Protein Translocation Acquires Substrate Selectivity Through ER Stress-Induced Reassembly of Translocon Auxiliary Components


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Molecular Biology
2.3. Cell Culture Analyses
2.4. Biochemistry
3. Results
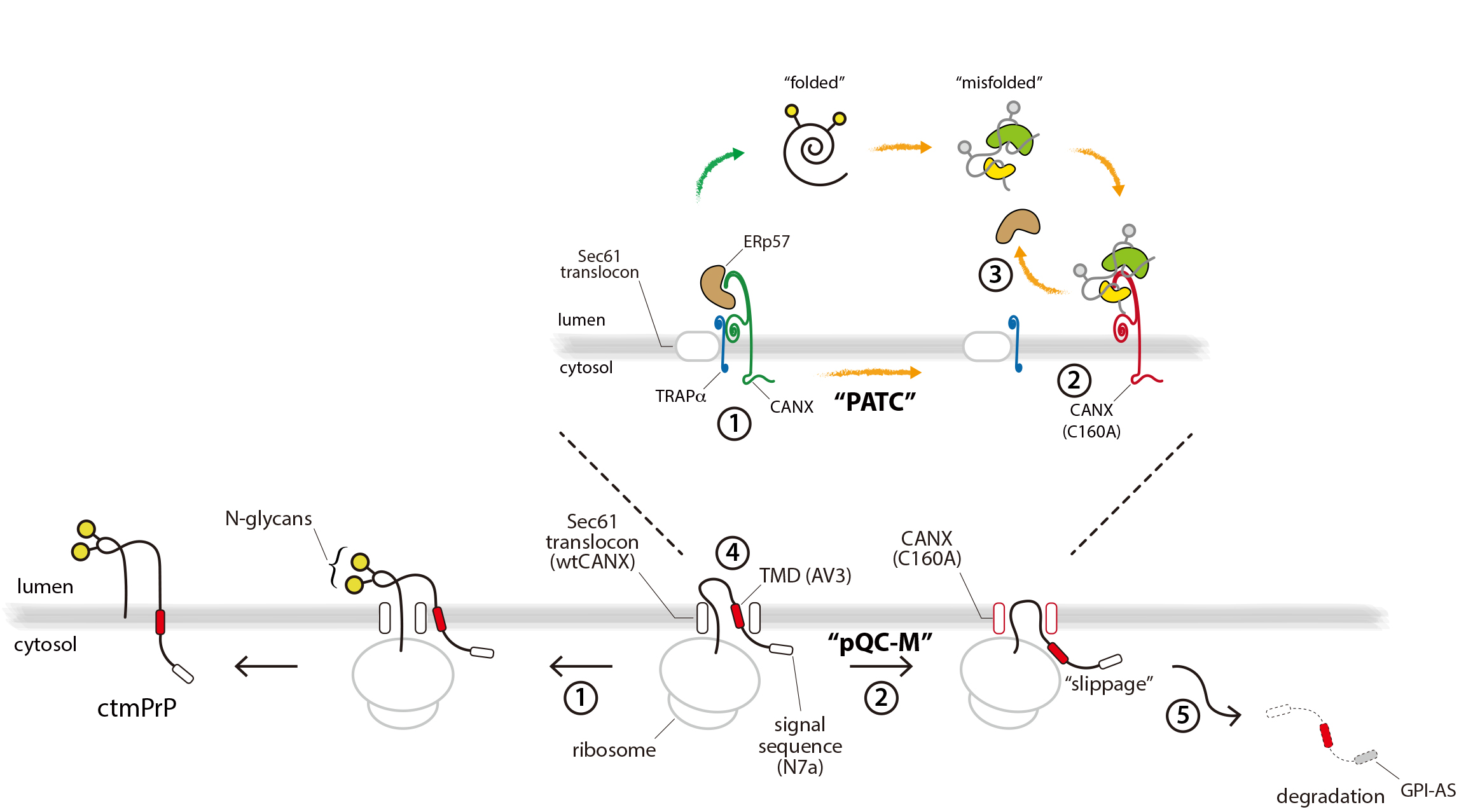
3.1. TRAPα Is Not the Key Element in PrP Synthesis
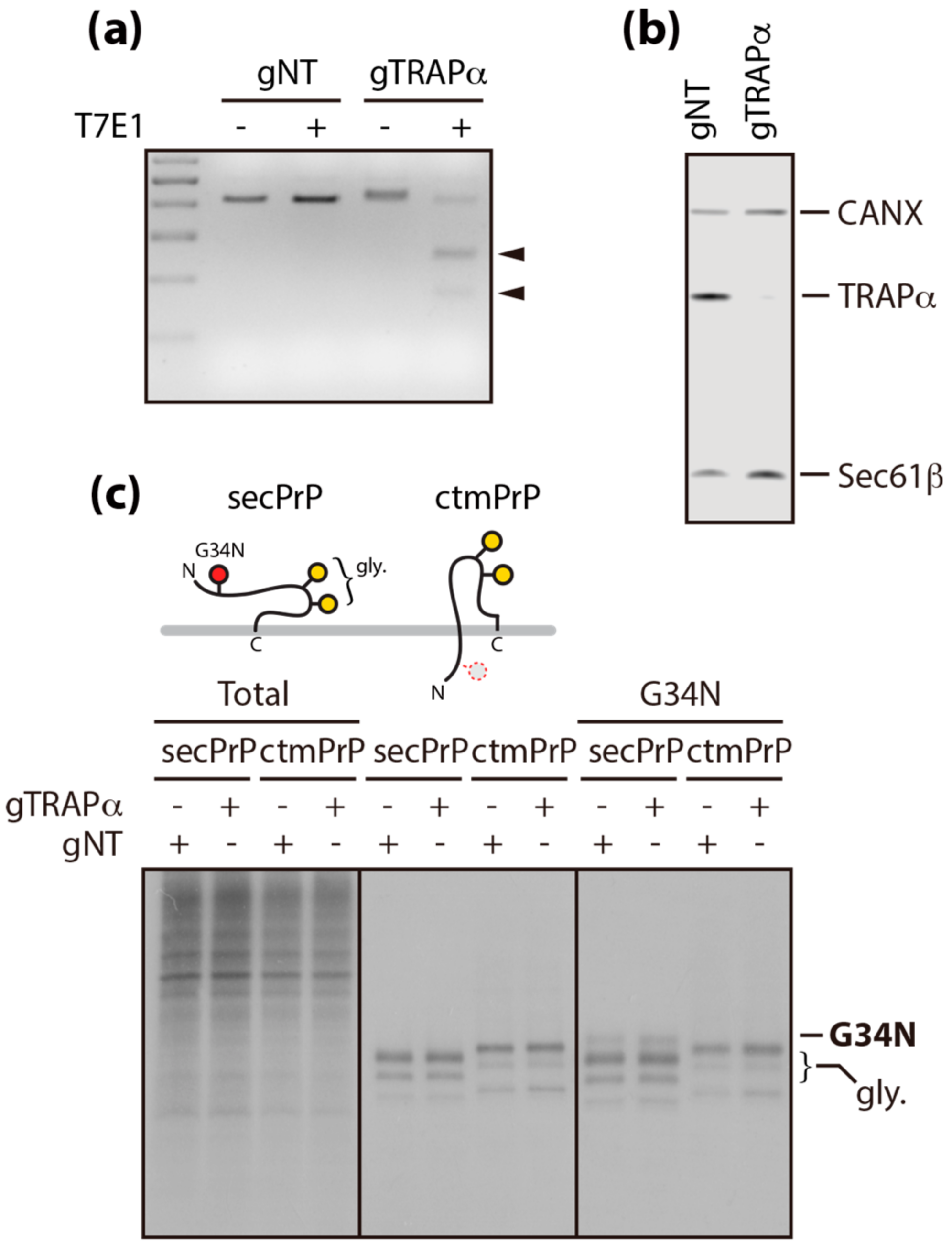
3.2. Calnexin is a Component of the TRAP Complex
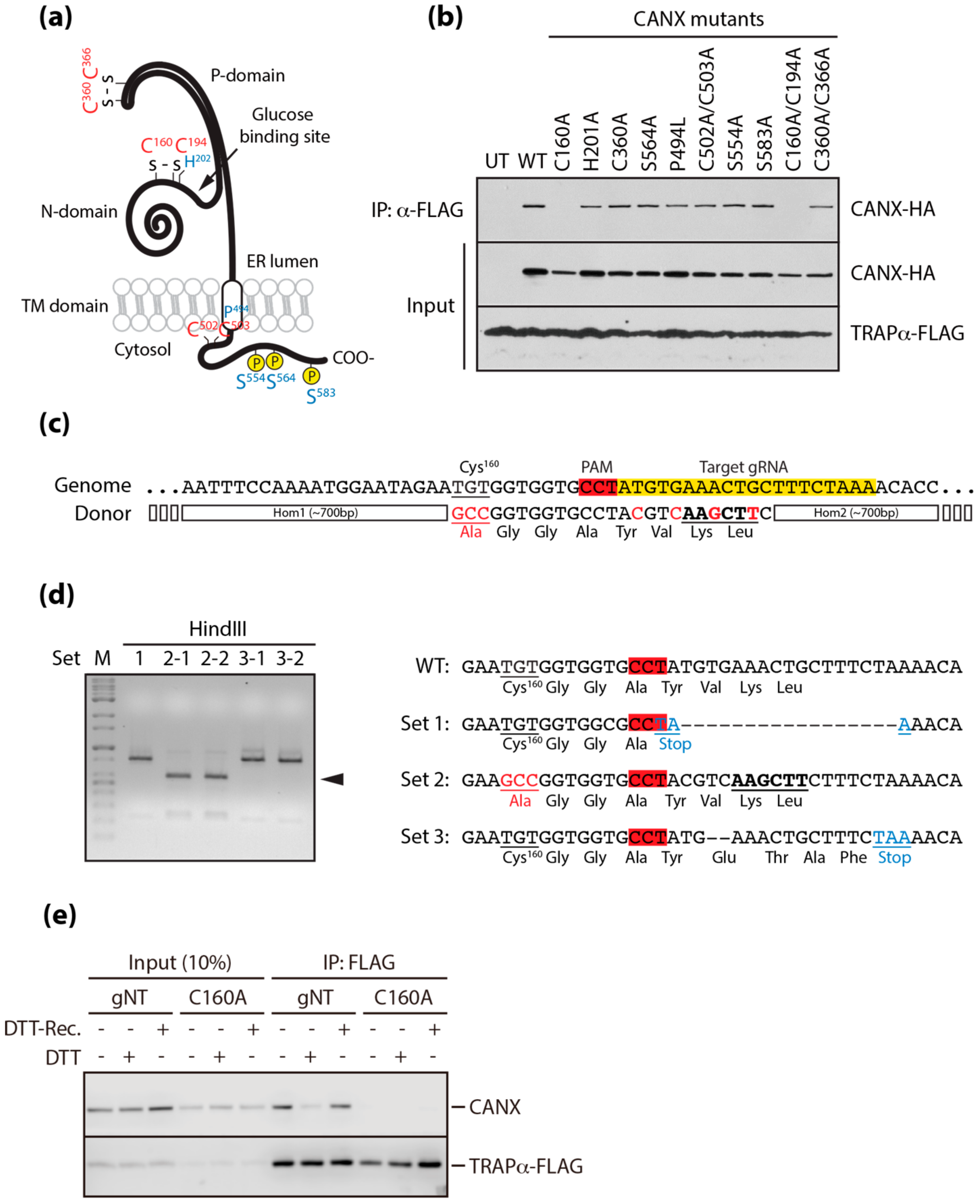
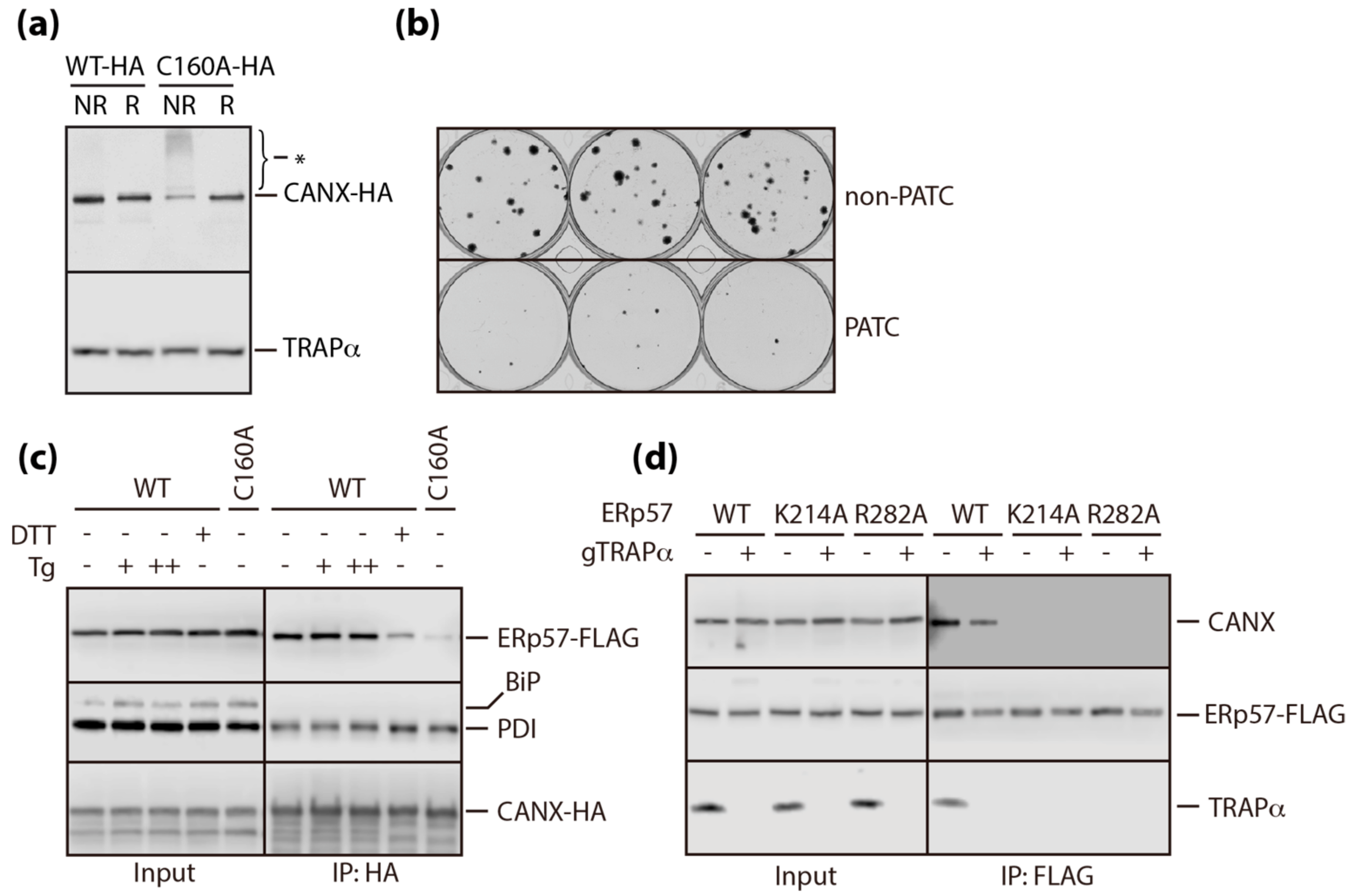
3.3. A Conserved Disulfide Bridge within CANX Provides the Interaction with TRAPα
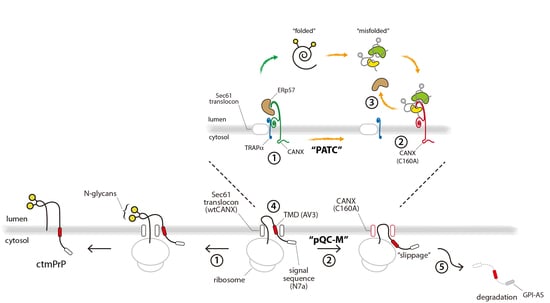
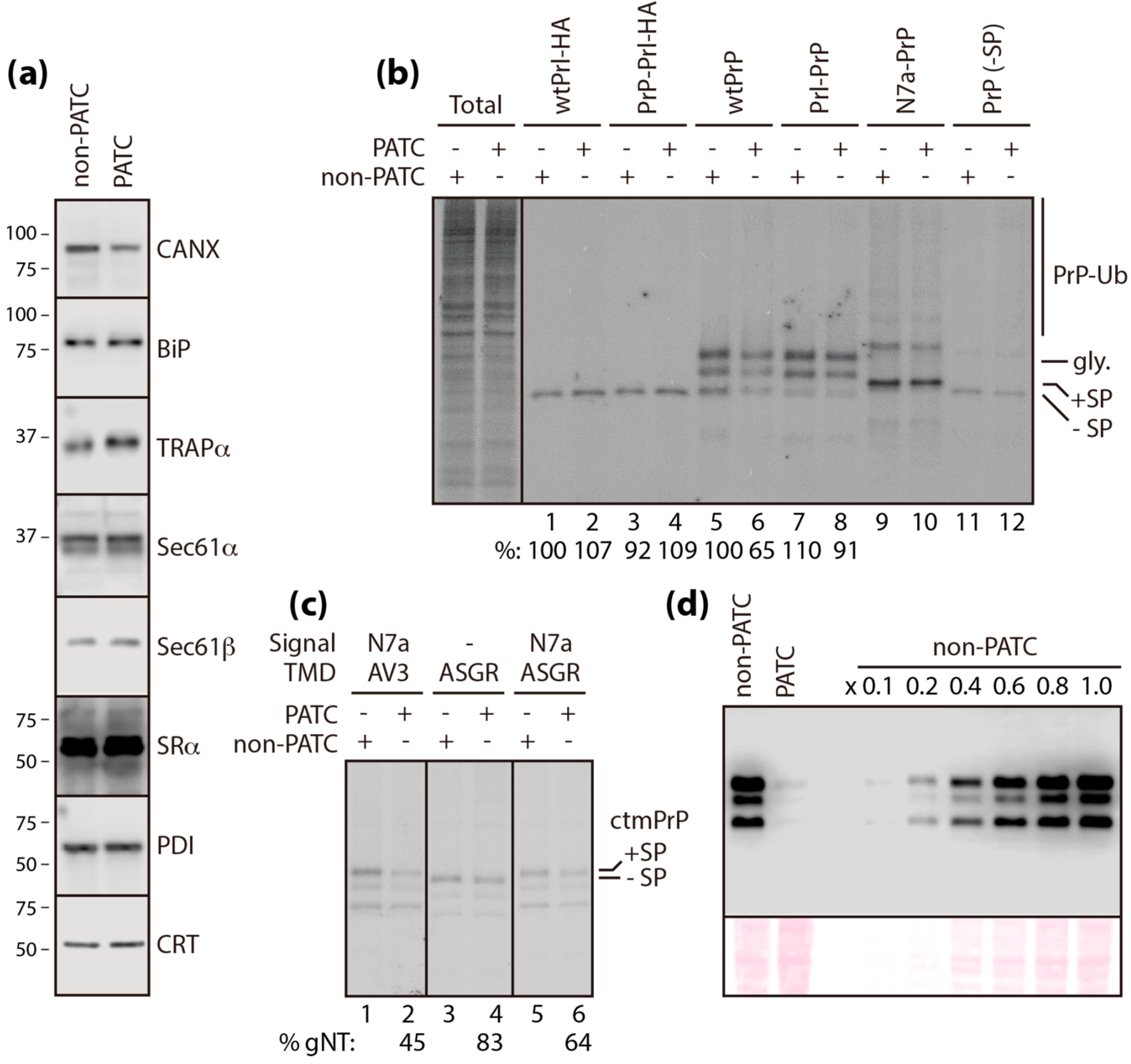
3.4. PATC Interferes with ctmPrP Synthesis
3.5. PATC Disrupts the Interaction of ERp57 with CANX
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wickner, W.; Schekman, R. Protein translocation across biological membranes. Science 2005, 310, 1452–1456. [Google Scholar] [CrossRef]
- Osborne, A.R.; Rapoport, T.A.; van den Berg, B. Protein translocation by the Sec61/SecY channel. Annu. Rev. Cell Dev. Biol. 2005, 21, 529–550. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, R.; Eyrisch, S.; Ahmad, M.; Helms, V. Protein translocation across the ER membrane. Biochim. Et Biophys. Acta 2011, 1808, 912–924. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Mitra, D.; Salerno, J.R.; Hegde, R.S. Signal sequences control gating of the protein translocation channel in a substrate-specific manner. Dev. Cell 2002, 2, 207–217. [Google Scholar] [CrossRef]
- Kim, S.J.; Rahbar, R.; Hegde, R.S. Combinatorial control of prion protein biogenesis by the signal sequence and transmembrane domain. J. Biol. Chem. 2001, 276, 26132–26140. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Rane, N.S.; Kim, S.J.; Garrison, J.L.; Taunton, J.; Hegde, R.S. Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell 2006, 127, 999–1013. [Google Scholar] [CrossRef]
- Lee, Y.; Eum, H.; Lee, D.; Lee, S.; Song, Y.; Kang, S.W. Mutant-selective topologic conversion facilitates selective degradation of a pathogenic prion isoform. Cell Death Differ. 2020, 27, 284–296. [Google Scholar] [CrossRef]
- Nyathi, Y.; Wilkinson, B.M.; Pool, M.R. Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim. Et Biophys. Acta 2013, 1833, 2392–2402. [Google Scholar] [CrossRef]
- Hegde, R.S.; Kang, S.W. The concept of translocational regulation. J. Cell Biol. 2008, 182, 225–232. [Google Scholar] [CrossRef]
- Fons, R.D.; Bogert, B.A.; Hegde, R.S. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J. Cell Biol. 2003, 160, 529–539. [Google Scholar] [CrossRef]
- Conti, B.J.; Devaraneni, P.K.; Yang, Z.; David, L.L.; Skach, W.R. Cotranslational stabilization of Sec62/63 within the ER Sec61 translocon is controlled by distinct substrate-driven translocation events. Mol. Cell 2015, 58, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Plumb, R.; Zhang, Z.R.; Appathurai, S.; Mariappan, M. A functional link between the co-translational protein translocation pathway and the UPR. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Noack, J.; Bergmann, T.J.; Cebollero Presmanes, E.; Pisoni, G.B.; Fasana, E.; Fregno, I.; Galli, C.; Loi, M.; Solda, T.; et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 2016, 18, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Menetret, J.F.; Neuhof, A.; Morgan, D.G.; Plath, K.; Radermacher, M.; Rapoport, T.A.; Akey, C.W. The structure of ribosome-channel complexes engaged in protein translocation. Mol. Cell 2000, 6, 1219–1232. [Google Scholar] [CrossRef]
- Pfeffer, S.; Dudek, J.; Schaffer, M.; Ng, B.G.; Albert, S.; Plitzko, J.M.; Baumeister, W.; Zimmermann, R.; Freeze, H.H.; Engel, B.D.; et al. Dissecting the molecular organization of the translocon-associated protein complex. Nat. Commun. 2017, 8, 14516. [Google Scholar] [CrossRef]
- Voorhees, R.M.; Fernandez, I.S.; Scheres, S.H.; Hegde, R.S. Structure of the Mammalian Ribosome-Sec61 Complex to 3.4 A Resolution. Cell 2014, 1016. [Google Scholar] [CrossRef]
- Sommer, N.; Junne, T.; Kalies, K.U.; Spiess, M.; Hartmann, E. TRAP assists membrane protein topogenesis at the mammalian ER membrane. Biochim. Et Biophys. Acta 2013, 1833, 3104–3111. [Google Scholar] [CrossRef]
- Snapp, E.L.; Sharma, A.; Lippincott-Schwartz, J.; Hegde, R.S. Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proc. Natl. Acad. Sci. USA 2006, 103, 6536–6541. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Ashok, A.; Hegde, R.S. Retrotranslocation of prion proteins from the endoplasmic reticulum by preventing GPI signal transamidation. Mol. Biol. Cell 2008, 19, 3463–3476. [Google Scholar] [CrossRef]
- Emerman, A.B.; Zhang, Z.R.; Chakrabarti, O.; Hegde, R.S. Compartment-restricted biotinylation reveals novel features of prion protein metabolism in vivo. Mol. Biol. Cell 2010, 21, 4325–4337. [Google Scholar] [CrossRef] [PubMed]
- Chevet, E.; Smirle, J.; Cameron, P.H.; Thomas, D.Y.; Bergeron, J.J. Calnexin phosphorylation: linking cytoplasmic signalling to endoplasmic reticulum lumenal functions. Semin. Cell Dev. Biol. 2010, 21, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Coe, H.; Schneider, J.D.; Dabrowska, M.; Groenendyk, J.; Jung, J.; Michalak, M. Role of cysteine amino acid residues in calnexin. Mol. Cell Biochem. 2012, 359, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Lakkaraju, A.K.; Abrami, L.; Lemmin, T.; Blaskovic, S.; Kunz, B.; Kihara, A.; Dal Peraro, M.; van der Goot, F.G. Palmitoylated calnexin is a key component of the ribosome-translocon complex. Embo J. 2012, 31, 1823–1835. [Google Scholar] [CrossRef]
- Schrag, J.D.; Bergeron, J.J.; Li, Y.; Borisova, S.; Hahn, M.; Thomas, D.Y.; Cygler, M. The Structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol. Cell 2001, 8, 633–644. [Google Scholar] [CrossRef]
- Ware, F.E.; Vassilakos, A.; Peterson, P.A.; Jackson, M.R.; Lehrman, M.A.; Williams, D.B. The molecular chaperone calnexin binds Glc1Man9GlcNAc2 oligosaccharide as an initial step in recognizing unfolded glycoproteins. J. Biol. Chem. 1995, 270, 4697–4704. [Google Scholar] [CrossRef]
- Ou, W.J.; Cameron, P.H.; Thomas, D.Y.; Bergeron, J.J. Association of folding intermediates of glycoproteins with calnexin during protein maturation. Nature 1993, 364, 771–776. [Google Scholar] [CrossRef]
- Lingappa, V.R.; Rutkowski, D.T.; Hegde, R.S.; Andersen, O.S. Conformational control through translocational regulation: a new view of secretory and membrane protein folding. Bioessays: News Rev. Mol. Cell. Dev. Biol. 2002, 24, 741–748. [Google Scholar] [CrossRef]
- Zhang, Y.; Kozlov, G.; Pocanschi, C.L.; Brockmeier, U.; Ireland, B.S.; Maattanen, P.; Howe, C.; Elliott, T.; Gehring, K.; Williams, D.B. ERp57 does not require interactions with calnexin and calreticulin to promote assembly of class I histocompatibility molecules, and it enhances peptide loading independently of its redox activity. J. Biol. Chem. 2009, 284, 10160–10173. [Google Scholar] [CrossRef]
- Kozlov, G.; Maattanen, P.; Schrag, J.D.; Pollock, S.; Cygler, M.; Nagar, B.; Thomas, D.Y.; Gehring, K. Crystal structure of the bb’ domains of the protein disulfide isomerase ERp57. Structure 2006, 14, 1331–1339. [Google Scholar] [CrossRef]
- Elliott, J.G.; Oliver, J.D.; High, S. The thiol-dependent reductase ERp57 interacts specifically with N-glycosylated integral membrane proteins. J. Biol. Chem. 1997, 272, 13849–13855. [Google Scholar] [CrossRef] [PubMed]
- Swanton, E.; High, S.; Woodman, P. Role of calnexin in the glycan-independent quality control of proteolipid protein. Embo J. 2003, 22, 2948–2958. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.S.; Mastrianni, J.A.; Scott, M.R.; DeFea, K.A.; Tremblay, P.; Torchia, M.; DeArmond, S.J.; Prusiner, S.B.; Lingappa, V.R. A transmembrane form of the prion protein in neurodegenerative disease. Science 1998, 279, 827–834. [Google Scholar] [CrossRef]
- Chakrabarti, O.; Hegde, R.S. Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell 2009, 137, 1136–1147. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Shin, Y.; Kim, K.; Song, Y.; Kim, Y.; Kang, S.-W. Protein Translocation Acquires Substrate Selectivity Through ER Stress-Induced Reassembly of Translocon Auxiliary Components. Cells 2020, 9, 518. https://doi.org/10.3390/cells9020518
Lee S, Shin Y, Kim K, Song Y, Kim Y, Kang S-W. Protein Translocation Acquires Substrate Selectivity Through ER Stress-Induced Reassembly of Translocon Auxiliary Components. Cells. 2020; 9(2):518. https://doi.org/10.3390/cells9020518
Chicago/Turabian StyleLee, Sohee, Yejin Shin, Kyunggon Kim, Youngsup Song, Yongsub Kim, and Sang-Wook Kang. 2020. "Protein Translocation Acquires Substrate Selectivity Through ER Stress-Induced Reassembly of Translocon Auxiliary Components" Cells 9, no. 2: 518. https://doi.org/10.3390/cells9020518
APA StyleLee, S., Shin, Y., Kim, K., Song, Y., Kim, Y., & Kang, S.-W. (2020). Protein Translocation Acquires Substrate Selectivity Through ER Stress-Induced Reassembly of Translocon Auxiliary Components. Cells, 9(2), 518. https://doi.org/10.3390/cells9020518