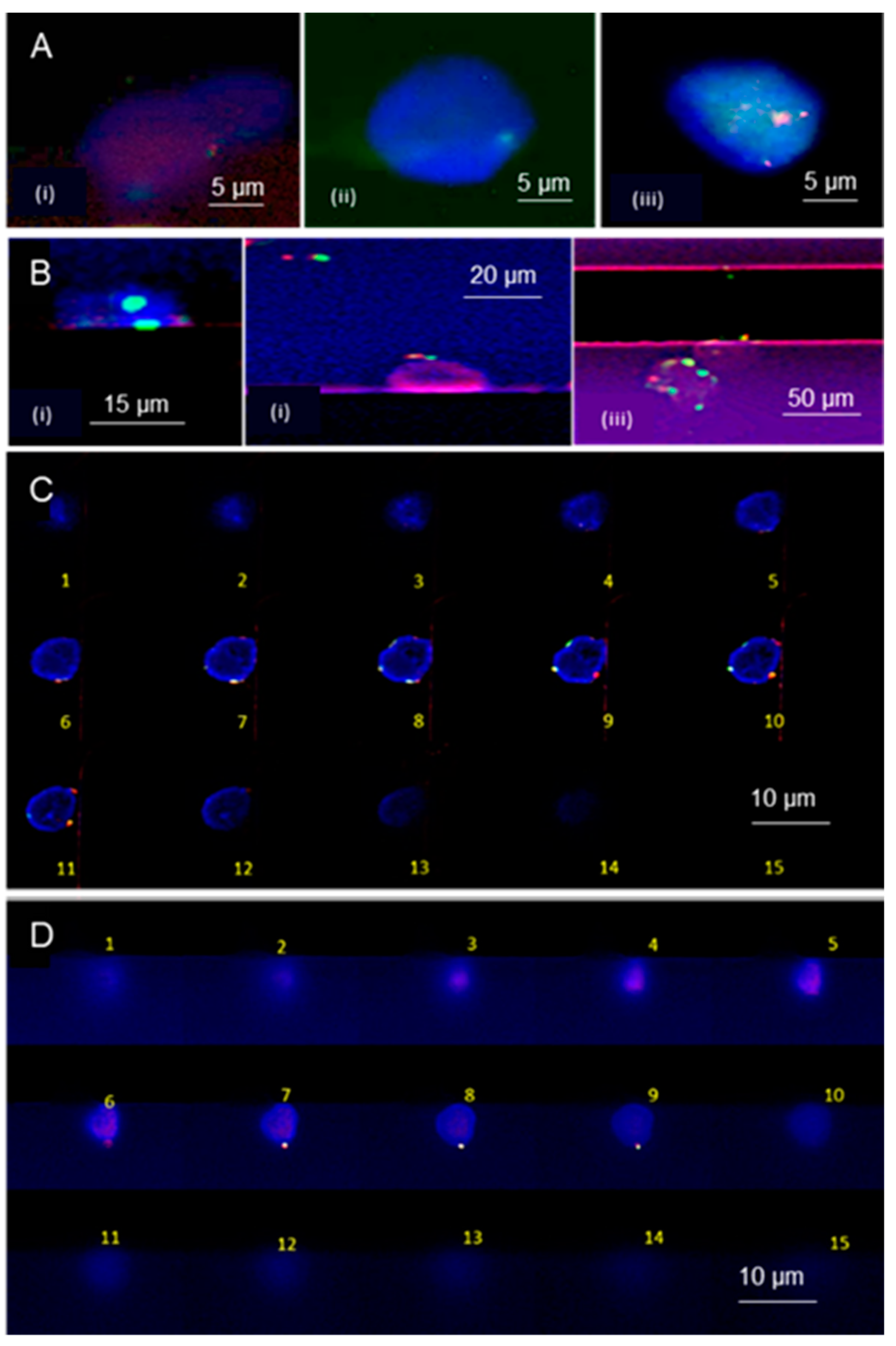
3.4. Microchip Processing and Imaging of a Large Number of Single Cells
3.5. On-Chip FISH
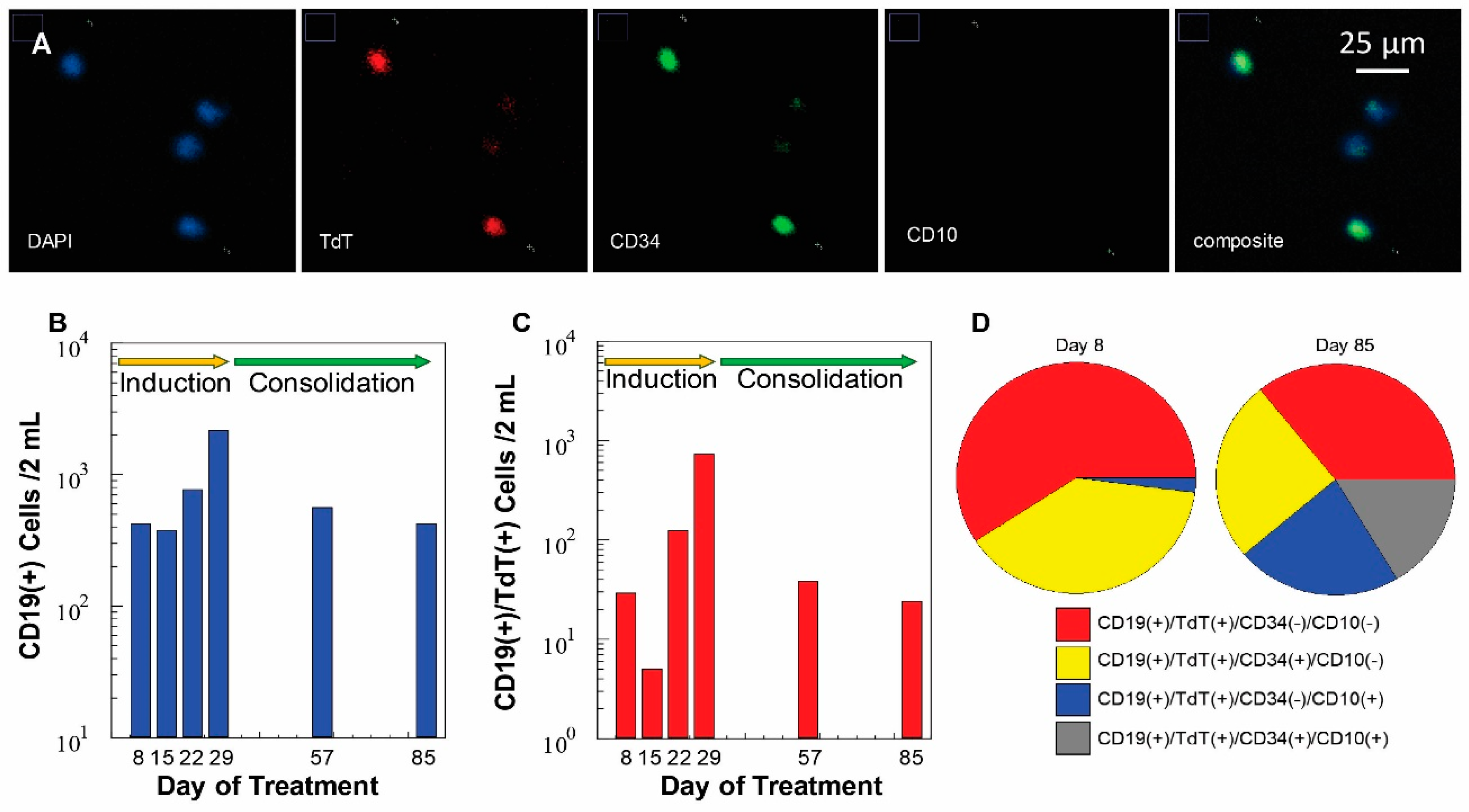
3.6. Measurement of MRD Status in Pediatric B-ALL Patients Using CLCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Macías, M.; Alegre, E.; Díaz-Lagares, A.; Patiño-García, A.; Pérez-Gracia, J.L.; Sanmamed, M.F.; López-López, R.; Varo, N.; Gonzalez, A. Liquid Biopsy: From Basic Research to Clinical Practice. In Advances in Virus Research; Elsevier BV: Bethesda, MD, USA, 2018; pp. 73–119. [Google Scholar]
- Jeffrey, S.S.; Toner, M. Liquid biopsy: A perspective for probing blood for cancer. Lab Chip 2019, 19, 548–549. [Google Scholar] [CrossRef]
- Kamande, J.W.; Lindell, M.A.M.; Witek, M.A.; Voorhees, P.M.; Soper, S.A. Isolation of circulating plasma cells from blood of patients diagnosed with clonal plasma cell disorders using cell selection microfluidics. Integr. Biol. 2018, 10, 82–91. [Google Scholar] [CrossRef]
- Jackson, J.M.; Taylor, J.B.; Witek, M.A.; Hunsucker, S.; Waugh, J.P.; Fedoriw, Y.; Shea, T.C.; Soper, S.A.; Armistead, P.M. Microfluidics for the detection of minimal residual disease in acute myeloid leukemia patients using circulating leukemic cells selected from blood. Analyst 2016, 141, 640–651. [Google Scholar] [CrossRef]
- Bauman, J.; Wiegant, J.; Borst, P.; Van Duijn, P. A new method for fluorescence microscopical localization of specific DNA sequences by in situ hybridization of fluorochrome-labelled RNA. Exp. Cell Res. 1980, 128, 485–490. [Google Scholar] [CrossRef]
- Langer-Safer, P.R.; Levine, M.; Ward, D.C. Immunological method for mapping genes on Drosophila polytene chromosomes. Proc. Natl. Acad. Sci. USA 1982, 79, 4381–4385. [Google Scholar] [CrossRef]
- Andreeff, M.; Pinkel, D. Introduction to Fluorescence in Situ Hybridization: Principles and Clinical Applications; Wiley-Liss: New York, NY, USA, 1999. [Google Scholar]
- Ye, C.; Stevens, J.; Liu, G.; Ye, K.; Yang, F.; Bremer, S.; Heng, H. Combined multicolor-FISH and immunostaining. Cytogenet. Genome Res. 2006, 114, 227–234. [Google Scholar] [CrossRef]
- Sato, K. Microdevice in Cellular Pathology: Microfluidic Platforms for Fluorescence in situ Hybridization and Analysis of Circulating Tumor Cells. Anal. Sci. 2015, 31, 867–873. [Google Scholar] [CrossRef]
- Dowd, A.; Homeida, S.; Elkarem, H.A. Detection of chromosome 13 (13q14) deletion among Sudanese patients with multiple myeloma using a molecular genetics fluorescent in situ hybridization technique (FISH). Malays. J. Pathol. 2015, 37, 95–100. [Google Scholar]
- Hu, Y.; Chen, L.; Sun, C.; She, X.-M.; Ai, L.; Qin, Y. Clinical significance of chromosomal abnormalities detected by interphase fluorescence in situ hybridization in newly diagnosed multiple myeloma patients. Chin. Med. J. 2011, 124, 2981–2985. [Google Scholar]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Schrauder, A.; Panzer-Grümayer, R.; Moricke, A.; Aricò, M.; Zimmermann, M.; Mann, G.; De Rossi, G.; et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef]
- Tai, C.-H.; Ho, C.-L.; Chen, Y.-L.; Chen, W.L.; Lee, G.-B. A novel integrated microfluidic platform to perform fluorescence in situ hybridization for chromosomal analysis. Microfluid. Nanofluidics 2013, 15, 745–752. [Google Scholar] [CrossRef]
- Sieben, V.J.; Debes-Marun, C.S.; Pilarski, L.M.; Backhouse, C.J. An integrated microfluidic chip for chromosome enumeration using fluorescence in situ hybridization. Lab Chip 2008, 8, 2151. [Google Scholar] [CrossRef]
- Chen, L.; Manz, A.; Day, P.J.R. Total nucleic acid analysis integrated on microfluidic devices. Lab Chip 2007, 7, 1413. [Google Scholar] [CrossRef]
- Packard, M.M.; Shusteff, M.; Alocilja, E. Microfluidic-Based Amplification-Free Bacterial DNA Detection by Dielectrophoretic Concentration and Fluorescent Resonance Energy Transfer Assisted in Situ Hybridization (FRET-ISH). Biosensors 2012, 2, 405–416. [Google Scholar] [CrossRef]
- Sieben, V.; Marun, C.D.; Pilarski, P.; Kaigala, G.; Pilarski, L.; Backhouse, C. FISH and chips: Chromosomal analysis on microfluidic platforms. IET Nanobiotechnol. 2007, 1, 27. [Google Scholar] [CrossRef]
- Zanardi, A.; Bandiera, D.; Bertolini, F.; Corsini, C.; Gregato, G.; Milani, P.; Barborini, E.; Carbone, R. Miniaturized FISH for screening of onco-hematological malignancies. Biotechniques 2010, 49, 497–504. [Google Scholar] [CrossRef]
- Liu, Y.; Kirkland, B.; Shirley, J.; Wang, Z.; Zhang, P.; Stembridge, J.; Wong, W.; Takebayashi, S.-I.; Gilbert, D.M.; Lenhert, S.; et al. Development of a single-cell array for large-scale DNA fluorescence in situ hybridization. Lab Chip 2013, 13, 1316–1324. [Google Scholar] [CrossRef][Green Version]
- Wang, X.; Takebayashi, S.-I.; Bernardin, E.; Gilbert, D.M.; Chella, R.; Guan, J. Microfluidic extraction and stretching of chromosomal DNA from single cell nuclei for DNA fluorescence in situ hybridization. Biomed. Microdevices 2012, 14, 443–451. [Google Scholar] [CrossRef][Green Version]
- Ciftlik, A.T.; Lehr, H.-A.; Gijs, M.A.M. Microfluidic processor allows rapid HER2 immunohistochemistry of breast carcinomas and significantly reduces ambiguous (2+) read-outs. Proc. Natl. Acad. Sci. USA 2013, 110, 5363–5368. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Trouillon, R.; Matsuoka, S.; Fiche, M.; De Leval, L.; Bisig, B.; Gijs, M.A. Microfluidics-assisted fluorescence in situ hybridization for advantageous human epidermal growth factor receptor 2 assessment in breast cancer. Lab. Investig. 2016, 97, 93–103. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Dupont, L.N.; Cuttaz, E.A.; Jean, A.M.; Trouillon, R.; Gijs, M.A.M. Breast cancer HER2 analysis by extra-short incubation microfluidics-assisted fluorescence in situ hybridization (ESIMA FISH). Microelectron. Eng. 2018, 189, 33–38. [Google Scholar] [CrossRef]
- Mayer, J.A.; Pham, T.; Wong, K.L.; Scoggin, J.; Sales, E.V.; Clarin, T.; Pircher, T.J.; Mikolajczyk, S.D.; Cotter, P.D.; Bischoff, F.Z. Fish-Based Determination of Her2 Status in Circulating Tumor Cells Isolated with the Microfluidic Cee™ Platform. Cancer Genet. 2011, 204, 589–595. [Google Scholar] [CrossRef]
- Mughal, F.; Baldock, S.J.; Karimiani, E.G.; Telford, N.; Goddard, N.J.; Day, P.J. Microfluidic channel-assisted screening of hematopoietic malignancies. Genes Chromosom. Cancer 2013, 53, 255–263. [Google Scholar] [CrossRef]
- Kwasny, D.; Mednova, O.; Vedarethinam, I.; Dimaki, M.; Silahtaroglu, A.; Tümer, Z.; Almdal, K.; Svendsen, W.E. A Semi-Closed Device for Chromosome Spreading for Cytogenetic Analysis. Micromachines 2014, 5, 158–170. [Google Scholar] [CrossRef]
- Perez-Toralla, K.; Guneri, E.T.; Champ, J.; Mottet, G.; Bidard, F.-C.; Pierga, J.-Y.; Klijanienko, J.; Draskovic, I.; Malaquin, L.; Viovy, J.-L.; et al. FISH in chips: Turning microfluidic fluorescence in situ hybridization into a quantitative and clinically reliable molecular diagnosis tool. Lab Chip 2015, 15, 811–822. [Google Scholar] [CrossRef]
- Vedarethinam, I.; Shah, P.; Dimaki, M.; Tümer, Z.; Tommerup, N.; Svendsen, W.E. Metaphase FISH on a Chip: Miniaturized Microfluidic Device for Fluorescence in situ Hybridization. Sensors 2010, 10, 9831–9846. [Google Scholar] [CrossRef]
- Baerlocher, G.M.; Vulto, I.; De Jong, G.; Lansdorp, P.M. Flow cytometry and FISH to measure the average length of telomeres (flow FISH). Nat. Protoc. 2006, 1, 2365–2376. [Google Scholar] [CrossRef]
- Arrigucci, R.; Bushkin, Y.; Radford, F.; Lakehal, K.; Vir, P.; Pine, R.; Martin, D.; Sugarman, J.; Zhao, Y.; Yap, G.S.; et al. FISH-Flow, a protocol for the concurrent detection of mRNA and protein in single cells using fluorescence in situ hybridization and flow cytometry. Nat. Protoc. 2017, 12, 1245–1260. [Google Scholar] [CrossRef]
- Kamande, J.W.; Hupert, M.L.; Witek, M.A.; Wang, H.; Torphy, R.J.; Dharmasiri, U.; Njoroge, S.K.; Jackson, J.M.; Aufforth, R.D.; Snavely, A.; et al. Modular Microsystem for the Isolation, Enumeration, and Phenotyping of Circulating Tumor Cells in Patients with Pancreatic Cancer. Anal. Chem. 2013, 85, 9092–9100. [Google Scholar] [CrossRef]
- Hupert, M.L.; Jackson, J.M.; Wang, H.; Witek, M.A.; Kamande, J.; Milowsky, M.I.; Whang, Y.E.; Soper, S.A. Arrays of High-Aspect Ratio Microchannels for High-Throughput Isolation of Circulating Tumor Cells (CTCs). Microsyst. Technol. 2013, 20, 1815–1825. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Nair, S.V.; Witek, M.A.; Jackson, J.M.; Lindell, M.A.M.; Hunsucker, S.; Sapp, T.; Perry, C.E.; Hupert, M.L.; Bae-Jump, V.; Gehrig, P.A.; et al. Enzymatic cleavage of uracil-containing single-stranded DNA linkers for the efficient release of affinity-selected circulating tumor cells. Chem. Commun. 2015, 51, 3266–3269. [Google Scholar] [CrossRef]
- Witek, M.A.; Aufforth, R.D.; Wang, H.; Kamande, J.W.; Jackson, J.M.; Pullagurla, S.R.; Hupert, M.L.; Usary, J.; Wysham, W.Z.; Hilliard, D.; et al. Discrete microfluidics for the isolation of circulating tumor cell subpopulations targeting fibroblast activation protein alpha and epithelial cell adhesion molecule. NPJ Precis. Oncol. 2017, 1, 24. [Google Scholar] [CrossRef]
- Bray, D. Cell Movements: From Molecules to Motility, 2nd ed.; Garland Science: New York, NY, USA, 1992; pp. 1–386. [Google Scholar] [CrossRef]
- Lauga, E.; Brenner, M.P.; Stone, H. Microfluidics: The No-Slip Boundary Condition. In Springer Handbook of Experimental Fluid Mechanics; Springer Science and Business Media LLC: New York, NY, USA, 2007; pp. 1219–1240. [Google Scholar]
- Rosser, J.; Thomas, D. Bioreactor processes for maturation of 3D bioprinted tissue. In 3D Bioprinting for Reconstructive Surgery; Elsevier BV: Duxford, UK, 2018; pp. 191–215. [Google Scholar]
- Lipowsky, H.H.; Kovalcheck, S.; Zweifach, B.W. The distribution of blood rheological parameters in the microvasculature of cat mesentery. Circ. Res. 1978, 43, 738–749. [Google Scholar] [CrossRef]
- Lipowsky, H.H.; Usami, S.; Chien, S. In vivo measurements of “apparent viscosity” and microvessel hematocrit in the mesentery of the cat. Microvasc. Res. 1980, 19, 297–319. [Google Scholar] [CrossRef]
- Varma, S.; Voldman, J. A cell-based sensor of fluid shear stress for microfluidics. Lab Chip 2015, 15, 1563–1573. [Google Scholar] [CrossRef]
- Papaioannou, T.G.; Stefanadis, C. Vascular wall shear stress: Basic principles and methods. Hell. J. Cardiol. 2005, 46, 9–15. [Google Scholar]
- Malek, A.M.; Alper, S.L.; Izumo, S. Hemodynamic shear stress and its role in atherosclerosis. JAMA 1999, 282, 2035–2042. [Google Scholar] [CrossRef]
- Strony, J.; Beaudoin, A.; Brands, D.; Adelman, B. Analysis of shear stress and hemodynamic factors in a model of coronary artery stenosis and thrombosis. Am. J. Physiol. Circ. Physiol. 1993, 265, H1787–H1796. [Google Scholar] [CrossRef]
- Barnes, J.M.; Nauseef, J.T.; Henry, M. Resistance to Fluid Shear Stress Is a Conserved Biophysical Property of Malignant Cells. PLoS ONE 2012, 7, e50973. [Google Scholar] [CrossRef]
- Pellat-Deceunynck, C.; Barillé, S.; Puthier, D.; Rapp, M.J.; Harousseau, J.L.; Bataille, R.; Amiot, M. Adhesion molecules on human myeloma cells: Significant changes in expression related to malignancy, tumor spreading, and immortalization. Cancer Res. 1995, 55, 3647–3653. [Google Scholar] [PubMed]
- Sahara, N.; Takeshita, A.; Shigeno, K.; Fujisawa, S.; Takeshita, K.; Naito, K.; Ihara, M.; Ono, T.; Tamashima, S.; Nara, K.; et al. Clinicopathological and prognostic characteristics of CD56-negative multiple myeloma. Br. J. Haematol. 2002, 117, 882–885. [Google Scholar] [CrossRef] [PubMed]
- Jamil, A.; Theil, K.S.; Kahwash, S.; Ruymann, F.B.; Klopfenstein, K.J. TEL/AML-1 fusion gene. Its frequency and prognostic significance in childhood acute lymphoblastic leukemia. Cancer Genet. Cytogenet. 2000, 122, 73–78. [Google Scholar] [CrossRef]
- Mikhail, F.M.; Serry, K.A.; Hatem, N.; Mourad, Z.I.; Farawela, H.M.; El Kaffash, D.M.; Coignet, L.; Nucifora, G. AML1 gene over-expression in childhood acute lymphoblastic leukemia. Leukemia 2002, 16, 658–668. [Google Scholar] [CrossRef][Green Version]
- Raynaud, S.; Cave, H.; Baens, M.; Bastard, C.; Cacheux, V.; Grosgeorge, J.; Guidal-Giroux, C.; Guo, C.; Vilmer, E.; Marynen, P.; et al. The 12;21 translocation involving TEL and deletion of the other TEL allele: Two frequently associated alterations found in childhood acute lymphoblastic leukemia. Blood 1996, 87, 2891–2899. [Google Scholar] [CrossRef]
- Chung, H.Y.; Kim, K.-H.; Jun, K.R.; Jang, S.; Park, C.-J.; Chi, H.-S.; Im, H.J.; Seo, J.J.; Seo, E.-J. Prognostic Significance ofTEL/AML1Rearrangement and Its Additional Genetic Changes in Korean Childhood Precursor B-Acute Lymphoblastic Leukemia. Korean J. Lab. Med. 2010, 30, 1. [Google Scholar] [CrossRef][Green Version]
- Mrózek, K.; Harper, D.P.; Aplan, P.D. Cytogenetics and Molecular Genetics of Acute Lymphoblastic Leukemia. Hematol. Clin. N. Am. 2009, 23, 991–1010. [Google Scholar] [CrossRef]
- Jaso, J.M.; Wang, S.; Jorgensen, J.L.; Lin, P. Multi-color flow cytometric immunophenotyping for detection of minimal residual disease in AML: Past, present and future. Bone Marrow Transplant. 2014, 49, 1129–1138. [Google Scholar] [CrossRef]
- Morrissette, J.J.; Bagg, A. Acute Myeloid Leukemia: Conventional Cytogenetics, FISH, and Moleculocentric Methodologies. Clin. Lab. Med. 2011, 31, 659–686. [Google Scholar] [CrossRef]
- Tschumper, R.C.; Asmann, Y.W.; Hossain, A.; Huddleston, P.; Wu, X.; Dispenzieri, A.; Eckloff, B.W.; Jelinek, D.F. Comprehensive Assessment of Potential Multiple Myeloma Immunoglobulin Heavy Chain V-D-J Intraclonal Variation Using Massively Parallel Pyrosequencing. Oncotarget 2012, 3, 502–513. [Google Scholar] [CrossRef]
- Hideshima, T.; Anderson, K.C. Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat. Rev. Cancer 2002, 2, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Wang, H.; Baladandayuthapani, V.; Lin, H.; He, J.; Jones, R.J.; Kuiatse, I.; Gu, N.; Wang, Z.; Ma, W.; et al. RNA Polymerase I Inhibition with CX-5461 as a Novel Therapeutic Strategy to Target MYC in Multiple Myeloma. Br. J. Haematol. 2017, 177, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Oken, M.M.; Harrington, D.; Bailey, R.J.; A Van Wier, S.; Henderson, K.J.; E Kay, N.; Van Ness, B.; Greipp, P.R.; Dewald, G.W. Deletions of chromosome 13 in multiple myeloma identified by interphase FISH usually denote large deletions of the q arm or monosomy. Leukemia 2001, 15, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Bullrich, F.; Fujii, H.; Calin, G.A.; Mabuchi, H.; Negrini, M.; Pekarsky, Y.; Rassenti, L.; Alder, H.; Reed, J.C.; Keating, M.J.; et al. Characterization of the 13q14 tumor suppressor locus in CLL: Identification of ALT1, an alternative splice variant of the LEU2 gene. Cancer Res. 2001, 61, 6640. [Google Scholar] [PubMed]
- Shaughnessy, J.; Erming, T.; Jeffrey, S.; Klaus, B.; Reid, L.; Ashraf, B.; Christopher, M.; Guido, T.; Joshua, E.; Bart, B. High Incidence of Chromosome 13 Deletion in Multiple Myeloma Detected by Multiprobe Interphase Fish. Blood 2000, 4, 1505–1511. [Google Scholar] [CrossRef]
- Bullrich, F.; Veronese, M.; Kitada, S.; Jurlander, J.; Caligiuri, M.; Reed, J.; Croce, C. Minimal region of loss at 13q14 in B-cell chronic lymphocytic leukemia. Blood 1996, 88, 3109–3115. [Google Scholar] [CrossRef]
- Childhood Acute Lymphoblastic Leukemia Treatment (PDQ®): Health Professional Version. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26389206 (accessed on 21 February 2020).
- Seibel, N.L. Treatment of Acute Lymphoblastic Leukemia in Children and Adolescents: Peaks and Pitfalls. Hematology 2008, 2008, 374–380. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Sancho, J.; Hancock, M.L.; Razzouk, B.I.; Ribeiro, R.C.; Rivera, G.K.; Rubnitz, J.; Sandlund, J.T.; Pui, C.-H.; Campana, D. Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood 2002, 100, 2399–2402. [Google Scholar] [CrossRef]
- Blunck, C.B.; Terra-Granado, E.; Noronha, E.P.; Wajnberg, G.; Passetti, F.; Pombo-De-Oliveira, M.S.; Emerenciano, M. CD9 predicts ETV6-RUNX1 in childhood B-cell precursor acute lymphoblastic leukemia. Hematol. Transfus. Cell Ther. 2019, 41, 205–211. [Google Scholar] [CrossRef]
- Van Delft, F.W.; Horsley, S.; Colman, S.; Anderson, K.; Bateman, C.; Kempski, H.; Zuna, J.; Eckert, C.; Saha, V.; Kearney, L.; et al. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 2011, 117, 6247–6254. [Google Scholar] [CrossRef][Green Version]
- Marshall, G.M.; Pozza, L.D.; Sutton, R.; Ng, A.; A De Groot-Kruseman, H.; Van Der Velden, V.H.; Venn, N.C.; Berg, H.V.D.; De Bont, E.S.J.M.; Egeler, R.M.; et al. High-risk childhood acute lymphoblastic leukemia in first remission treated with novel intensive chemotherapy and allogeneic transplantation. Leukemia 2013, 27, 1497–1503. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Young, M.; Wimalendra, M.; Tunstall, O. PO-0168 Leukaemia Cutis: An Unusual Paediatric Presentation Of Acute Lymphoblastic Leukaemia. Arch. Dis. Child. 2014, 99, 1030–1043. [Google Scholar] [CrossRef][Green Version]
- Borkhardt, A.; Cazzaniga, G.; Viehmann, S.; Valsecchi, M.G.; Ludwig, W.D.; Mangioni, S.; Schrappe, M.; Riehm, H.; Lampert, F.; Basso, G.; et al. Incidence and Clinical Relevance of Tel/Aml1 Fusion Genes in Children with Acute Lymphoblastic Leukemia Enrolled in the German and Italian Multicenter Therapy Trials. Blood 1997, 2, 571–577. [Google Scholar] [CrossRef]
- Mosad, E.; Hamed, H.; Bakry, R.; Ezz-Eldin, A.M.; Khalifa, N.M. Persistence of TEL-AML1 fusion gene as minimal residual disease has no additive prognostic value in CD 10 positive B-acute lymphoblastic leukemia: A FISH study. J. Hematol. Oncol. 2008, 1, 17. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Link, M.P.; Shuster, J.J.; Carroll, A.J.; Hakami, N.; Frankel, L.S.; Pullen, D.J.; Cleary, M.L. Frequency and Prognostic Significance of Hrx Rearrangements in Infant Acute Lymphoblastic Leukemia: A Pediatric Oncology Group Study. Blood 1994, 2, 570–573. [Google Scholar] [CrossRef]
- Secker-Walker, L.M.; Moorman, A.V.; Bain, B.J.; Mehta, A.B. Secondary acute leukemia and myelodysplastic syndrome with 11q23 abnormalities. Leukemia 1998, 12, 840–844. [Google Scholar] [CrossRef]
- Rowley, J.D. The Critical Role of Chromosome Translocation in Human Leukemias. Annu. Rev. Genet. 1998, 32, 495–519. [Google Scholar] [CrossRef]
- Naumovski, L.; Morgan, R.; Hecht, F.; Link, M.P.; E Glader, B.; Smith, S.D. Philadelphia chromosome-positive acute lymphoblastic leukemia cell lines without classical breakpoint cluster region rearrangement. Cancer Res. 1988, 48, 2876. [Google Scholar]
- Harata, M.; Soda, Y.; Tani, K.; Ooi, J.; Takizawa, T.; Chen, M.; Bai, Y.; Izawa, K.; Kobayashi, S.; Tomonari, A.; et al. CD19-targeting liposomes containing imatinib efficiently kill Philadelphia chromosome-positive acute lymphoblastic leukemia cells. Blood 2004, 104, 1442–1449. [Google Scholar] [CrossRef]
- Groffen, J.; Stephenson, J.R.; Heisterkamp, N.; De Klein, A.; Bartram, C.R.; Grosveld, G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984, 36, 93–99. [Google Scholar] [CrossRef]
- Vij, R.; Mazumder, A.; Klinger, M.; O’Dea, D.; Paasch, J.; Martin, T.; Weng, L.; Park, J.; Fiala, M.; Faham, M.; et al. Deep Sequencing Reveals Myeloma Cells in Peripheral Blood in Majority of Multiple Myeloma Patients. Clin. Lymphoma Myeloma Leuk. 2014, 14, 131–1390. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Rajkumar, S.V.; Greipp, P.R.; Witzig, T.E. Cell proliferation of myeloma plasma cells: Comparison of the blood and marrow compartments. Am. J. Hematol. 2004, 77, 7–11. [Google Scholar]
- Aghvami, S.A.; Opathalage, A.; Zhang, Z.; Ludwig, M.; Heymann, M.; Norton, M.; Wilkins, N.; Fraden, S. Rapid prototyping of cyclic olefin copolymer (COC) microfluidic devices. Sens. Actuators B Chem. 2017, 247, 940–949. [Google Scholar] [CrossRef]
- Jackson, J.M.; Witek, M.A.; Hupert, M.L.; Brady, C.; Pullagurla, S.; Kamande, J.; Aufforth, R.D.; Tignanelli, C.; Torphy, R.J.; Yeh, J.J.; et al. UV activation of polymeric high aspect ratio microstructures: Ramifications in antibody surface loading for circulating tumor cell selection. Lab Chip 2014, 14, 106–117. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Cells (Antigen Used for Selection) | Target Cells (mL−1) | Non-Aberrant Cells (mL−1) | Cell Diameter (µm) | Reference |
|---|---|---|---|---|
| CLCs (AML, CD33, CD34, CD117) | 11–2684 | 10–2450 | 11–16 | [4] |
| CLCs (B-ALL, CD19) | 40–840 | 400–2050 | 6–12 | this work |
| CPCs (CD138) | 10–5900 | 43–1875 | 12–16 | [3] |
| CTCs in epithelial tumors (EpCAM) | 1–800 | 3–10 | 10–23 | [35] |
| Flow Rate (μL/min) | Shear Rate (1/s) | Shear Stress (dynes/cm2) |
|---|---|---|
| 1 | 6042 | 53.8 |
| 3 | 18,206 | 162.0 |
| 5 | 30,454 | 271.0 |
| 10 | 63,750 | 567.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
M. Weerakoon-Ratnayake, K.; Vaidyanathan, S.; Larkey, N.; Dathathreya, K.; Hu, M.; Jose, J.; Mog, S.; August, K.; K. Godwin, A.; L. Hupert, M.; et al. Microfluidic Device for On-Chip Immunophenotyping and Cytogenetic Analysis of Rare Biological Cells. Cells 2020, 9, 519. https://doi.org/10.3390/cells9020519
M. Weerakoon-Ratnayake K, Vaidyanathan S, Larkey N, Dathathreya K, Hu M, Jose J, Mog S, August K, K. Godwin A, L. Hupert M, et al. Microfluidic Device for On-Chip Immunophenotyping and Cytogenetic Analysis of Rare Biological Cells. Cells. 2020; 9(2):519. https://doi.org/10.3390/cells9020519
Chicago/Turabian StyleM. Weerakoon-Ratnayake, Kumuditha, Swarnagowri Vaidyanathan, Nicholas Larkey, Kavya Dathathreya, Mengjia Hu, Jilsha Jose, Shalee Mog, Keith August, Andrew K. Godwin, Mateusz L. Hupert, and et al. 2020. "Microfluidic Device for On-Chip Immunophenotyping and Cytogenetic Analysis of Rare Biological Cells" Cells 9, no. 2: 519. https://doi.org/10.3390/cells9020519
APA StyleM. Weerakoon-Ratnayake, K., Vaidyanathan, S., Larkey, N., Dathathreya, K., Hu, M., Jose, J., Mog, S., August, K., K. Godwin, A., L. Hupert, M., A. Witek, M., & A. Soper, S. (2020). Microfluidic Device for On-Chip Immunophenotyping and Cytogenetic Analysis of Rare Biological Cells. Cells, 9(2), 519. https://doi.org/10.3390/cells9020519