Pluripotent-Stem-Cell-Derived Hepatic Cells: Hepatocytes and Organoids for Liver Therapy and Regeneration

 ,
,  ,
,
Abstract
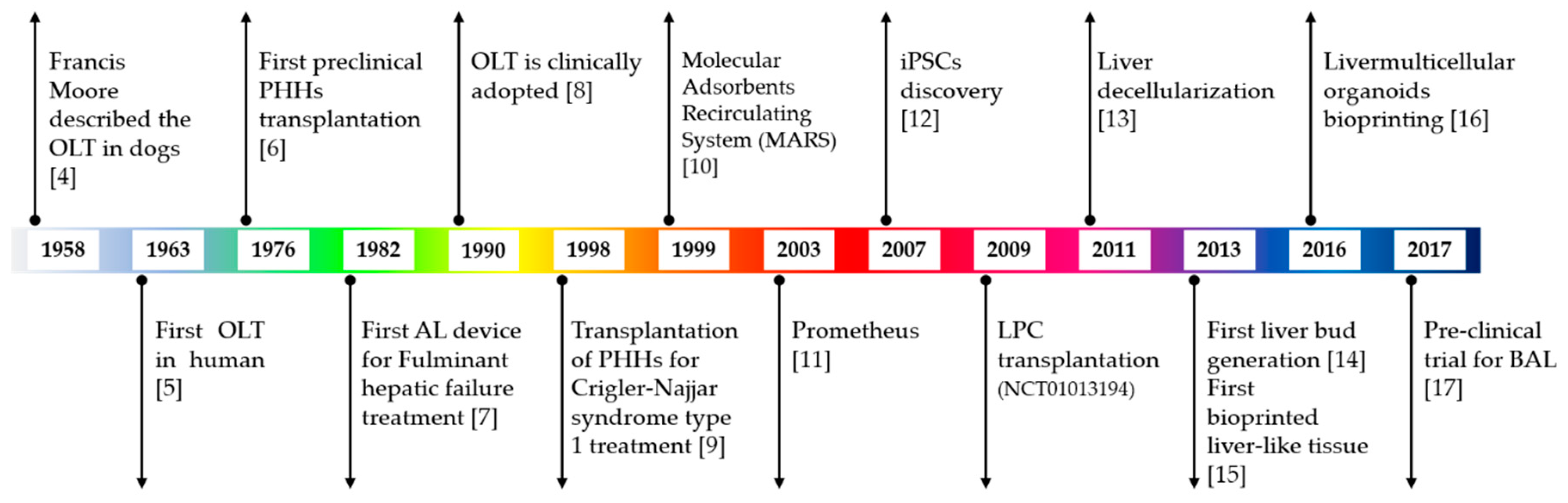
1. Introduction
2. Liver Therapy and Regeneration Approaches: Pros and Cons
2.1. Orthotopic Liver Transplantation (OLT)
2.2. Artificial Liver Devices
2.3. Cell Therapy: Transplantation of Isolated Cell
2.4. Cell Therapy: Bioengineering Approaches
2.4.1. Liver Organoids
2.4.2. Bio Artificial Liver (BAL) Devices
2.4.3. Decellularized/Cellularized Liver Scaffolds
2.4.4. Liver Biofabrication and Bioprinting
3. Cell Sources for Hepatocyte Transplantation and Liver Repair
3.1. Primary Human Hepatocytes (PHHs)
3.2. Fetal Liver Progenitors (FLPs)
3.3. Adult Human Liver Stem Cells (AdHLSCs)
3.4. Hematopoietic Stem Cells (HSCs) and Mesenchymal Stem Cells (MSCs)
3.5. Human Pluripotent Stem Cells (hPSCs)
3.6. Overview on Embryogenesis and Published Differentiation Protocols for hiPSC Differentiation into HLCs
3.7. Genetic Integrity
3.8. Epigenetics in hPSCs
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Adam, R.; Karam, V.; Cailliez, V.; O Grady, J.G.; Mirza, D.; Cherqui, D.; Klempnauer, J.; Salizzoni, M.; Pratschke, J.; Jamieson, N.; et al. 2018 Annual Report of the European Liver Transplant Registry (ELTR)-50-year evolution of liver transplantation. Transpl. Int. 2018, 31, 1293–1317. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Palakkan, A.A.; Hay, D.C.; Pr, A.K.; Tv, K.; Ross, J.A. Liver tissue engineering and cell sources: Issues and challenges. Liver Int. 2013, 33, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Moore, F.D.; Smith, L.L.; Burnap, T.K.; Dallenbach, F.D.; Dammin, G.J.; Gruber, U.F.; Shoemaker, W.C.; Steenburg, R.W.; Ball, M.R.; Belko, J.S. One-stage homotransplantation of the liver following total hepatectomy in dogs. Plast. Reconstr. Surg. 1959, 23, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Starzl, T.E.; Marchioro, T.L.; Vonkaulla, K.N.; Hermann, G.; Brittain, R.S.; Waddell, W.R. Homotransplantation of the liver in humans. Surg. Gynecol. Obstet. 1963, 117, 659–676. [Google Scholar] [PubMed]
- Puppi, J.; Strom, S.C.; Hughes, R.D.; Bansal, S.; Castell, J.V.; Dagher, I.; Ellis, E.C.S.; Nowak, G.; Ericzon, B.-G.; Fox, I.J.; et al. Improving the Techniques for Human Hepatocyte Transplantation: Report from a Consensus Meeting in London. Cell Transplant. 2012, 21, 1–10. [Google Scholar] [CrossRef]
- Gu, J.; Shi, X.; Ren, H.; Xu, Q.; Wang, J.; Xiao, J.; Ding, Y. Systematic review: Extracorporeal bio-artificial liver-support system for liver failure. Hepatol. Int. 2012, 6, 670–683. [Google Scholar] [CrossRef]
- Meirelles Júnior, R.F.; Salvalaggio, P.; de Rezende, M.B.; Evangelista, A.S.; Guardia, B.D.; Matielo, C.E.L.; Neves, D.B.; Pandullo, F.L.; Felga, G.E.G.; Alves, J.A.D.S.; et al. Liver transplantation: History, outcomes and perspectives. Einstein São Paulo 2015, 13, 149–152. [Google Scholar] [CrossRef]
- Fox, I.J.; Chowdhury, J.R.; Kaufman, S.S.; Goertzen, T.C.; Chowdhury, N.R.; Warkentin, P.I.; Dorko, K.; Sauter, B.V.; Strom, S.C. Treatment of the Crigler–Najjar Syndrome Type I with Hepatocyte Transplantation. N. Engl. J. Med. 1998, 338, 1422–1427. [Google Scholar] [CrossRef]
- Zabulica, M.; Srinivasan, R.C.; Vosough, M.; Hammarstedt, C.; Wu, T.; Gramignoli, R.; Ellis, E.; Kannisto, K.; Collin de l’Hortet, A.; Takeishi, K.; et al. Guide to the Assessment of Mature Liver Gene Expression in Stem Cell-Derived Hepatocytes. Stem Cells Dev. 2019, 28, 907–919. [Google Scholar] [CrossRef]
- Rifai, K.; Ernst, T.; Kretschmer, U.; Bahr, M.J.; Schneider, A.; Hafer, C.; Haller, H.; Manns, M.P.; Fliser, D. Prometheus®–A new extracorporeal system for the treatment of liver failure☆. J. Hepatol. 2003, 39, 984–990. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Baptista, P.M.; Siddiqui, M.M.; Lozier, G.; Rodriguez, S.R.; Atala, A.; Soker, S. The use of whole organ decellularization for the generation of a vascularized liver organoid. Hepatology 2011, 53, 604–617. [Google Scholar] [CrossRef]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.-R.; Ueno, Y.; Zheng, Y.-W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef]
- Faulkner-Jones, A.; Greenhough, S.; A King, J.; Gardner, J.; Courtney, A.; Shu, W. Development of a valve-based cell printer for the formation of human embryonic stem cell spheroid aggregates. Biofabrication 2013, 5, 015013. [Google Scholar] [CrossRef]
- Ma, X.; Qu, X.; Zhu, W.; Li, Y.-S.; Yuan, S.; Zhang, H.; Liu, J.; Wang, P.; Lai, C.S.E.; Zanella, F.; et al. Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting. Proc. Natl. Acad. Sci. USA 2016, 113, 2206–2211. [Google Scholar] [CrossRef]
- Selden, C.; Bundy, J.; Erro, E.; Puschmann, E.; Miller, M.; Kahn, D.; Hodgson, H.; Fuller, B.; Gonzalez-Molina, J.; Le Lay, A.; et al. A clinical-scale BioArtificial Liver, developed for GMP, improved clinical parameters of liver function in porcine liver failure. Sci. Rep. 2017, 7, 14518. [Google Scholar] [CrossRef]
- Kim, W.R.; Lake, J.R.; Smith, J.M.; Schladt, D.P.; Skeans, M.A.; Noreen, S.M.; Robinson, A.M.; Miller, E.; Snyder, J.J.; Israni, A.K.; et al. OPTN/SRTR 2017 Annual Data Report: Liver. Am. J. Transplant. 2019, 19, 184–283. [Google Scholar] [CrossRef]
- Carpentier, B.; Gautier, A.; Legallais, C. Artificial and bioartificial liver devices: Present and future. Gut 2009, 58, 1690–1702. [Google Scholar] [CrossRef]
- Rademacher, S.; Oppert, M.; Jörres, A. Artificial extracorporeal liver support therapy in patients with severe liver failure. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 591–599. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, X.; Liang, L.; Li, J.; Demirci, U.; Wang, S. A decade of progress in liver regenerative medicine. Biomaterials 2018, 157, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, H.J.; Choi, D. Cell Sources, Liver Support Systems and Liver Tissue Engineering: Alternatives to Liver Transplantation. Int. J. Stem Cells 2015, 8, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Böttger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef]
- Ogawa, S.; Miyagawa, S. Potentials of regenerative medicine for liver disease. Surg. Today 2009, 39, 1019–1025. [Google Scholar] [CrossRef]
- Neuberger, J. An update on liver transplantation: A critical review. J. Autoimmun. 2016, 66, 51–59. [Google Scholar] [CrossRef]
- Sutherland, D.E.; Numata, M.; Matas, A.J.; Simmons, R.L.; Najarian, J.S. Hepatocellular transplantation in acute liver failure. Surgery 1977, 82, 124–132. [Google Scholar]
- Yoshida, Y.; Tokusashi, Y.; Lee, G.; Ogawa, K. Intrahepatic transplantation of normal hepatocytes prevents Wilson’s disease in Long-Evans cinnamon rats. Gastroenterology 1996, 111, 1654–1660. [Google Scholar] [CrossRef]
- Horslen, S.P.; McCowan, T.C.; Goertzen, T.C.; Warkentin, P.I.; Cai, H.B.; Strom, S.C.; Fox, I.J. Isolated Hepatocyte Transplantation in an Infant With a Severe Urea Cycle Disorder. PEDIATRICS 2003, 111, 1262–1267. [Google Scholar] [CrossRef]
- Sokal, E.M.; Smets, F.; Bourgois, A.; Van Maldergem, L.; Buts, J.-P.; Reding, R.; Bernard Otte, J.; Evrard, V.; Latinne, D.; Vincent, M.F.; et al. Hepatocyte transplantation in a 4-year-old girl with peroxisomal biogenesis disease: Technique, safety, and metabolic follow-up1. Transplantation 2003, 76, 735–738. [Google Scholar] [CrossRef]
- Dhawan, A.; Mitry, R.R.; Hughes, R.D.; Lehec, S.; Terry, C.; Bansal, S.; Arya, R.; Wade, J.J.; Verma, A.; Heaton, N.D.; et al. Hepatocyte Transplantation for Inherited Factor VII Deficiency. Transplantation 2004, 78, 1812–1814. [Google Scholar] [CrossRef]
- Ribes-Koninckx, C.; Ibars, E.P.; Agrasot, M.Á.C.; Bonora-Centelles, A.; Miquel, B.P.; Carbó, J.J.V.; Aliaga, E.D.; Pallardó, J.M.; Gómez-Lechón, M.J.; Castell, J.V. Clinical Outcome of Hepatocyte Transplantation in Four Pediatric Patients with Inherited Metabolic Diseases. Cell Transplant. 2012, 21, 2267–2282. [Google Scholar] [CrossRef]
- Alwahsh, S.M.; Rashidi, H.; Hay, D.C. Liver cell therapy: Is this the end of the beginning? Cell. Mol. Life Sci. 2018, 75, 1307–1324. [Google Scholar] [CrossRef]
- Soltys, K.A.; Soto-Gutiérrez, A.; Nagaya, M.; Baskin, K.M.; Deutsch, M.; Ito, R.; Shneider, B.L.; Squires, R.; Vockley, J.; Guha, C.; et al. Barriers to the successful treatment of liver disease by hepatocyte transplantation. J. Hepatol. 2010, 53, 769–774. [Google Scholar] [CrossRef]
- Dagher, I.; Boudechiche, L.; Branger, J.; Coulomb-Lhermine, A.; Parouchev, A.; Sentilhes, L.; Lin, T.; Groyer-Picard, M.-T.; Vons, C.; Hadchouel, M.; et al. Efficient Hepatocyte Engraftment in a Nonhuman Primate Model After Partial Portal Vein Embolization. Transplantation 2006, 82, 1067–1073. [Google Scholar] [CrossRef]
- Dagher, I.; Nguyen, T.H.; Groyer-Picard, M.-T.; Lainas, P.; Mainot, S.; Guettier, C.; Pariente, D.; Franco, D.; Weber, A. Efficient hepatocyte engraftment and long-term transgene expression after reversible portal embolization in nonhuman primates. Hepatology 2009, 49, 950–959. [Google Scholar] [CrossRef]
- Pourcher, G.; El-Kehdy, H.; Kanso, F.; Groyer-Picard, M.-T.; Gaillard, M.; Trassard, O.; Blazsek, I.; Agostini, H.; Dubart-Kupperschmitt, A.; Dagher, I. Volumetric Portal Embolization: A New Concept to Improve Liver Regeneration and Hepatocyte Engraftment. Transplantation 2016, 100, 344–354. [Google Scholar] [CrossRef]
- Gaillard, M.; Tranchart, H.; Lainas, P.; Trassard, O.; Remy, S.; Dubart-Kupperschmitt, A.; Dagher, I. Improving Hepatocyte Engraftment Following Hepatocyte Transplantation Using Repeated Reversible Portal Vein Embolization in Rats. Liver Transpl. 2019, 25, 98–110. [Google Scholar] [CrossRef]
- Soltys, K.A.; Setoyama, K.; Tafaleng, E.N.; Soto Gutiérrez, A.; Fong, J.; Fukumitsu, K.; Nishikawa, T.; Nagaya, M.; Sada, R.; Haberman, K.; et al. Host conditioning and rejection monitoring in hepatocyte transplantation in humans. J. Hepatol. 2017, 66, 987–1000. [Google Scholar] [CrossRef]
- Green, C.J.; Charlton, C.A.; Wang, L.-M.; Silva, M.; Morten, K.J.; Hodson, L. The isolation of primary hepatocytes from human tissue: Optimising the use of small non-encapsulated liver resection surplus. Cell Tissue Bank. 2017, 18, 597–604. [Google Scholar] [CrossRef]
- Elaut, G.; Henkens, T.; Papeleu, P.; Snykers, S.; Vinken, M.; Vanhaecke, T.; Rogiers, V. Molecular Mechanisms Underlying the Dedifferentiation Process of Isolated Hepatocytes and Their Cultures. Curr. Drug Metab. 2006, 7, 629–660. [Google Scholar] [CrossRef]
- Forbes, S.J.; Gupta, S.; Dhawan, A. Cell therapy for liver disease: From liver transplantation to cell factory. J. Hepatol. 2015, 62, S157–S169. [Google Scholar] [CrossRef] [PubMed]
- Nevi, L.; Carpino, G.; Costantini, D.; Cardinale, V.; Riccioni, O.; Di Matteo, S.; Melandro, F.; Berloco, P.B.; Reid, L.; Gaudio, E.; et al. Hyaluronan coating improves liver engraftment of transplanted human biliary tree stem/progenitor cells. Stem Cell Res. Ther. 2017, 8, 68. [Google Scholar] [CrossRef]
- Chang, S.H.; Huang, H.H.; Kang, P.L.; Wu, Y.C.; Chang, M.-H.; Kuo, S.M. In vitro and in vivo study of the application of volvox spheres to co-culture vehicles in liver tissue engineering. Acta Biomater. 2017, 63, 261–273. [Google Scholar] [CrossRef]
- Tong, X.-F.; Zhao, F.-Q.; Ren, Y.-Z.; Zhang, Y.; Cui, Y.-L.; Wang, Q.-S. Injectable hydrogels based on glycyrrhizin, alginate, and calcium for three-dimensional cell culture in liver tissue engineering: Injectable hydrogels for liver tissue engineering. J. Biomed. Mater. Res. A 2018, 106, 3292–3302. [Google Scholar] [CrossRef]
- Cui, J.; Wang, H.; Shi, Q.; Sun, T.; Huang, Q.; Fukuda, T. Multicellular Co-Culture in Three-Dimensional Gelatin Methacryloyl Hydrogels for Liver Tissue Engineering. Molecules 2019, 24, 1762. [Google Scholar] [CrossRef]
- Brassard, J.A.; Lutolf, M.P. Engineering Stem Cell Self-organization to Build Better Organoids. Cell Stem Cell 2019, 24, 860–876. [Google Scholar] [CrossRef]
- Takeichi, M. Self-Organization of Animal Tissues: Cadherin-Mediated Processes. Dev. Cell 2011, 21, 24–26. [Google Scholar] [CrossRef]
- Lou, Y.-R.; Leung, A.W. Next generation organoids for biomedical research and applications. Biotechnol. Adv. 2018, 36, 132–149. [Google Scholar] [CrossRef]
- Calpe, S.; Correia, A.; El Khattabi, M.; Zimberlin, C.; Medema, J.P.; Verrips, T.; Krishnadath, K.K. Inhibition of BMP2 and BMP4 by a Novel Llama-Derived Nanobody Sustains Intestinal Stem Cells in Organoid Cultures. Gastroenterology 2015, 148, S105. [Google Scholar] [CrossRef]
- Yip, H.Y.K.; Tan, C.W.; Hirokawa, Y.; Burgess, A.W. Colon organoid formation and cryptogenesis are stimulated by growth factors secreted from myofibroblasts. PLoS ONE 2018, 13, e0199412. [Google Scholar] [CrossRef]
- Sasai, Y. Cytosystems dynamics in self-organization of tissue architecture. Nature 2013, 493, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Bratt-Leal, A.M.; Carpenedo, R.L.; McDevitt, T.C. Engineering the embryoid body microenvironment to direct embryonic stem cell differentiation. Biotechnol. Prog. 2009, 25, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Brusatin, G.; Panciera, T.; Gandin, A.; Citron, A.; Piccolo, S. Biomaterials and engineered microenvironments to control YAP/TAZ-dependent cell behaviour. Nat. Mater. 2018, 17, 1063–1075. [Google Scholar] [CrossRef]
- Takebe, T.; Enomura, M.; Yoshizawa, E.; Kimura, M.; Koike, H.; Ueno, Y.; Matsuzaki, T.; Yamazaki, T.; Toyohara, T.; Osafune, K.; et al. Vascularized and Complex Organ Buds from Diverse Tissues via Mesenchymal Cell-Driven Condensation. Cell Stem Cell 2015, 16, 556–565. [Google Scholar] [CrossRef]
- Dianat, N.; Steichen, C.; Vallier, L.; Weber, A.; Dubart-Kupperschmitt, A. Human Pluripotent Stem Cells for Modelling Human Liver Diseases and Cell Therapy. Curr. Gene Ther. 2013, 13, 120–132. [Google Scholar] [CrossRef]
- Handa, K.; Matsubara, K.; Fukumitsu, K.; Guzman-Lepe, J.; Watson, A.; Soto-Gutierrez, A. Assembly of Human Organs from Stem Cells to Study Liver Disease. Am. J. Pathol. 2014, 184, 348–357. [Google Scholar] [CrossRef]
- Wu, L.-J.; Chen, Z.-Y.; Wang, Y.; Zhao, J.-G.; Xie, X.-Z.; Chen, G. Organoids of liver diseases: From bench to bedside. World J. Gastroenterol. 2019, 25, 1913–1927. [Google Scholar] [CrossRef]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Kim, J.H.; Jang, Y.J.; An, S.Y.; Son, J.; Lee, J.; Lee, G.; Park, J.Y.; Park, H.-J.; Hwang, D.-Y.; Kim, J.-H.; et al. Enhanced Metabolizing Activity of Human ES Cell-Derived Hepatocytes Using a 3D Culture System with Repeated Exposures to Xenobiotics. Toxicol. Sci. Off. J. Soc. Toxicol. 2015, 147, 190–206. [Google Scholar] [CrossRef]
- Sengupta, S.; Johnson, B.P.; Swanson, S.A.; Stewart, R.; Bradfield, C.A.; Thomson, J.A. Aggregate Culture of Human Embryonic Stem Cell-Derived Hepatocytes in Suspension Are an Improved In Vitro Model for Drug Metabolism and Toxicity Testing. Toxicol. Sci. 2014, 140, 236–245. [Google Scholar] [CrossRef]
- Takayama, K.; Kawabata, K.; Nagamoto, Y.; Kishimoto, K.; Tashiro, K.; Sakurai, F.; Tachibana, M.; Kanda, K.; Hayakawa, T.; Furue, M.K.; et al. 3D spheroid culture of hESC/hiPSC-derived hepatocyte-like cells for drug toxicity testing. Biomaterials 2013, 34, 1781–1789. [Google Scholar] [CrossRef]
- Du, Y.; Wang, J.; Jia, J.; Song, N.; Xiang, C.; Xu, J.; Hou, Z.; Su, X.; Liu, B.; Jiang, T.; et al. Human Hepatocytes with Drug Metabolic Function Induced from Fibroblasts by Lineage Reprogramming. Cell Stem Cell 2014, 14, 394–403. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer–derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Hendriks, D.F.G.; Bell, C.C.; Andersson, T.B.; Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chem. Res. Toxicol. 2016, 29, 1936–1955. [Google Scholar] [CrossRef]
- Augustyniak, J.; Bertero, A.; Coccini, T.; Baderna, D.; Buzanska, L.; Caloni, F. Organoids are promising tools for species-specific in vitro toxicological studies. J. Appl. Toxicol. 2019, 39, 1610–1622. [Google Scholar] [CrossRef]
- Mittal, R.; Woo, F.W.; Castro, C.S.; Cohen, M.A.; Karanxha, J.; Mittal, J.; Chhibber, T.; Jhaveri, V.M. Organ-on-chip models: Implications in drug discovery and clinical applications. J. Cell. Physiol. 2019, 234, 8352–8380. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Deng, P.; Chen, W.; Guo, Y.; Tao, T.; Qin, J. In situ differentiation and generation of functional liver organoids from human iPSCs in a 3D perfusable chip system. Lab. Chip 2018, 18, 3606–3616. [Google Scholar] [CrossRef]
- Pan, X.-P.; Li, L.-J. Advances in cell sources of hepatocytes for bioartificial liver. Hepatobiliary Pancreat. Dis. Int. 2012, 11, 594–605. [Google Scholar] [CrossRef]
- Hoffman, D. The ultraflex esophageal and diamond biliary stents. Gastrointest. Endosc. Clin. N. Am. 1999, 9, 383–393. [Google Scholar] [CrossRef]
- Tysoe, O.C.; Justin, A.W.; Brevini, T.; Chen, S.E.; Mahbubani, K.T.; Frank, A.K.; Zedira, H.; Melum, E.; Saeb-Parsy, K.; Markaki, A.E.; et al. Isolation and propagation of primary human cholangiocyte organoids for the generation of bioengineered biliary tissue. Nat. Protoc. 2019, 14, 1884–1925. [Google Scholar] [CrossRef]
- Sampaziotis, F.; Tysoe, O.; Brevini, T.; Vallier, L. Use of Biliary Organoids in Cholestasis Research. Methods Mol. Biol. Clifton N. J. 2019, 1981, 373–382. [Google Scholar]
- Ovsianikov, A.; Khademhosseini, A.; Mironov, V. The Synergy of Scaffold-Based and Scaffold-Free Tissue Engineering Strategies. Trends Biotechnol. 2018, 36, 348–357. [Google Scholar] [CrossRef]
- Rajab, T.K.; Tchantchaleishvili, V. Can tissue engineering produce bioartificial organs for transplantation? Artif. Organs 2019, 43, 536–541. [Google Scholar] [CrossRef]
- Bao, J.; Shi, Y.; Sun, H.; Yin, X.; Yang, R.; Li, L.; Chen, X.; Bu, H. Construction of a Portal Implantable Functional Tissue-Engineered Liver Using Perfusion-Decellularized Matrix and Hepatocytes in Rats. Cell Transplant. 2011, 20, 753–766. [Google Scholar] [CrossRef]
- Crawford, A.R.; Lin, X.-Z.; Crawford, J.M. The normal adult human liver biopsy: A quantitative reference standard. Hepatology 1998, 28, 323–331. [Google Scholar] [CrossRef]
- Matai, I.; Kaur, G.; Seyedsalehi, A.; McClinton, A.; Laurencin, C.T. Progress in 3D bioprinting technology for tissue/organ regenerative engineering. Biomaterials 2020, 226, 119536. [Google Scholar] [CrossRef]
- Nakamura, M.; Kobayashi, A.; Takagi, F.; Watanabe, A.; Hiruma, Y.; Ohuchi, K.; Iwasaki, Y.; Horie, M.; Morita, I.; Takatani, S. Biocompatible Inkjet Printing Technique for Designed Seeding of Individual Living Cells. Tissue Eng. 2005, 11, 1658–1666. [Google Scholar] [CrossRef]
- Li, C.; Faulkner-Jones, A.; Dun, A.R.; Jin, J.; Chen, P.; Xing, Y.; Yang, Z.; Li, Z.; Shu, W.; Liu, D.; et al. Rapid Formation of a Supramolecular Polypeptide-DNA Hydrogel for In Situ Three-Dimensional Multilayer Bioprinting. Angew. Chem. Int. Ed. 2015, 54, 3957–3961. [Google Scholar] [CrossRef]
- Binder, K.W.; Zhao, W.; Aboushwareb, T.; Dice, D.; Atala, A.; Yoo, J.J. In situ bioprinting of the skin for burns. J. Am. Coll. Surg. 2010, 211, S76. [Google Scholar] [CrossRef]
- Pardo, L.; Wilson, W.C.; Boland, T. Characterization of Patterned Self-Assembled Monolayers and Protein Arrays Generated by the Ink-Jet Method †. Langmuir 2003, 19, 1462–1466. [Google Scholar] [CrossRef]
- Melchels, F.P.W.; Domingos, M.A.N.; Klein, T.J.; Malda, J.; Bartolo, P.J.; Hutmacher, D.W. Additive manufacturing of tissues and organs. Prog. Polym. Sci. 2012, 37, 1079–1104. [Google Scholar] [CrossRef]
- Xu, T.; Jin, J.; Gregory, C.; Hickman, J.J.; Boland, T. Inkjet printing of viable mammalian cells. Biomaterials 2005, 26, 93–99. [Google Scholar] [CrossRef]
- Wang, W.; Huang, Y.; Grujicic, M.; Chrisey, D.B. Study of Impact-Induced Mechanical Effects in Cell Direct Writing Using Smooth Particle Hydrodynamic Method. J. Manuf. Sci. Eng. 2008, 130, 021012. [Google Scholar] [CrossRef]
- Gruene, M.; Unger, C.; Koch, L.; Deiwick, A.; Chichkov, B. Dispensing pico to nanolitre of a natural hydrogel by laser-assisted bioprinting. Biomed. Eng. OnLine 2011, 10, 19. [Google Scholar] [CrossRef]
- Faulkner-Jones, A.; Fyfe, C.; Cornelissen, D.-J.; Gardner, J.; King, J.; Courtney, A.; Shu, W. Bioprinting of human pluripotent stem cells and their directed differentiation into hepatocyte-like cells for the generation of mini-livers in 3D. Biofabrication 2015, 7, 044102. [Google Scholar] [CrossRef]
- Mahieu-Caputo, D.; Allain, J.-E.; Branger, J.; Coulomb, A.; Delgado, J.-P.; Andreoletti, M.; Mainot, S.; Frydman, R.; Leboulch, P.; Di Santo, J.P.; et al. Repopulation of Athymic Mouse Liver by Cryopreserved Early Human Fetal Hepatoblasts. Hum. Gene Ther. 2004, 15, 1219–1228. [Google Scholar] [CrossRef]
- Weber, A.; Delgado, J.-P.; Parouchev, A.; Branger, J.; Mainot, S.; Coulomb, A.; Mahieu, D. Primate hepatic foetal progenitor cells and their therapeutic potential. Pathol. Biol. 2006, 54, 58–63. [Google Scholar] [CrossRef]
- Irudayaswamy, A.; Muthiah, M.; Zhou, L.; Hung, H.; Jumat, N.H.B.; Haque, J.; Teoh, N.; Farrell, G.; Riehle, K.J.; Lin, J.S.; et al. Long-Term Fate of Human Fetal Liver Progenitor Cells Transplanted in Injured Mouse Livers: Transplant of Human Fetal Liver Progenitor Cells. STEM CELLS 2018, 36, 103–113. [Google Scholar] [CrossRef]
- Oertel, M.; Menthena, A.; Dabeva, M.D.; Shafritz, D.A. Cell Competition Leads to a High Level of Normal Liver Reconstitution by Transplanted Fetal Liver Stem/Progenitor Cells. Gastroenterology 2006, 130, 507–520. [Google Scholar] [CrossRef]
- Herrera, M.B.; Bruno, S.; Buttiglieri, S.; Tetta, C.; Gatti, S.; Deregibus, M.C.; Bussolati, B.; Camussi, G. Isolation and Characterization of a Stem Cell Population from Adult Human Liver. Stem Cells 2006, 24, 2840–2850. [Google Scholar] [CrossRef]
- Najimi, M.; Khuu, D.N.; Lysy, P.A.; Jazouli, N.; Abarca, J.; Sempoux, C.; Sokal, E.M. Adult-Derived Human Liver Mesenchymal-Like Cells as a Potential Progenitor Reservoir of Hepatocytes? Cell Transplant. 2007, 16, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Park, H.-J.; Kim, Y.-A.; Lee, D.-H.; Noh, J.-K.; Kwon, C.H.D.; Jung, S.-M.; Lee, S.-K. The Phenotypic Characteristic of Liver-Derived Stem Cells From Adult Human Deceased Donor Liver. Transplant. Proc. 2012, 44, 1110–1112. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N. Liver regeneration and repair: Hepatocytes, progenitor cells, and stem cells. Hepatology 2004, 39, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Shafritz, D.A.; Oertel, M.; Menthena, A.; Nierhoff, D.; Dabeva, M.D. Liver stem cells and prospects for liver reconstitution by transplanted cells. Hepatology 2006, 43, S89–S98. [Google Scholar] [CrossRef] [PubMed]
- Khuu, D.N.; Nyabi, O.; Maerckx, C.; Sokal, E.; Najimi, M. Adult Human Liver Mesenchymal Stem/Progenitor Cells Participate in Mouse Liver Regeneration after Hepatectomy. Cell Transplant. 2013, 22, 1369–1380. [Google Scholar] [CrossRef] [PubMed]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transplant. 2016, 25, 829–848. [Google Scholar] [CrossRef]
- Wang, X.; Willenbring, H.; Akkari, Y.; Torimaru, Y.; Foster, M.; Al-Dhalimy, M.; Lagasse, E.; Finegold, M.; Olson, S.; Grompe, M. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 2003, 422, 897–901. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, X.; Xiong, X.I.; Yang, Z.; Li, P.; Wang, J.; Sun, Y.U.; Yang, Z.; Hoffman, R.M. Bone Marrow Mesenchymal Stem Cells Reverse Liver Damage in a Carbon Tetrachloride-induced Mouse Model of Chronic Liver Injury. Vivo Athens Greece 2016, 30, 187–193. [Google Scholar]
- Shi, D.; Zhang, J.; Zhou, Q.; Xin, J.; Jiang, J.; Jiang, L.; Wu, T.; Li, J.; Ding, W.; Li, J.; et al. Quantitative evaluation of human bone mesenchymal stem cells rescuing fulminant hepatic failure in pigs. Gut 2017, 66, 955–964. [Google Scholar] [CrossRef]
- Russo, F.P.; Alison, M.R.; Bigger, B.W.; Amofah, E.; Florou, A.; Amin, F.; Bou–Gharios, G.; Jeffery, R.; Iredale, J.P.; Forbes, S.J. The Bone Marrow Functionally Contributes to Liver Fibrosis. Gastroenterology 2006, 130, 1807–1821. [Google Scholar] [CrossRef]
- Iansante, V.; Chandrashekran, A.; Dhawan, A. Cell-based liver therapies: Past, present and future. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170229. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.C.; Zhao, D.; Fletcher, J.; Hewitt, Z.A.; McLean, D.; Urruticoechea-Uriguen, A.; Black, J.R.; Elcombe, C.; Ross, J.A.; Wolf, R.; et al. Efficient Differentiation of Hepatocytes from Human Embryonic Stem Cells Exhibiting Markers Recapitulating Liver Development In Vivo. Stem Cells 2008, 26, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Touboul, T.; Hannan, N.R.F.; Corbineau, S.; Martinez, A.; Martinet, C.; Branchereau, S.; Mainot, S.; Strick-Marchand, H.; Pedersen, R.; Di Santo, J.; et al. Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology 2010, 51, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, G.J.; Hay, D.C.; Park, I.-H.; Fletcher, J.; Hannoun, Z.; Payne, C.M.; Dalgetty, D.; Black, J.R.; Ross, J.A.; Samuel, K.; et al. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology 2010, 51, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Invest. 2010, 120, 3127–3136. [Google Scholar] [CrossRef]
- Hannoun, Z.; Steichen, C.; Dianat, N.; Weber, A.; Dubart-Kupperschmitt, A. The potential of induced pluripotent stem cell derived hepatocytes. J. Hepatol. 2016, 65, 182–199. [Google Scholar] [CrossRef]
- Baxter, M.; Withey, S.; Harrison, S.; Segeritz, C.-P.; Zhang, F.; Atkinson-Dell, R.; Rowe, C.; Gerrard, D.T.; Sison-Young, R.; Jenkins, R.; et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 2015, 62, 581–589. [Google Scholar] [CrossRef]
- Douarin, N.M. An experimental analysis of liver development. Med. Biol. 1975, 53, 427–455. [Google Scholar]
- Gualdi, R.; Bossard, P.; Zheng, M.; Hamada, Y.; Coleman, J.R.; Zaret, K.S. Hepatic specification of the gut endoderm in vitro: Cell signaling and transcriptional control. Genes Dev. 1996, 10, 1670–1682. [Google Scholar] [CrossRef]
- Wandzioch, E.; Zaret, K.S. Dynamic Signaling Network for the Specification of Embryonic Pancreas and Liver Progenitors. Science 2009, 324, 1707–1710. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S. Regulatory phases of early liver development: Paradigms of organogenesis. Nat. Rev. Genet. 2002, 3, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Sauer, V.; Roy-Chowdhury, N.; Guha, C.; Roy-Chowdhury, J. Induced Pluripotent Stem Cells as a Source of Hepatocytes. Curr. Pathobiol. Rep. 2014, 2, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ober, E.A.; Lemaigre, F.P. Development of the liver: Insights into organ and tissue morphogenesis. J. Hepatol. 2018, 68, 1049–1062. [Google Scholar] [CrossRef]
- Duncan, S.A. Mechanisms controlling early development of the liver. Mech. Dev. 2003, 120, 19–33. [Google Scholar] [CrossRef]
- Li, J.; Ning, G.; Duncan, S.A. Mammalian hepatocyte differentiation requires the transcription factor HNF-4alpha. Genes Dev. 2000, 14, 464–474. [Google Scholar]
- Jones, E.A.; Tosh, D.; Wilson, D.I.; Lindsay, S.; Forrester, L.M. Hepatic Differentiation of Murine Embryonic Stem Cells. Exp. Cell Res. 2002, 272, 15–22. [Google Scholar] [CrossRef]
- Rambhatla, L.; Chiu, C.-P.; Kundu, P.; Peng, Y.; Carpenter, M.K. Generation of Hepatocyte-Like Cells from Human Embryonic Stem Cells. Cell Transplant. 2003, 12, 1–11. [Google Scholar] [CrossRef]
- Matsuno, K.; Mae, S.-I.; Okada, C.; Nakamura, M.; Watanabe, A.; Toyoda, T.; Uchida, E.; Osafune, K. Redefining definitive endoderm subtypes by robust induction of human induced pluripotent stem cells. Differentiation 2016, 92, 281–290. [Google Scholar] [CrossRef]
- Touboul, T.; Chen, S.; To, C.C.; Mora-Castilla, S.; Sabatini, K.; Tukey, R.H.; Laurent, L.C. Stage-specific regulation of the WNT/β-catenin pathway enhances differentiation of hESCs into hepatocytes. J. Hepatol. 2016, 64, 1315–1326. [Google Scholar] [CrossRef]
- Lv, L.; Han, Q.; Chu, Y.; Zhang, M.; Sun, L.; Wei, W.; Jin, C.; Li, W. Self-renewal of hepatoblasts under chemically defined conditions by iterative growth factor and chemical screening. Hepatology 2015, 61, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Caron, J.; Pène, V.; Tolosa, L.; Villaret, M.; Luce, E.; Fourrier, A.; Heslan, J.-M.; Saheb, S.; Bruckert, E.; Gómez-Lechón, M.J.; et al. Low-density lipoprotein receptor-deficient hepatocytes differentiated from induced pluripotent stem cells allow familial hypercholesterolemia modeling, CRISPR/Cas-mediated genetic correction, and productive hepatitis C virus infection. Stem Cell Res. Ther. 2019, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, R.D.; Lai, W.G.; Wong, Y.N. Application of a Substrate Cocktail Approach in the Assessment of Cytochrome P450 Induction Using Cultured Human Hepatocytes. J. Biomol. Screen. 2013, 18, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Tang, C.; Rao, M.S.; Weissman, I.L.; Wu, J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013, 19, 998–1004. [Google Scholar] [CrossRef]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef]
- Neofytou, E.; O’Brien, C.G.; Couture, L.A.; Wu, J.C. Hurdles to clinical translation of human induced pluripotent stem cells. J. Clin. Invest. 2015, 125, 2551–2557. [Google Scholar] [CrossRef]
- Steichen, C.; Hannoun, Z.; Luce, E.; Hauet, T.; Dubart-Kupperschmitt, A. Genomic integrity of human induced pluripotent stem cells: Reprogramming, differentiation and applications. World J. Stem Cells 2019, 11, 729–747. [Google Scholar] [CrossRef]
- Nussbaum, J.; Minami, E.; Laflamme, M.A.; Virag, J.A.I.; Ware, C.B.; Masino, A.; Muskheli, V.; Pabon, L.; Reinecke, H.; Murry, C.E. Transplantation of undifferentiated murine embryonic stem cells in the heart: Teratoma formation and immune response. FASEB J. 2007, 21, 1345–1357. [Google Scholar] [CrossRef]
- Cunningham, J.J.; Ulbright, T.M.; Pera, M.F.; Looijenga, L.H.J. Lessons from human teratomas to guide development of safe stem cell therapies. Nat. Biotechnol. 2012, 30, 849–857. [Google Scholar] [CrossRef]
- Murry, C.E.; Keller, G. Differentiation of Embryonic Stem Cells to Clinically Relevant Populations: Lessons from Embryonic Development. Cell 2008, 132, 661–680. [Google Scholar] [CrossRef]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Sackett, S.D.; Brown, M.E.; Tremmel, D.M.; Ellis, T.; Burlingham, W.J.; Odorico, J.S. Modulation of human allogeneic and syngeneic pluripotent stem cells and immunological implications for transplantation. Transplant. Rev. 2016, 30, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.; Widera, A.; Schmidt-Heck, W.; Campos, G.; Meyer, C.; Cadenas, C.; Reif, R.; Stöber, R.; Hammad, S.; Pütter, L.; et al. Gene network activity in cultivated primary hepatocytes is highly similar to diseased mammalian liver tissue. Arch. Toxicol. 2016, 90, 2513–2529. [Google Scholar] [CrossRef]
- Unzu, C.; Planet, E.; Brandenberg, N.; Fusil, F.; Cassano, M.; Perez-Vargas, J.; Friedli, M.; Cosset, F.; Lutolf, M.P.; Wildhaber, B.E.; et al. Pharmacological Induction of a Progenitor State for the Efficient Expansion of Primary Human Hepatocytes. Hepatology 2019, 69, 2214–2231. [Google Scholar] [CrossRef]
- Li, T.; Zhou, Z.-W.; Ju, Z.; Wang, Z.-Q. DNA Damage Response in Hematopoietic Stem Cell Ageing. Genomics Proteomics Bioinform. 2016, 14, 147–154. [Google Scholar] [CrossRef]
- Gazdic, M.; Volarevic, V.; Arsenijevic, N.; Stojkovic, M. Mesenchymal Stem Cells: A Friend or Foe in Immune-Mediated Diseases. Stem Cell Rev. Rep. 2015, 11, 280–287. [Google Scholar] [CrossRef]
- Fu, X.; Xu, Y. Challenges to the clinical application of pluripotent stem cells: Towards genomic and functional stability. Genome Med. 2012, 4, 55. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Liu, C.-L.; Ting, C.-Y.; Chiu, Y.-T.; Cheng, Y.-C.; Nicholson, M.W.; Hsieh, P.C.H. Human iPSC banking: Barriers and opportunities. J. Biomed. Sci. 2019, 26, 87. [Google Scholar] [CrossRef]
- Ronen, D.; Benvenisty, N. Genomic stability in reprogramming. Curr. Opin. Genet. Dev. 2012, 22, 444–449. [Google Scholar] [CrossRef]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Methods for Organoid Generation | |
|---|---|
| Scaffold-Based Methods |
|
| Scaffold-Free Methods |
|
| Advantages | Disadvantages | |
|---|---|---|
| Orthotopic Liver Transplantation (OLT) | 88% patient survival Clinically defined | Shortage of donors Post-surgery complications Life-long immunosuppressive treatment Organ rejection |
| Artificial Liver (AL) Device | Detoxification ability Bridge patients to OLT | Selective removal/detoxification of toxins Ineffective against encephalopathy Temporary support device |
| Cell Transplantation | Surgical procedure safer and less invasive than OLT Partial correction of liver metabolic disorders | Shortage of cells Transitory improvement of patients’ conditions |
| Bio Artificial Liver (BAL) Device | Improved detoxification ability due to biological components Bridge patients to OLT | Shortage of cells Clinical trials suspended or incomplete Complex set-up and scale-up |
| Decellularized/CellularizedLiver Scaffolds (Pre-Clinical Development) | Improvement of hepatic cells functions with respect to classic scaffold-based culture approaches Liver-like tissue bio-construction transplantable | Shortage of cells Partial cell repopulation of the scaffolds Slow maturation of the construct Poor viability in pre-clinical studies |
| Liver Biofabrication and Bioprinting (Pre-Clinical Development) | Easy scale-up of the 3D liver constructs Improvement of hepatic cell maturations with respect to 3D classic culture approaches | Shortage of cells Slow maturation of the construct Contradictory published data on construct viability |
| Cell | Source | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|
| PHHs | Cadaveric liver Partial hepatectomy | No ethical/political concerns Mature functional cells No risk of teratoma formation Clinically established cells | Immunogenicity Not proliferative in vitro Rapid loss of functionality Not available at large scale Cell aging DNA damage | [23,134] |
| FLPs | Aborted fetus | Highly proliferative Lower immunogenicity than PHHs Transdifferentiation into mature hepatocytes | Ethical concern Low number of cells per fetal liver leading to multiple donors Difficult supply | [135] |
| AdLSCs | Adult liver | Proliferative Bi-potent | Immunogenicity Not available at large scale Cell aging DNA damage | [90,91,92] |
| HSCs | Bone marrow Blood | No ethical concern Highly proliferative Non-invasive collection procedures Abundant supplies (bone marrow) Low viral contamination No risk of teratoma formation Contribution to liver regeneration | Poorly effective: cell fusion with resident hepatocytes/trophic effects Limited number per single cord blood unit (multiple donors) Cell aging DNA damage | [136] |
| MSCs | Bone marrow Umbilical cord Adipose tissue Blood | Highly proliferative Multipotency Immunomodulatory effects Antifibrotic effects | Downregulation of apoptotic genes Downregulation of DNA repair genes Heteroplasmic point mutations Viral transmission Cell aging DNA damage | [137] |
| ESCs | Embryos | Self-renewal Pluripotency | Ethical concern Tumorigenicity Safety concerns (genetic stability) Immunogenicity | [126] |
| iPSCs | Reprogramming of somatic cells | Self-renewal Pluripotency Possibly autologous | Safety concerns Tumorigenicity | [126,138] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messina, A.; Luce, E.; Hussein, M.; Dubart-Kupperschmitt, A. Pluripotent-Stem-Cell-Derived Hepatic Cells: Hepatocytes and Organoids for Liver Therapy and Regeneration. Cells 2020, 9, 420. https://doi.org/10.3390/cells9020420
Messina A, Luce E, Hussein M, Dubart-Kupperschmitt A. Pluripotent-Stem-Cell-Derived Hepatic Cells: Hepatocytes and Organoids for Liver Therapy and Regeneration. Cells. 2020; 9(2):420. https://doi.org/10.3390/cells9020420
Chicago/Turabian StyleMessina, Antonietta, Eléanor Luce, Marwa Hussein, and Anne Dubart-Kupperschmitt. 2020. "Pluripotent-Stem-Cell-Derived Hepatic Cells: Hepatocytes and Organoids for Liver Therapy and Regeneration" Cells 9, no. 2: 420. https://doi.org/10.3390/cells9020420
APA StyleMessina, A., Luce, E., Hussein, M., & Dubart-Kupperschmitt, A. (2020). Pluripotent-Stem-Cell-Derived Hepatic Cells: Hepatocytes and Organoids for Liver Therapy and Regeneration. Cells, 9(2), 420. https://doi.org/10.3390/cells9020420