Arteriogenesis of the Spinal Cord—The Network Challenge

 , ,
, , {kind=link}
Abstract
1. Introduction
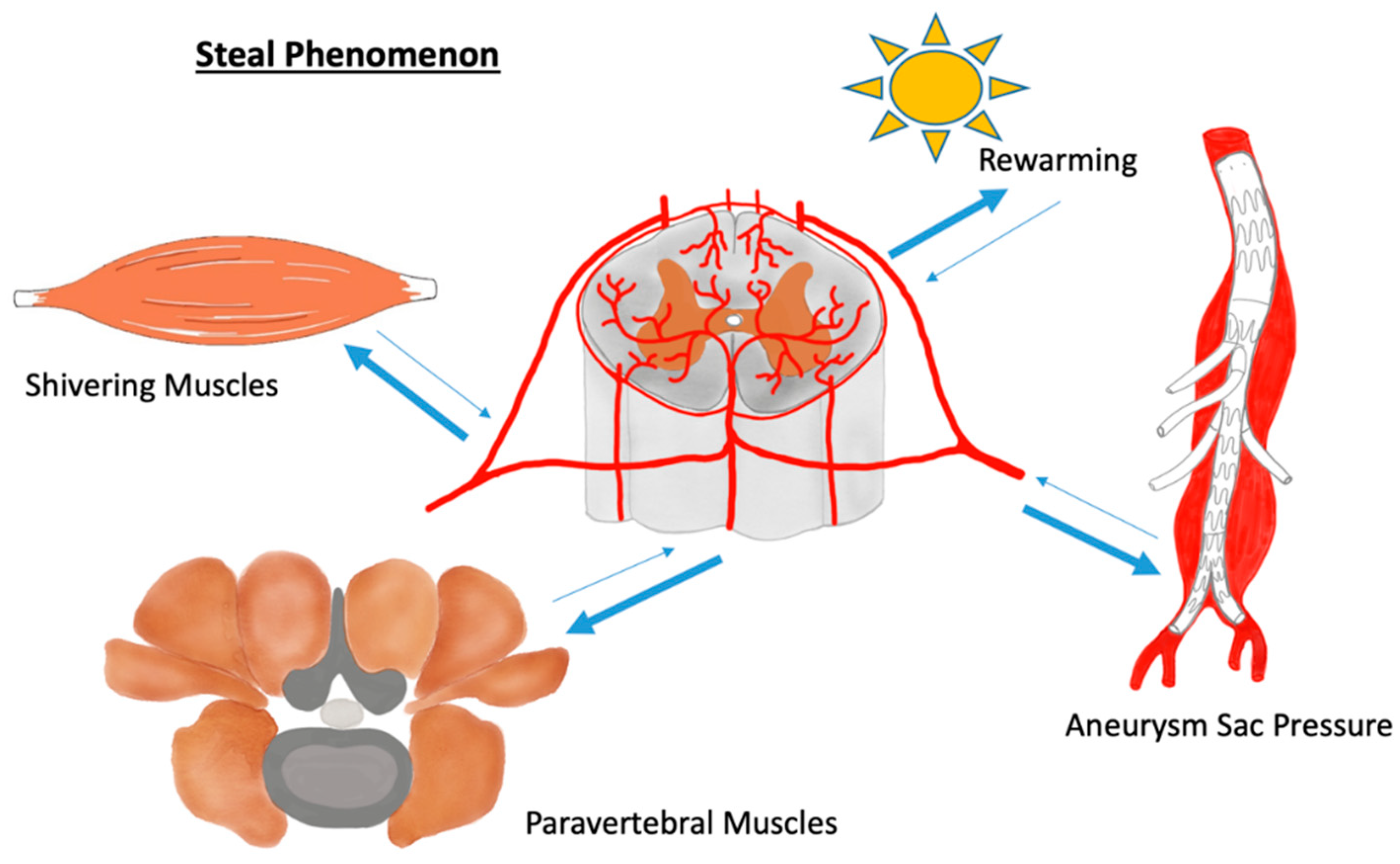
2. Blood Supply of the Spinal Cord
3. Stress-Related Changes in Blood Vessels
4. Signaling Pathways
5. Conclusions
Funding
Conflicts of Interest
References
- Coselli, J.S.; LeMaire, S.A.; Preventza, O.; de la Cruz, K.I.; Cooley, D.A.; Price, M.D. Outcomes of 3309 thoracoabdominal aortic aneurysm repairs. J. Thorac. Cardiovasc. Surg. 2016, 151, 1323–1337. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.S. Thoraco-abdominal and abdominal aortic aneurysms involving renal, superior mesenteric, celiac arteries. Ann. Surg. 1974, 179, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Svensson, L.G.; Crawford, E.S. Aortic dissection and aortic aneurysm surgery: Clinical observations, experimental investigations, and statistical analyses. Part III. Curr. Probl. Surg. 1993, 30, 1–163. [Google Scholar] [CrossRef]
- Greenberg, R.K.; Lu, Q.; Roselli, E.E.; Svensson, L.G.; Moon, M.C.; Hernandez, A.V. Contemporary analysis of descending thoracic and thoracoabdominal aneurysm repair: A comparison of endovascular and open techniques. Circulation 2008, 118, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Fattori, R.; Cao, P.; De Rango, P.; Czerny, M.; Evangelista, A.; Nienaber, C.; Rousseau, H.; Schepens, M. Interdisciplinary expert consensus document on management of type B aortic dissection. J. Am. Coll. Cardiol. 2013, 61, 1661–1678. [Google Scholar] [CrossRef] [PubMed]
- Luebke, T.; Brunkwall, J. Outcome of patients with open and endovascular repair in acute complicated type B aortic dissection: A systematic review and meta-analysis of case series and comparative studies. J. Cardiovasc. Surg. (Torino) 2010, 51, 613–632. [Google Scholar]
- Riambau, V.; Böckler, D.; Brunkwall, J. Editor’s Choice - Management of Descending Thoracic Aorta Diseases: Clinical Practice Guidelines of the European Society for Vascular Surgery (ESVS). Eur. J. Vasc. Endovasc. Surg. 2017, 53, 4–52. [Google Scholar] [CrossRef]
- Wortmann, M.; Böckler, D.; Geisbüsch, P. Perioperative cerebrospinal fluid drainage for the prevention of spinal ischemia after endovascular aortic repair. Gefasschirurgie 2017, 22, 35–40. [Google Scholar] [CrossRef]
- Lettinga-van de Poll, T.; Schurink, G.W.; De Haan, M.W.; Verbruggen, J.P.; Jacobs, M.J. Endovascular treatment of traumatic rupture of the thoracic aorta. Br. J. Surg. 2007, 94, 525–533. [Google Scholar] [CrossRef]
- Cheng, D.; Martin, J.; Shennib, H. Endovascular aortic repair versus open surgical repair for descending thoracic aortic disease a systematic review and meta-analysis of comparative studies. J. Am. Coll. Cardiol. 2010, 55, 986–1001. [Google Scholar] [CrossRef]
- Hiramoto, J.S.; Fernandez, C.; Gasper, W.; Vartanian, S.; Reilly, L.; Chuter, T. Lower extremity weakness is associated with elevated blood and cerebrospinal fluid glucose levels following multibranched endovascular aortic aneurysm repair. J. Vasc. Surg. 2017, 65, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Peppelenbosch, N.; Cuypers, P.W.; Vahl, A.C.; Vermassen, F.; Buth, J. Emergency endovascular treatment for ruptured abdominal aortic aneurysm and the risk of spinal cord ischemia. J. Vasc. Surg. 2005, 42, 608–614. [Google Scholar] [CrossRef]
- Acher, C.W.; Wynn, M.M.; Mell, M.W.; Tefera, G.; Hoch, J.R. A quantitative assessment of the impact of intercostal artery reimplantation on paralysis risk in thoracoabdominal aortic aneurysm repair. Ann. Surg. 2008, 248, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Acher, C. It is not just assisted circulation, hypothermic arrest, or clamp and sew. J. Thorac. Cardiovasc. Surg. 2010, 140, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Erbel, R.; Aboyans, V.; Boileau, C. ESC Committee for Practice Guidelines. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2873–2926. [Google Scholar] [PubMed]
- Jacobs, M.J.; Meylaerts, S.A.; de Haan, P.; de Mol, B.A.; Kalkman, C.J. Strategies to prevent neurologic deficit based on motor-evoked potentials in type I and II thoracoabdominal aortic aneurysm repair. J. Vasc. Surg. 1999, 29, 48–57. [Google Scholar] [CrossRef]
- Etz, C.D.; Luehr, M.; Kari, F.A.; Bodian, C.A.; Smego, D.; Plestis, K.A. Paraplegia after extensive thoracic and thoracoabdominal aortic aneurysm repair: Does critical spinal cord ischemia occur postoperatively? J. Thorac. Cardiovasc. Surg. 2008, 135, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Pouw, M.H.; Hosman, A.J.; van Middendorp, J.J.; Verbeek, M.M.; Vos, P.E.; van de Meent, H. Biomarkers in spinal cord injury. Spinal. Cord. 2009, 47, 519–525. [Google Scholar] [CrossRef]
- Winnerkvist, A.; Anderson, R.E.; Hansson, L.O.; Rosengren, L.; Estrera, A.E.; Huynh, T.T. Multilevel somatosensory evoked potentials and cerebrospinal proteins: Indicators of spinal cord injury in thoracoabdominal aortic aneurysm surgery. Eur J. Cardiothorac. Surg. 2007, 31, 637–642. [Google Scholar] [CrossRef]
- Khaladj, N.; Teebken, O.E.; Hagl, C.; Wilhelmi, M.H.; Tschan, C.; Weissenborn, K. The role of cerebrospinal fluid S100 and lactate to predict clinically evident spinal cord ischaemia in thoraco-abdominal aortic surgery. Eur. J. Vasc. Endovasc. Surg. 2008, 36, 11–19. [Google Scholar] [CrossRef]
- Lases, E.C.; Schepens, M.A.; Haas, F.J.; Aarts, L.P.; ter Beek, H.T.; van Dongen, E.P. Clinical prospective study of biochemical markers and evoked potentials for identifying adverse neurological outcome after thoracic and thoracoabdominal aortic aneurysm surgery. Br. J. Anaesth. 2005, 95, 651–661. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Harky, A.; Fok, M.; Fraser, H.; Howard, C.; Rimmer, L.; Bashir, M. Could Cerebrospinal Fluid Biomarkers Offer Better Predictive Value for Spinal Cord Ischaemia Than Current Neuromonitoring Techniques During Thoracoabdominal Aortic Aneurysm Repair - A Systematic Review. Braz. J. Cardiovasc. Surg. 2019, 34, 464–471. [Google Scholar] [CrossRef]
- Fuchs, E.; Weber, K. Intermediate filaments: Structure, dynamics, function, and disease. Annu. Rev. Biochem. 1994, 63, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Eng, L.F.; Ghirnikar, R.S.; Lee, Y.L. Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000). Neurochem. Res. 2000, 25, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.E.; Winnerkvist, A.; Hansson, L.O.; Nilsson, O.; Rosengren, L.; Settergren, G. Biochemical markers of cerebrospinal ischemia after repair of aneurysms of the descending and thoracoabdominal aorta. J. Cardiothorac. Vasc. Anesth. 2003, 17, 598–603. [Google Scholar] [CrossRef]
- Backes, W.H.; Nijenhuis, R.J.; Mess, W.H. Magnetic resonance angiography of collateral blood supply to spinal cord in thoracic and thoracoabdominal aortic aneurysm patients. J. Vasc. Surg. 2008, 48, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Nijenhuis, R.J.; Backes, W.H. Optimal preopera- tive imaging of spinal cord blood supply. AJNR Am. J. Neuroradiol. 2009, 30, 38–39. [Google Scholar] [CrossRef]
- Lazorthes, G.; Poulhes, J.; Bastide, G.; Roulleau, J.; Chancholle, A.R. Research on the arterial vascularization of the medulla; applications to medullary pathology. Bull. Acad. Natl. Med. 1957, 141, 464–477. [Google Scholar]
- Lazorthes, G.; Poulhes, J.; Bastide, G.; Roulleau, J.; Chancholle, A.R. Arterial vascularization of the spine; anatomic research and applications in pathology of the spinal cord and aorta. Neurochirurgie 1958, 4, 3–19. [Google Scholar]
- Lazorthes, G.; Gouaze, A.; Zadeh, J.O.; Santini, J.J.; Lazorthes, Y.; Burdin, P. Arterial vascularization of the spinal cord. J. Neurosurg. 1971, 35, 253–262. [Google Scholar] [CrossRef]
- Adamkiewicz, A. Die Blutgefäße des menschlichen Rückenmarks. II. Teil. Die Gefäße der Rückenmarksoberfläche; Sitz der Akad Wiss: Berlin, Germany, 1882; pp. 101–130. [Google Scholar]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D. Neuroscience, 2nd ed.; Sinauer Associates: Oxford, MS, USA, 2001; pp. 145–165. [Google Scholar]
- Griepp, R.B.; Griepp, E.B. Spinal cord perfusion and protection during descending thoracic and thoracoabdominal aortic surgery: The collateral network concept. Ann. Thorac. Surg. 2007, 83, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Etz, C.D.; Kari, F.A.; Mueller, C.S. The collateral network concept: A reassessment of the anatomy of spinal cord perfusion. J. Thorac. Cardiovasc. Surg. 2011, 141, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Meffert, P.; Bischoff, M.S.; Brenner, R.; Siepe, M.; Beyersdorf, F.; Kari, F.A. Significance and function of different spinal collateral compartments following thoracic aortic surgery: Immediate versus long-term flow compensation. Eur. J. Cardio-Thorac. Surg. 2014, 45, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Kari, F.A.; Wittmann, K.; Saravi, B.; Puttfarcken, L.; Krause, S.; Förster, K.; Maier, S.; Göbel, U.; Beyersdorf, F. Immediate Spinal Cord Collateral Blood Flow During Thoracic Aortic Procedures: The Role of Epidural Arcades. Semin. Thorac. Cardiovasc. Surg. 2016, 28, 378–387. [Google Scholar] [CrossRef]
- Strauch, J.T.; Spielvogel, D.; Lauten, A.; Zhang, N.; Shiang, H.; Weisz, D. Importance of extrasegmental vessels for spinal cord blood supply in a chronic porcine model. Eur J. Cardiothorac. Surg. 2003, 24, 817–824. [Google Scholar] [CrossRef]
- Hiatt, W.R. Pathophysiology of Peripheral Artery Disease, Intermittent Claudication, and Critical Limb Ischemia, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 223–230. [Google Scholar]
- Safi, H.J.; Miller, C.C.; Carr, C.; Iliopoulos, D.C.; Dorsay, D.A.; Baldwin, J.C. Importance of intercostal artery reattachment during thoracoabdominal aortic aneurysm repair. J. Vasc. Surg. 1998, 27, 58–66. [Google Scholar] [CrossRef]
- Coselli, J.S.; LeMaire, S.A.; de Figueiredo, L.P.; Kirby, R.P. Paraplegia after thoracoabdominal aortic aneurysm repair: Is dissection a risk factor? Ann. Thorac. Surg. 1997, 63, 28–36. [Google Scholar] [CrossRef]
- Williams, G.M.; Roseborough, G.S.; Webb, T.H.; Perler, B.A.; Krosnick, T. Preoperative selective intercostal angiography in patients undergoing thoracoabdominal aneurysm repair. J. Vasc. Surg. 2004, 39, 314–321. [Google Scholar] [CrossRef]
- Etz, C.D.; Homann, T.M.; Plestis, K.A.; Zhang, N.; Luehr, M.; Weisz, D.J. Spinal cord perfusion after extensive segmental artery sacrifice: Can paraplegia be prevented? Eur. J. Cardiothorac. Surg. 2007, 31, 643–648. [Google Scholar] [CrossRef][Green Version]
- Etz, C.D.; Di Luozzo, G.; Zoli, S.; Lazala, R.; Plestis, K.A.; Bodian, C.A. Direct spinal cord perfusion pressure monitoring in extensive distal aortic aneurysm repair. Ann. Thorac. Surg. 2009, 87, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Schurink, G.W.; De Haan, M.W.; Peppelenbosch, A.G. Spinal cord function monitoring during endovascular treatment of thoracoabdominal aneurysms: Implications for staged procedures. J. Cardiovasc. Surg. (Torino) 2013, 54, 117–124. [Google Scholar]
- Bischoff, M.S.; Di Luozzo, G.; Griepp, E.B.; Griepp, R.B. Spinal cord preservation in thoracoabdominal aneurysm repair. Perspect. Vasc. Surg. Endovasc. 2011, 23, 214–222. [Google Scholar] [CrossRef] [PubMed]
- De Haan, P.; Kalkman, C.J.; Jacobs, M.J. Pharmacologic neuroprotection in experimental spinal cord ischemia: A systematic review. J. Neurosurg. Anesth. 2001, 13, 3–12. [Google Scholar] [CrossRef]
- Heil, M.; Schaper, W. Influence of mechanical, cellular, and molecular factors on collateral artery growth (arteriogenesis). Circ. Res. 2004, 95, 449–458. [Google Scholar] [CrossRef]
- Heil, M.; Schaper, W. Pathophysiology of collateral development. Coron. Artery Dis. 2004, 15, 373–378. [Google Scholar] [CrossRef]
- Nijenhuis, R.J.; Jacobs, M.J.; Schurink, G.W. Magnetic resonance angiography and neuro- monitoring to assess spinal cord blood supply in thoracic and thoracoabdominal aortic aneurysm surgery. J. Vasc. Surg. 2007, 45, 71–77. [Google Scholar] [CrossRef]
- Potente, M.; Makinen, T. Vascular heterogeneity and specialization in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 477–494. [Google Scholar] [CrossRef]
- Kashiwagi, S.; Izumi, Y.; Gohongi, T. NO mediates mural cell recruitment and vessel morphogenesis in murine melanomas and tissue-engineered blood vessels. J. Clin. Investig. 2005, 115, 1816–1827. [Google Scholar] [CrossRef]
- Dar, A.; Domev, H.; Ben-Yosef, O. Multipotent vasculogenic pericytes from human pluripotent stem cells promote recovery of murine ischemic limb. Circulation 2011, 125, 87–99. [Google Scholar] [CrossRef]
- Grundmann, S.; Piek, J.J.; Pasterkamp, G.; Hoefer, I.E. Arteriogenesis: Basic mechanisms and therapeutic stimulation. Eur. J. Clin. Investig. 2007, 37, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Helisch, A.; Schaper, W. Arteriogenesis: The development and growth of collateral arteries. Microcirculation 2003, 10, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Etz, C.D.; Kari, F.A.; Mueller, C.S.; Brenner, R.M.; Lin, H.M.; Griepp, R.B. The collateral network concept: Remodeling of the arterial collateral network after experimental segmental artery sacrifice. J. Thorac. Cardiovasc. Surg. 2011, 141, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, I.; Pries, A.; Styp-Rekowska, B. Pulsatile shear and Gja5 modulate arterial identity and remodeling events during flow-driven arteriogenesis. Development 2010, 137, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Kawano, H.; Motoyama, T.; Hirai, N.; Kugiyama, K.; Yasue, H.; Ogawa, H. Endothelial dysfunction in hyperchoelsterolemia is improved by L-arginine admnistration: Possible role of oxidative stress. Atherosclerosis 2002, 161, 375–380. [Google Scholar] [CrossRef]
- Lloyd, P.G.; Yang, H.T.; Terjung, R.L. Arteriogenesis and angiogenesis in rat ischemic hindlimb: Role of nitric oxide. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, 2528–2538. [Google Scholar] [CrossRef]
- Van Dijk, C.G.M.; Nieuweboer, F.E.; Pei, J.Y. The complex mural cell: Pericyte function in health and disease. Int. J. Cardiol. 2015, 190, 75–89. [Google Scholar] [CrossRef]
- Hashimoto, T.; Tsuneki, M.; Foster, T.R.; Santana, J.M.; Bai, H.; Wang, M. Membrane-mediated regulation of vascular identity. Birth Defects Res. Part C Embryo Today 2016, 108, 65–84. [Google Scholar] [CrossRef]
- Zhang, H.; Faber, J.E. De-novo collateral formation following acute myocardial infarction: Dependence on CCR2+ bone marrow cells. J. Mol. Cell. Cardiol. 2015, 87, 4–16. [Google Scholar] [CrossRef]
- Scholz, D.; Ito, W.; Fleming, I. Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis). Virchows Archiv. 2000, 436, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Pipp, F.; Boehm, S.; Cai, W.J.; Adili, F.; Ziegler, B.; Karanovic, G.; Ritter, R.; Balzer, J.; Scheler, C.; Schaper, W. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arter. Thromb. Vasc. Biol. 2004, 24, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Hakimzadeh, N.; Verberne, H.J.; Siebes, M.; Piek, J.J. The future of collateral artery research. Curr. Cardiol. Rev. 2014, 10, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389. [Google Scholar] [CrossRef] [PubMed]
- Deindl, E.; Schaper, W. The art of arteriogenesis. Cell Biochem. Biophys. 2005, 43, 1–15. [Google Scholar] [CrossRef]
- Jazwa, A.; Florczyk, U.; Grochot-Przeczek, A.; Krist, B.; Loboda, A.; Jozkowicz, A.; Dulak, J. Limb ischemia and vessel regeneration: Is there a role for VEGF? Vasc. Pharmacol. 2016, 86, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Stabile, E.; Kinnaird, T. Temporal patterns of gene expression after acute hindlimb ischemia in mice. JACC 2004, 43, 474–482. [Google Scholar] [CrossRef]
- Behm, C.Z.; Kaufmann, B.A.; Carr, C. Molecular imaging of endothelial vascular cell adhesion molecule-1 expression and inflammatory cell recruitment during vasculogenesis and ischemia-mediated arteriogenesis. Circulation 2008, 117, 2902–2911. [Google Scholar] [CrossRef]
- Risau, W. Development and differentiation of endothelium. Kidney Int. 1998, 54, 3–6. [Google Scholar] [CrossRef]
- Schaper, W.; Flameng, W.; Winkler, B. Quantification of collateral resistance in acute and chronic experimental coronary occlusion in the dog. Circ. Res. 1976, 39, 371–377. [Google Scholar] [CrossRef]
- Hoefer, I.E.; van Royen, N.; Rectenwald, J.E. Arteriogenesis proceeds via ICAM-1/Mac-1-mediated mechanisms. Circ. Res. 2004, 94, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Castro, P.R.; Barbosa, A.S.; Pereira, J.M. Cellular and Molecular Heterogeneity Associated with Vessel Formation Processes. Biomed. Res. Int. 2018, 6740408. [Google Scholar] [CrossRef] [PubMed]
- Van Royen, N. Stimulation of arteriogenesis; a new concept for the treatment of arterial occlusive disease. Cardiovasc. Res. 2001, 49, 543–553. [Google Scholar] [CrossRef]
- Arras, M.; Ito, W.D.; Scholz, D.; Winkler, B.; Schaper, J.; Schaper, W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J. Clin. Investig. 1998, 101, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.J.; Kocsis, E.; Wu, X. Remodeling of the vascular tunica media is essential for development of collateral vessels in the canine heart. Mol. Cell. Biochem. 2004, 264, 201–210. [Google Scholar] [CrossRef]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A. Perivascular Mast Cells Govern Shear Stress-Induced Arteriogenesis by Orchestrating Leukocyte Function. Cell Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef]
- Dodd, T.; Jadhav, R.; Wiggins, L. MMPs 2 and 9 are essential for coronary collateral growth and are prominently regulated by p38 MAPK. J. Mol. Cell. Cardiol. 2011, 51, 1015–1025. [Google Scholar] [CrossRef]
- Cai, W.J.; Koltai, S.; Kocsis, E. Remodeling of the adventitia during coronary arteriogenesis. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, 31–40. [Google Scholar] [CrossRef][Green Version]
- Wolf, C.; Cai, W.J.; Vosschulte, R. Vascular remodeling and altered protein expression during growth of coronary collateral arteries. J. Mol. Cell. Cardiol. 1998, 30, 2291–2305. [Google Scholar] [CrossRef]
- Hoefer, I.E.; van Royen, N.; Buschmann, I.R.; Piek, J.J.; Schaper, W. Time course of arteriogenesis following femoral artery occlusion in the rabbit. Cardiovasc. Res. 2001, 49, 609–617. [Google Scholar] [CrossRef]
- Seiler, C.; Stoller, M.; Pitt, B.; Meier, P. The human coronary collateral circulation: Development and clinical importance. Eur. Heart J. 2013, 34, 2674–2682. [Google Scholar] [CrossRef]
- Zhang, H.; Prabhakar, P.; Sealock, R.; Faber, J.E. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J. Cereb. Blood Flow Metab. 2010, 30, 923–934. [Google Scholar] [CrossRef]
- Heil, M.; Eitenmüller, I.; Schmitz-Rixen, T.; Schaper, W. Arteriogenesis versus angiogenesis: Similarities and differences. J. Cell. Mol. Med. 2006, 10, 45–55. [Google Scholar] [CrossRef]
- Chalothorn, D.; Clayton, J.A.; Zhang, H.; Pomp, D.; Faber, J.E. Collateral density, remodeling, and VEGF-A expression differ widely between mouse strains. Physiol. Genomics. 2007, 30, 179–191. [Google Scholar] [CrossRef]
- Bischoff, M.S.; Scheumann, J.; Brenner, R.M.; Ladage, D.; Bodian, C.A.; Kleinman, G. Staged approach prevents spinal cord injury in hybrid surgical endovascular thoracoabdominal aortic aneurysm repair: An experimental model. Ann. Thorac. Surg. 2011, 92, 138–146. [Google Scholar] [CrossRef]
- Zoli, S.; Etz, C.D.; Roder, F.; Brenner, R.M.; Bodian, C.A.; Kleinman, G. Experimental two-stage simulated repair of extensive thoracoabdominal aneurysms reduces paraplegia risk. Ann. Thorac. Surg. 2010, 90, 722–729. [Google Scholar] [CrossRef]
- Geisbusch, S.; Stefanovic, A.; Koruth, J.S.; Lin, H.M.; Morgello, S.; Weisz, D.J. Endovascular coil embolization of segmental arteries prevents paraplegia after subsequent thoracoabdominal aneurysm repair: An experimental model. J. Thorac. Cardiovasc. Surg. 2014, 147, 220–226. [Google Scholar] [CrossRef]
- Griepp, E.B.; Di Luozzo, G.; Schray, D.; Stefanovic, A.; Geisbüsch, S.; Griepp, R.B. The anatomy of the spinal cord collateral circulation. Ann. Cardiothorac. Surg. 2012, 1, 350–357. [Google Scholar]
- Etz, C.D.; Zoli, S.; Mueller, C.S.; Bodian, C.A.; Di Luozzo, G.; Lazala, R. Staged repair significantly reduces paraplegia rate after extensive thoracoabdominal aortic aneurysm repair. J. Thorac. Cardiovasc. Surg. 2010, 139, 1464–1472. [Google Scholar] [CrossRef]
- Etz, C.D.; Debus, E.S.; Mohr, F.W.; Kölbel, T. First-in-man endovascular preconditioning of the paraspinal collateral network by segmental artery coil embolization to prevent ischemic spinal cord injury. J. Thorac. Cardiovasc. Surg. 2015, 149, 1074–1079. [Google Scholar] [CrossRef]
- Ufnal, M.; Skrzypecki, J. Blood borne hormones in a crosstalk between peripheral and brain mechanisms regulating blood pressure, the role of circumventricular organs. Neuropeptides 2014, 48, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Lautz, T.; Lasch, M.; Borgolte, J.; Troidl, K.; Pagel, J.I.; Caballero-Martinez, A.; Kleinert, E.C.; Walzog, B.; Deindl, E. Midkine Controls Arteriogenesis by Regulating the Bioavailability of Vascular Endothelial Growth Factor A and the Expression of Nitric Oxide Synthase 1 and 3. EBioMedicine 2018, 27, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Pagel, J.I.; Borgolte, J.; Hoefer, I.; Fernandez, B.; Schaper, W.; Deindl, E. Involvement of neuronal NO synthase in collateral artery growth. Indian J. Biochem. Biophys. 2011, 48, 270–274. [Google Scholar]
- Troidl, K.; Tribulova, S.; Cai, W.J.; Ruding, I.; Apfelbeck, H.; Schierling, W.; Troidl, C.; Schmitz-Rixen, T.; Schaper, W. Effects of endogenous nitric oxide and of DETA NONOate in arteriogenesis. J. Cardiovasc. Pharm. 2010, 55, 153–160. [Google Scholar] [CrossRef]
- Son, H.; Hawkins, R.D.; Martin, K.; Kiebler, M.; Huang, P.L.; Fishman, M.C.; Kandel, E.R. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell 1996, 87, 1015–1023. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Michell, B.J.; Griffiths, J.E.; Mitchelhill, K.I.; Rodriguez-Crespo, I.; Tiganis, T.; Bozinovski, S.; de Montellano, P.R.; Kemp, B.E.; Pearson, R.B. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr. Biol. 1999, 9, 845–848. [Google Scholar] [CrossRef]
- Ho, F.M.; Lin, W.W.; Chen, B.C.; Chao, C.M.; Yang, C.R.; Lin, L.Y.; Lai, C.C.; Liu, S.H.; Lian, C.S. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-κB and c-Jun NH2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cell Signal. 2006, 18, 391–399. [Google Scholar] [CrossRef]
- Gao, F.; Gao, E.; Yue, T.L.; Ohlstein, E.H.; Lopez, B.L.; Christopher, T.A.; Ma, X.L. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: The roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation 2002, 105, 1497–1502. [Google Scholar] [CrossRef]
- Lanahan, A.A.; Lech, D.; Dubrac, A.; Zhang, J.; Zhuang, Z.W.; Eichmann, A.; Simons, M. Ptp1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation 2014, 130, 902–909. [Google Scholar] [CrossRef]
- Pipp, F.; Heil, M.; Issbrucker, K.; Ziegelhoe_er, T.; Martin, S.; Van Den Heuvel, J.; Weich, H.; Fernandez, B.; Golomb, G.; Carmeliet, P.; et al. Vegfr-1-selective vegf homologue plgf is arteriogenic: Evidence for a monocyte-mediated mechanism. Circ. Res. 2003, 92, 378–385. [Google Scholar] [CrossRef]
- Heil, M.; Ziegelhoe_er, T.; Pipp, F.; Kostin, S.; Martin, S.; Clauss, M.; Schaper, W. Blood monocyte concentration is critical for enhancement of collateral artery growth. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2411–H2419. [Google Scholar] [CrossRef]
- Ziegelhoeffer, T.; Fernandez, B.; Kostin, S.; Heil, M.; Voswinckel, R.; Helisch, A.; Schaper, W. Bone marrow-derived cells do not incorporate into the adult growing vasculature. Circ. Res. 2004, 94, 230–238. [Google Scholar] [CrossRef]
- Ricard, N.; Zhang, J.; Zhuang, W.; Simon, M. Isoform-Specific Roles of ERK1 and ERK2 in Arteriogenesis. Cells 2019, 9, 38. [Google Scholar] [CrossRef]
- Voyvodic, P.L.; Min, D.; Liu, R.; Williams, E.; Chitalia, V.; Dunn, A.K.; Baker, A.B. Loss of syndecan-1 induces a pro-inflammatory phenotype in endothelial cells with a dysregulated response to atheroprotective flow. J. Biol. Chem. 2014, 289, 9547–9559. [Google Scholar] [CrossRef]
- Siebel, C.; Lendahl, U. Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef]
- Krishnasamy, K.; Limbourg, A.; Kapanadze, T. Blood vessel control of macrophage maturation promotes arteriogenesis in ischemia. Nat. Commun. 2017, 8, 1. [Google Scholar] [CrossRef]
- Yang, C.; Guo, Y.; Jadlowiec, C.C.; Li, X.; Lv, W.; Model, L. Vascular endothelial growth factor-A inhibits EphB4 and stimulates delta-like ligand 4 expression in adult endothelial cells. J. Surg. Res. 2013, 183, 478–486. [Google Scholar] [CrossRef]
- Kerr, B.A.; West, X.Z.; Kim, Y.W. Stability and function of adult vasculature is sustained by Akt/Jagged1 signalling axis in endothelium. Nat. Commun. 2016, 7, 10960. [Google Scholar] [CrossRef]
- Kang, J.; Yoo, J.; Lee, S.; Tang, W.; Aguilar, B.; Ramu, S. An exquisite cross-control mechanism among endothelial cell fate regulators directs the plasticity and heterogeneity of lymphatic endothelial cells. Blood 2010, 116, 140–150. [Google Scholar] [CrossRef]
- Morrow, D.; Cullen, J.P.; Liu, W. Sonic hedgehog induces notch target gene expression in vascular smooth muscle cells via VEGF-A. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1112–1118. [Google Scholar] [CrossRef]
- Tian, D.Y.; Jin, X.R.; Zeng, X.; Wang, Y. Notch signaling in endothelial cells: Is it the therapeutic target for vascular neointimal hyperplasia? Int. J. Mol. Sci. 2017, 18, 1615. [Google Scholar] [CrossRef]
- Balligand, J.L.; Feron, O.; Dessy, C. eNOS activation by physical forces: From short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol. Rev. 2009, 89, 481–534. [Google Scholar] [CrossRef]
- Bennett, B.D.; Zeigler, F.C.; Gu, Q.; Fendly, B.; Goddard, A.D.; Gillet, N. Molecular cloning of a ligand for the EPH related receptor protein-tyrosine kinase Htk. Proc. Natl. Acad. Sci. USA 1995, 92, 1866–1870. [Google Scholar] [CrossRef]
- Bae, J.H.; Schlessinger, J. Asymmetric tyrosine kinase arrangements in activation or autophosphorylation of receptor tyrosine kinases. Mol. Cells 2010, 29, 443–448. [Google Scholar] [CrossRef]
- Pitulescu, M.E.; Adams, R.H. Regulation of signaling interactions and receptor endocytosis in growing blood vessels. Cell Adhes. Migr. 2014, 8, 366–377. [Google Scholar] [CrossRef]
- Nakayama, A.; Nakayama, M.; Turner, C.J.; Höing, S.; Lepore, J.J.; Adams, R.H. Ephrin-B2 controls PDGFRb internalization and signaling. Genes Dev. 2013, 27, 2576–2589. [Google Scholar] [CrossRef]
- Himanen, J.P. Ectodomain structures of Eph receptors. Semin. Cell Dev. Biol. 2012, 23, 35–42. [Google Scholar] [CrossRef]
- Lisabeth, E.M.; Falivelli, G.; Pasquale, E.B. Eph receptor signaling and ephrins. Cold Spring Harb. Perspect. Biol. 2013, 5, a009159. [Google Scholar] [CrossRef]
- Deindl, E.; Buschmann, I.; Hoefer, I.E.; Podzuweit, T.; Boengler, K.; Vogel, S.; van Royen, N.; Fernandez, B.; Schaper, W. Role of ischemia and of hypoxia-inducible genes in arteriogenesis after femoral artery occlusion in the rabbit. Circ. Res. 2001, 89, 779–786. [Google Scholar] [CrossRef]
- Horbelt, D.; Denkis, A.; Knaus, P. A portrait of Transforming Growth Factor β superfamily signalling: Background matters. Int. J. Biochem. Cell. Biol. 2012, 44, 469–474. [Google Scholar] [CrossRef]
- Evans, R.a.; Tian, Y.C.; Steadman, R.; Phillips, A.O. TGF-β1-mediated fibroblast–myofibroblast terminal differentiation—the role of smad proteins. Exp. Cell. Res. 2003, 282, 90–100. [Google Scholar] [CrossRef]
- Kadomatsu, K.; Tomomura, M.; Muramatsu, T. cDNA cloning and sequencing of a new gene intensely expressed in early differentiation stages of embryonal carcinoma cells and in mid-gestation period of mouse embryogenesis. Biochem. Biophys. Res. Commun. 1988, 151, 1312–1318. [Google Scholar] [CrossRef]
- Weckbach, L.T.; Groesser, L.; Borgolte, J.; Pagel, J.I.; Pogoda, F.; Schymeinsky, J.; Muller-Hocker, J.; Shakibaei, M.; Muramatsu, T.; Deindl, E. Midkine acts as proangiogenic cytokine in hypoxia-induced angiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, 429–438. [Google Scholar] [CrossRef]
- Weckbach, L.T.; Preissner, K.T.; Deindl, E. The Role of Midkine in Arteriogenesis, Involving Mechanosensing, Endothelial Cell Proliferation, and Vasodilation. Int. J. Mol. Sci. 2018, 19, 2559. [Google Scholar] [CrossRef]
- Novotny, W.F.; Maffi, T.; Mehta, R.L.; Milner, P.G. Identification of novel heparin-releasable proteins, as well as the cytokines midkine and pleiotrophin, in human postheparin plasma. Arter. Thromb. 1993, 13, 1798–1805. [Google Scholar] [CrossRef]
- Weckbach, L.T.; Muramatsu, T.; Walzog, B. Midkine in inflammation. Sci. World J. 2011, 11, 2491–2505. [Google Scholar] [CrossRef]
- Gungor, C.; Zander, H.; Effenberger, K.E.; Vashist, Y.K.; Kalinina, T.; Izbicki, J.R.; Yekebas, E.; Bockhorn, M. Notch signaling activated by replication stress-induced expression of midkine drives epithelial-mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res. 2011, 71, 5009–5019. [Google Scholar] [CrossRef]
- Huang, Y.; Hoque, M.O.; Wu, F.; Trink, B.; Sidransky, D.; Ratovitski, E.A. Midkine induces epithelial-mesenchymal transition through Notch2/Jak2-Stat3 signaling in human keratinocytes. Cell Cycle 2008, 7, 1613–1622. [Google Scholar] [CrossRef]
- Orr, A.W.; Sanders, J.M.; Bevard, M.; Coleman, E.; Sarembock, I.J.; Schwartz, M.A. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: A potential role in atherosclerosis. J. Cell Biol. 2005, 169, 191–202. [Google Scholar] [CrossRef]
- Horiba, M.; Kadomatsu, K.; Nakamura, E.; Muramatsu, H.; Ikematsu, S.; Sakuma, S.; Hayashi, K.; Yuzawa, Y.; Matsuo, S.; Kuzuya, M. Neointima formation in a restenosis model is suppressed in midkine-deficient mice. J. Clin. Investig. 2000, 105, 489–495. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, F.; Wagenhäuser, M.U.; Busch, A.; Schelzig, H.; Gombert, A. Arteriogenesis of the Spinal Cord—The Network Challenge. Cells 2020, 9, 501. https://doi.org/10.3390/cells9020501
Simon F, Wagenhäuser MU, Busch A, Schelzig H, Gombert A. Arteriogenesis of the Spinal Cord—The Network Challenge. Cells. 2020; 9(2):501. https://doi.org/10.3390/cells9020501
Chicago/Turabian StyleSimon, Florian, Markus Udo Wagenhäuser, Albert Busch, Hubert Schelzig, and Alexander Gombert. 2020. "Arteriogenesis of the Spinal Cord—The Network Challenge" Cells 9, no. 2: 501. https://doi.org/10.3390/cells9020501
APA StyleSimon, F., Wagenhäuser, M. U., Busch, A., Schelzig, H., & Gombert, A. (2020). Arteriogenesis of the Spinal Cord—The Network Challenge. Cells, 9(2), 501. https://doi.org/10.3390/cells9020501