Impairment of Membrane Lipid Homeostasis by Bichalcone Analog TSWU-BR4 Attenuates Function of GRP78 in Regulation of the Oxidative Balance and Invasion of Cancer Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Chemicals, Reagents, and Plasmids
2.3. Antibodies
2.4. Cell Viability Assay
2.5. Assays for the Detection of Caspase-3 Activity and Early Apoptotic Cells
2.6. Density-Based Membrane Flotation Technique
2.7. Isolation of ER and Cytosolic Fractions
2.8. Subcellular Fractionation
2.9. Western Blot and Co-Immunoprecipitation
2.10. Cell Surface Biotinylation
2.11. Measurement of Cell Surface or Intracellular GRP78 by Flow Cytometry
2.12. Rac1 Activation Assay
2.13. Determination of Cholesterol, Sphingomyelin, and Ceramide
2.14. Measurement ofMMP-2Promoter Activity
2.15. In Vitro Invasion Assay
2.16. Plasmid Transfection
2.17. NF-κB Promoter Activity
2.18. Statistical Analysis
3. Results
3.1. Delocalization of Cell Surface GRP78 by TSWU-BR4 Induces the Lipid Raft Membrane Localization of Unphosphorylated PTEN to Affect Cancer Cell Invasion
3.2. Recruitment of Unphosphorylated PTEN to the Lipid Raft Membranes to Form a Complex with p85α in TSWU-BR4-Treated Cells
3.3. TSWU-BR4-Induced Dissociation of GRP78 and p85α Leads to the Complex Formation of Lipid Raft Membrane-Associated p85α–Unphosphorylated PTEN and Subsequently Causes Increased ROS Accumulation and Decreased Levels of Cholesterol and Invaded Cells
3.4. Activation of ASM by TSWU-BR4 Deregulates Membrane Trafficking of GRP78, Causes the Lipid Raft Membrane-Associated p85α–Unphosphorylated PTEN Complex Formation, and Thereby Attenuates GRP78-Modulated Oxidative Stress Balance and Cell Invasion
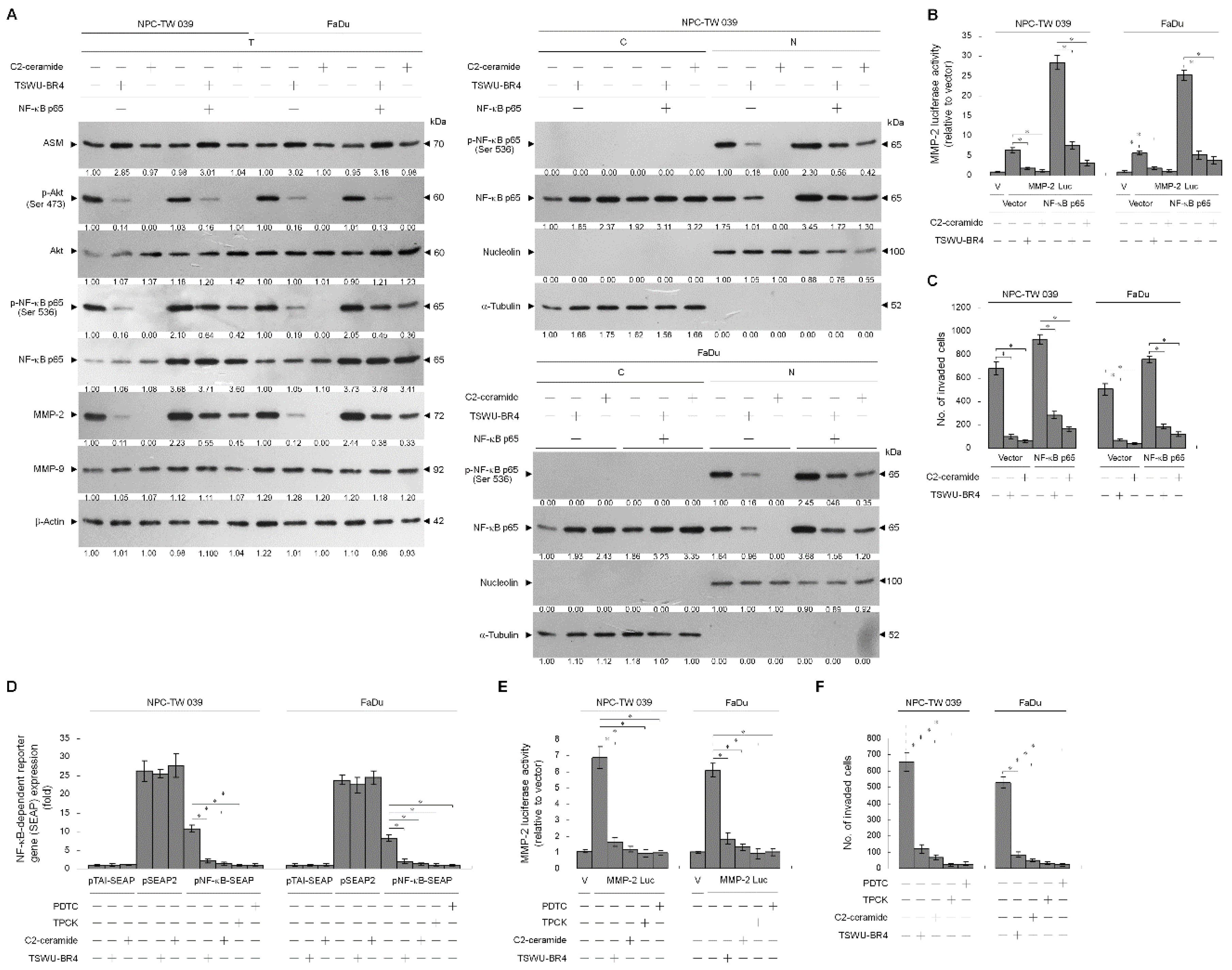
3.5. TSWU-BR4-Induced Ceramide Generation Attenuates Cell Invasion by Suppressing the PI3K–Akt-Regulated NF-κB-Mediated MMP-2 Expression Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Acid sphingomyelinase | ASM |
| Endoplasmic reticulum | ER |
| Glucose regulated protein 78 | GRP78 |
| Matrix metalloproteinase-2 | MMP-2 |
| Nasopharyngeal carcinoma | NPC |
| Nuclear factor erythroid-2-related factor 2 | Nrf2 |
| Nuclear factor-kappa B | NF-κB |
| Phosphatase and tensin homolog deleted from chromosome 10 | PTEN |
| Phosphatidylinositol 3-kinase | PI3K |
| Phosphatidylinositol-4,5-bisphosphate | PIP2 |
| Phosphatidylinositol-3,4,5-trisphosphate | PIP3 |
| Protein kinase B | Akt |
| Protein kinase RNA-like endoplasmic reticulum kinase | PERK |
| Ras-related C3 botulinum toxin substrate 1 | Rac1 |
| Reactive oxygen species | ROS |
| Short hairpin RNA | shRNA |
| Human pharyngeal squamous carcinoma | PSC |
References
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Drevot, P.; Langlet, C.; Guo, X.J.; Bernard, A.M.; Colard, O.; Chauvin, J.P.; Lasserre, R.; He, H.T. TCR signal initiation machinery is pre-assembled and activated in a subset of membrane rafts. EMBO J. 2002, 21, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lowry, P.R.; Zhou, X.; Depry, C.; Wei, Z.; Wong, G.W.; Zhang, J. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc. Natl. Acad. Sci. USA 2011, 108, 14509–14514. [Google Scholar] [CrossRef]
- Payapilly, A.; Malliri, A. Compartmentalisation of RAC1 signalling. Curr. Opin. Cell Biol. 2018, 54, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Cheung, L.W.; Walkiewicz, K.W.; Besong, T.M.; Guo, H.; Hawke, D.H.; Arold, S.T.; Mills, G.B. Regulation of the PI3K pathway through a p85alpha monomer-homodimer equilibrium. eLife 2015, 4, e06866. [Google Scholar] [CrossRef]
- Raftopoulou, M.; Etienne-Manneville, S.; Self, A.; Nicholls, S.; Hall, A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science 2004, 303, 1179–1181. [Google Scholar] [CrossRef]
- Megha; London, E. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): Implications for lipid raft structure and function. J. Biol. Chem. 2004, 279, 9997–10004. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tseng, C.C.; Tsai, Y.L.; Fu, X.; Schiff, R.; Lee, A.S. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PLoS ONE 2013, 8, e80071. [Google Scholar] [CrossRef] [PubMed]
- Arap, M.A.; Lahdenranta, J.; Mintz, P.J.; Hajitou, A.; Sarkis, A.S.; Arap, W.; Pasqualini, R. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell 2004, 6, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lillo, A.M.; Steiniger, S.C.; Liu, Y.; Ballatore, C.; Anichini, A.; Mortarini, R.; Kaufmann, G.F.; Zhou, B.; Felding-Habermann, B.; et al. Targeting heat shock proteins on cancer cells: Selection, characterization, and cell-penetrating properties of a peptidic GRP78 ligand. Biochemistry 2006, 45, 9434–9444. [Google Scholar] [CrossRef]
- Liu, Y.; Steiniger, S.C.; Kim, Y.; Kaufmann, G.F.; Felding-Habermann, B.; Janda, K.D. Mechanistic studies of a peptidic GRP78 ligand for cancer cell-specific drug delivery. Mol. Pharm. 2007, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef]
- Cook, K.L.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Warri, A.; Hilakivi-Clarke, L.; Roberts, D.D.; Clarke, R. Endoplasmic Reticulum Stress Protein GRP78 Modulates Lipid Metabolism to Control Drug Sensitivity and Antitumor Immunity in Breast Cancer. Cancer Res. 2016, 76, 5657–5670. [Google Scholar] [CrossRef]
- Choy, K.W.; Murugan, D.; Mustafa, M.R. Natural products targeting ER stress pathway for the treatment of cardiovascular diseases. Pharmacol. Res. 2018, 132, 119–129. [Google Scholar] [CrossRef]
- Suyama, K.; Watanabe, M.; Sakabe, K.; Otomo, A.; Okada, Y.; Terayama, H.; Imai, T.; Mochida, J. GRP78 suppresses lipid peroxidation and promotes cellular antioxidant levels in glial cells following hydrogen peroxide exposure. PLoS ONE 2014, 9, e86951. [Google Scholar] [CrossRef]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Durden, B.; Hadad, R.; Banerjee, S.; Dudeja, V.; Saluja, A.; Banerjee, S. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining “stemness”. Cell Death Dis. 2019, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef]
- Reddy, M.V.; Shen, Y.C.; Yang, J.S.; Hwang, T.L.; Bastow, K.F.; Qian, K.; Lee, K.H.; Wu, T.S. New bichalcone analogs as NF-kappaB inhibitors and as cytotoxic agents inducing Fas/CD95-dependent apoptosis. Bioorganic Med. Chem. 2011, 19, 1895–1906. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sahni, S.; Hickok, J.R.; Thomas, D.D. Nitric oxide reduces oxidative stress in cancer cells by forming dinitrosyliron complexes. Nitric Oxid. Biol. Chem. 2018, 76, 37–44. [Google Scholar] [CrossRef]
- Caneba, C.A.; Yang, L.; Baddour, J.; Curtis, R.; Win, J.; Hartig, S.; Marini, J.; Nagrath, D. Nitric oxide is a positive regulator of the Warburg effect in ovarian cancer cells. Cell Death Dis. 2014, 5, e1302. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.T.; Wong, C.I.; Chan, W.Y.; Tzung, K.W.; Ho, J.K.; Hsu, M.M.; Chuang, S.M. Establishment and characterization of two nasopharyngeal carcinoma cell lines. Lab. Investig. J. Tech. Methods Pathol. 1990, 62, 713–724. [Google Scholar]
- Lin, M.L.; Lu, Y.C.; Chung, J.G.; Li, Y.C.; Wang, S.G.; NG, S.H.; Wu, C.Y.; Su, H.L.; Chen, S.S. Aloe-emodin induces apoptosis of human nasopharyngeal carcinoma cells via caspase-8-mediated activation of the mitochondrial death pathway. Cancer Lett. 2010, 291, 46–58. [Google Scholar] [CrossRef]
- Lin, M.L.; Lu, Y.C.; Chen, H.Y.; Lee, C.C.; Chung, J.G.; Chen, S.S. Suppressing the formation of lipid raft-associated Rac1/PI3K/Akt signaling complexes by curcumin inhibits SDF-1alpha-induced invasion of human esophageal carcinoma cells. Mol. Carcinog. 2014, 53, 360–379. [Google Scholar] [CrossRef]
- Zong, W.X.; Li, C.; Hatzivassiliou, G.; Lindsten, T.; Yu, Q.C.; Yuan, J.; Thompson, C.B. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 2003, 162, 59–69. [Google Scholar] [CrossRef]
- Taha, M.S.; Nouri, K.; Milroy, L.G.; Moll, J.M.; Herrmann, C.; Brunsveld, L.; Piekorz, R.P.; Ahmadian, M.R. Subcellular fractionation and localization studies reveal a direct interaction of the fragile X mental retardation protein (FMRP) with nucleolin. PLoS ONE 2014, 9, e91465. [Google Scholar] [CrossRef]
- Wu, C.W.; Wang, S.G.; Lin, M.L.; Chen, S.S. Downregulation of miR-144 by triptolide enhanced p85alpha-PTEN complex formation causing S phase arrest of human nasopharyngeal carcinoma cells. Eur. J. Pharmacol. 2019, 855, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.L.; Chen, S.S.; Ng, S.H. CHM-1 Suppresses Formation of Cell Surface-associated GRP78-p85alpha Complexes, Inhibiting PI3K-AKT Signaling and Inducing Apoptosis of Human Nasopharyngeal Carcinoma Cells. Anticancer Res. 2015, 35, 5359–5368. [Google Scholar] [PubMed]
- Lin, M.L.; Lu, Y.C.; Su, H.L.; Lin, H.T.; Lee, C.C.; Kang, S.E.; Lai, T.C.; Chung, J.G.; Chen, S.S. Destabilization of CARP mRNAs by aloe-emodin contributes to caspase-8-mediated p53-independent apoptosis of human carcinoma cells. J. Cell. Biochem. 2011, 112, 1176–1191. [Google Scholar] [CrossRef] [PubMed]
- Dobrowsky, R.T.; Kolesnick, R.N. Analysis of sphingomyelin and ceramide levels and the enzymes regulating their metabolism in response to cell stress. Methods Cell Biol. 2001, 66, 135–165. [Google Scholar]
- Lu, H.L.; Chen, S.S.; Hsu, W.T.; Lu, Y.C.; Lee, C.C.; Wu, T.S.; Lin, M.L. Suppression of phospho-p85alpha-GTP-Rac1 lipid raft interaction by bichalcone analog attenuates cancer cell invasion. Mol. Carcinog. 2016, 55, 2106–2120. [Google Scholar] [CrossRef]
- Wheelock, M.J.; Johnson, K.R. Cadherins as modulators of cellular phenotype. Annu. Rev. Cell Dev. Biol. 2003, 19, 207–235. [Google Scholar] [CrossRef]
- Goswami, R.; Singh, D.; Phillips, G.; Kilkus, J.; Dawson, G. Ceramide regulation of the tumor suppressor phosphatase PTEN in rafts isolated from neurotumor cell lines. J. Neurosci. Res. 2005, 81, 541–550. [Google Scholar] [CrossRef]
- Oninla, V.O.; Breiden, B.; Babalola, J.O.; Sandhoff, K. Acid sphingomyelinase activity is regulated by membrane lipids and facilitates cholesterol transfer by NPC2. J. Lipid Res. 2014, 55, 2606–2619. [Google Scholar] [CrossRef]
- Bai, A.; Kokkotou, E.; Zheng, Y.; Robson, S.C. Role of acid sphingomyelinase bioactivity in human CD4+ T-cell activation and immune responses. Cell Death Dis. 2015, 6, e1828. [Google Scholar] [CrossRef]
- Nakanishi, C.; Toi, M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat. Rev. Cancer 2005, 5, 297–309. [Google Scholar] [CrossRef]
- Perona, R.; Montaner, S.; Saniger, L.; Sanchez-Perez, I.; Bravo, R.; Lacal, J.C. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997, 11, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef] [PubMed]
- Murai, T. The role of lipid rafts in cancer cell adhesion and migration. Int. J. Cell Biol. 2012, 2012, 763283. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, N.; Fialho, A.M. Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies. Int. J. Mol. Sci. 2018, 19, 3871. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef]
- Sozen, E.; Ozer, N.K. Impact of high cholesterol and endoplasmic reticulum stress on metabolic diseases: An updated mini-review. Redox Biol. 2017, 12, 456–461. [Google Scholar] [CrossRef]
- Bellezza, I.; Scarpelli, P.; Pizzo, S.V.; Grottelli, S.; Costanzi, E.; Minelli, A. ROS-independent Nrf2 activation in prostate cancer. Oncotarget 2017, 8, 67506–67518. [Google Scholar] [CrossRef]
- Li, Y.C.; Park, M.J.; Ye, S.K.; Kim, C.W.; Kim, Y.N. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am. J. Pathol. 2006, 168, 1107–1118. [Google Scholar] [CrossRef]
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef]
- Grassme, H.; Jendrossek, V.; Bock, J.; Riehle, A.; Gulbins, E. Ceramide-rich membrane rafts mediate CD40 clustering. J. Immunol. 2002, 168, 298–307. [Google Scholar] [CrossRef]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic beta-cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef] [PubMed]
- Chagpar, R.B.; Links, P.H.; Pastor, M.C.; Furber, L.A.; Hawrysh, A.D.; Chamberlain, M.D.; Anderson, D.H. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 5471–5476. [Google Scholar] [CrossRef] [PubMed]
- Otsu, M.; Hiles, I.; Gout, I.; Fry, M.J.; Ruiz-Larrea, F.; Panayotou, G.; Thompson, A.; Dhand, R.; Hsuan, J.; Totty, N.; et al. Characterization of two 85 kd proteins that associate with receptor tyrosine kinases, middle-T/pp60c-src complexes, and PI3-kinase. Cell 1991, 65, 91–104. [Google Scholar] [CrossRef]
- Shepherd, P.R.; Withers, D.J.; Siddle, K. Phosphoinositide 3-kinase: The key switch mechanism in insulin signalling. Biochem. J. 1998, 333, 471–490. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Spangle, J.M.; Ohlson, C.E.; Cheng, H.; Roberts, T.M.; Cantley, L.C.; Zhao, J.J. PI3K-p110alpha mediates the oncogenic activity induced by loss of the novel tumor suppressor PI3K-p85alpha. Proc. Natl. Acad. Sci. USA 2017, 114, 7095–7100. [Google Scholar] [CrossRef]
- Ueki, K.; Fruman, D.A.; Brachmann, S.M.; Tseng, Y.H.; Cantley, L.C.; Kahn, C.R. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol. Cell. Biol. 2002, 22, 965–977. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Ho-Fun Lee, V.; Wong, A.M.; Kwong, D.L.; Zhu, Y.H.; Dong, S.S.; Kong, K.L.; Chen, J.; Tsao, S.W.; Guan, X.Y.; et al. MicroRNA-144 promotes cell proliferation, migration and invasion in nasopharyngeal carcinoma through repression of PTEN. Carcinogenesis 2013, 34, 454–463. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, T.-L.; Wang, S.-G.; Chan, W.-L.; Lee, C.-H.; Wu, T.-S.; Lin, M.-L.; Chen, S.-S. Impairment of Membrane Lipid Homeostasis by Bichalcone Analog TSWU-BR4 Attenuates Function of GRP78 in Regulation of the Oxidative Balance and Invasion of Cancer Cells. Cells 2020, 9, 371. https://doi.org/10.3390/cells9020371
Lee T-L, Wang S-G, Chan W-L, Lee C-H, Wu T-S, Lin M-L, Chen S-S. Impairment of Membrane Lipid Homeostasis by Bichalcone Analog TSWU-BR4 Attenuates Function of GRP78 in Regulation of the Oxidative Balance and Invasion of Cancer Cells. Cells. 2020; 9(2):371. https://doi.org/10.3390/cells9020371
Chicago/Turabian StyleLee, Tsung-Lin, Shyang-Guang Wang, Wen-Ling Chan, Ching-Hsiao Lee, Tian-Shung Wu, Meng-Liang Lin, and Shih-Shun Chen. 2020. "Impairment of Membrane Lipid Homeostasis by Bichalcone Analog TSWU-BR4 Attenuates Function of GRP78 in Regulation of the Oxidative Balance and Invasion of Cancer Cells" Cells 9, no. 2: 371. https://doi.org/10.3390/cells9020371
APA StyleLee, T.-L., Wang, S.-G., Chan, W.-L., Lee, C.-H., Wu, T.-S., Lin, M.-L., & Chen, S.-S. (2020). Impairment of Membrane Lipid Homeostasis by Bichalcone Analog TSWU-BR4 Attenuates Function of GRP78 in Regulation of the Oxidative Balance and Invasion of Cancer Cells. Cells, 9(2), 371. https://doi.org/10.3390/cells9020371