Poison-Exon Inclusion in DHX9 Reduces Its Expression and Sensitizes Ewing Sarcoma Cells to Chemotherapeutic Treatment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Drug Treatment
2.2. Transfections
2.3. SDS–PAGE and Western Blot Analyses
2.4. Real-time Quantitative PCR Analyses (RT-qPCR)
2.5. CLIP Assays
2.6. MTS Proliferation Assay
2.7. Colony Formation Assay
2.8. Patient Datasets
2.9. Bioinformatic Analysis
2.10. Statistical Analysis
3. Results
3.1. DHX9 Expression Correlates with Worse Prognosis in Ewing Sarcoma Patients
3.2. A siRNA Library Identifies Regulators of DHX9 Alternative Splicing
3.3. CLIP Assay Unveils Direct Binding of Specific SR Proteins and HnRNPs to DHX9 Pre-mRNA
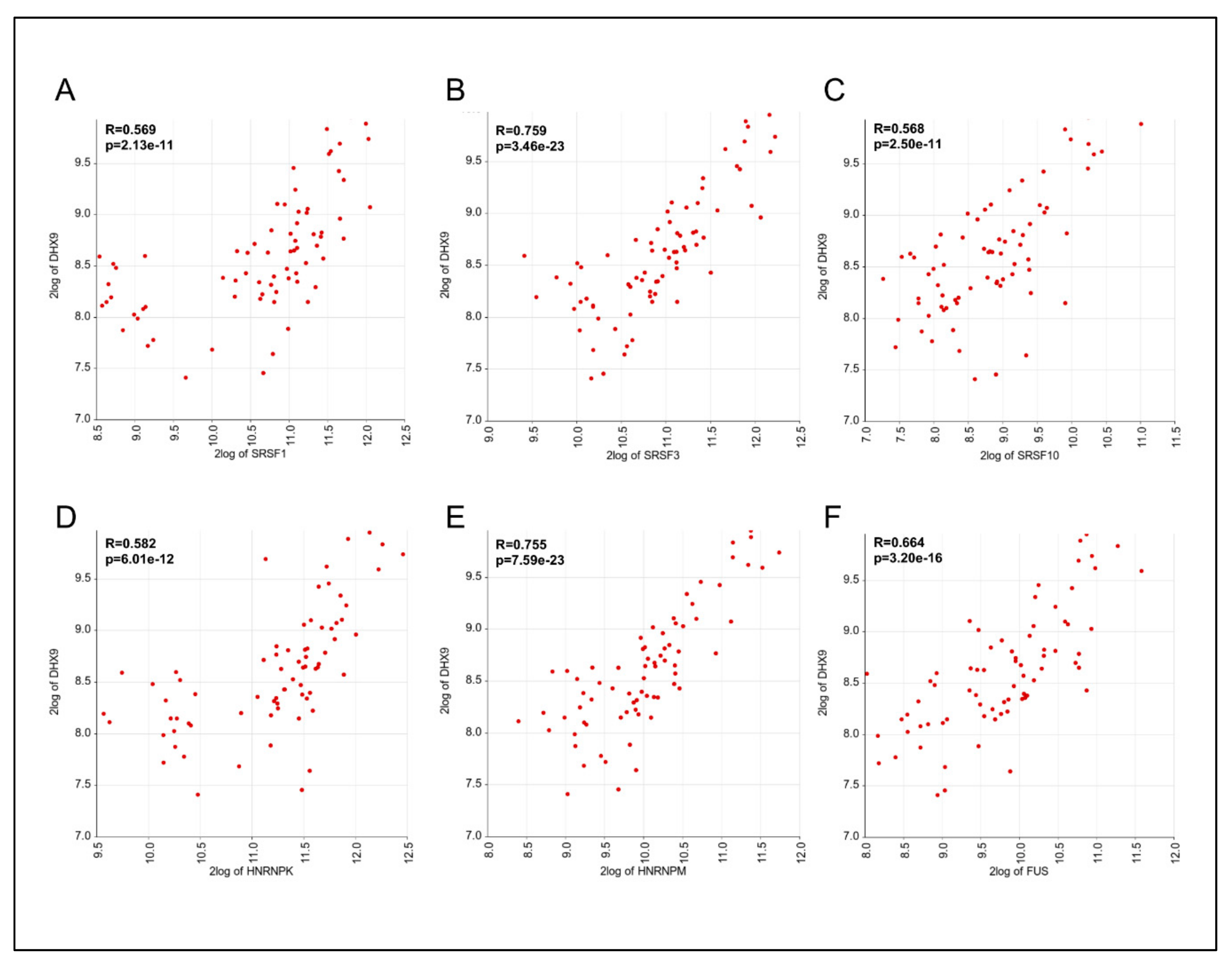
3.4. Expression of DHX9, HNRNPM, and SRSF3 is Positively Correlated in Ewing Sarcoma Patients
3.5. Depletion of SRSF3 and HnRNPM Affects the Expression of EWS-FLI1 Target Genes
3.6. Depletion of SRSF3 and hnRNPM Increases Doxorubicin Sensitivity of Ewing Sarcoma Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Graveley, B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Passacantilli, I.; Sette, C. Alternative splicing and cell survival: From tissue homeostasis to disease. Cell Death Differ. 2016, 23, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002, 30, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.E.; Parker, R. Nonsense-mediated mRNA decay: Terminating erroneous gene expression. Curr. Opin. Cell Biol. 2004, 16, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Lareau, L.F.; Inada, M.; Green, R.E.; Wengrod, J.C.; Brenner, S.E. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 2007, 446, 926–929. [Google Scholar] [CrossRef]
- Ruiz-Echevarría, M.J.; González, C.I.; Peltz, S.W. Identifying the right stop: Determining how the surveillance complex recognizes and degrades an aberrant mRNA. EMBO J. 1998, 17, 575–589. [Google Scholar] [CrossRef]
- Valacca, C.; Bonomi, S.; Buratti, E.; Pedrotti, S.; Baralle, F.E.; Sette, C.; Ghigna, C.; Biamonti, G. Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J. Cell Biol. 2010, 191, 87–99. [Google Scholar] [CrossRef]
- Jacob, A.G.; Smith, C.W.J. Intron retention as a component of regulated gene expression programs. Hum. Genet. 2017, 136, 1043–1057. [Google Scholar] [CrossRef]
- Ip, J.Y.; Schmidt, D.; Pan, Q.; Ramani, A.K.; Fraser, A.G.; Odom, D.T.; Blencowe, B.J. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res. 2011, 21, 390–401. [Google Scholar] [CrossRef]
- Fidaleo, M.; Svetoni, F.; Volpe, E.; Miñana, B.; Caporossi, D.; Paronetto, M.P. Genotoxic stress inhibits Ewing sarcoma cell growth by modulating alternative pre-mRNA processing of the RNA helicase DHX9. Oncotarget 2015, 6, 31740–31757. [Google Scholar] [CrossRef] [PubMed]
- Fidaleo, M.; De Paola, E.; Paronetto, M.P. The RNA helicase A in malignant transformation. Oncotarget 2016, 7, 28711–28723. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.F.; Schlegel, B.P.; Nakajima, T.; Wolpin, E.S.; Parvin, J.D. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat. Genet. 1998, 19, 254–256. [Google Scholar] [CrossRef]
- Aratani, S.; Fujii, R.; Oishi, T.; Fujita, H.; Amano, T.; Ohshima, T.; Hagiwara, M.; Fukamizu, A.; Nakajima, T. Dual roles of RNA helicase A in CREB-dependent transcription. Mol. Cell Biol. 2001, 21, 4460–4469. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Bacolla, A.; Chakraborty, P.; Grosse, F.; Vasquez, K.M. Human DHX9 helicase unwinds triple-helical DNA structures. Biochemistry 2010, 49, 6992–6999. [Google Scholar] [CrossRef]
- Jain, A.; Bacolla, A.; Del Mundo, I.M.; Zhao, J.; Wang, G.; Vasquez, K.M. DHX9 helicase is involved in preventing genomic instability induced by alternatively structured DNA in human cells. Nucleic Acids Res. 2013, 41, 10345–10357. [Google Scholar] [CrossRef]
- Chakraborty, P.; Grosse, F. WRN helicase unwinds Okazaki fragment-like hybrids in a reaction stimulated by the human DHX9 helicase. Nucleic Acids Res. 2010, 38, 4722–4730. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Erkizan, V.; Levenson, A.; Abaan, O.D.; Parvin, J.D.; Cripe, T.P.; Rice, A.M.; Lee, S.B.; Uren, A. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res. 2006, 66, 5574–5581. [Google Scholar] [CrossRef]
- Palombo, R.; Frisone, P.; Fidaleo, M.; Mercatelli, N.; Sette, C.; Paronetto, M.P. The Promoter-Associated Noncoding RNA. Cancer Res. 2019, 79, 3570–3582. [Google Scholar] [CrossRef]
- Mercatelli, N.; Fortini, D.; Palombo, R.; Paronetto, M.P. Small molecule inhibition of Ewing sarcoma cell growth via targeting the long non coding RNA HULC. Cancer Lett. 2019. [Google Scholar] [CrossRef]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Youbi, S.E.; Hong, S.P.; Kallakury, B.; Monroe, P.; Erkizan, H.V.; Barber-Rotenberg, J.S.; Houghton, P.; Üren, A.; Toretsky, J.A. Pharmacokinetic modeling optimizes inhibition of the ‘undruggable’ EWS-FLI1 transcription factor in Ewing Sarcoma. Oncotarget 2014, 5, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Zöllner, S.K.; Selvanathan, S.P.; Graham, G.T.; Commins, R.M.T.; Hong, S.H.; Moseley, E.; Parks, S.; Haladyna, J.N.; Erkizan, H.V.; Dirksen, U.; et al. Inhibition of the oncogenic fusion protein EWS-FLI1 causes G. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Passacantilli, I.; Frisone, P.; De Paola, E.; Fidaleo, M.; Paronetto, M.P. hnRNPM guides an alternative splicing program in response to inhibition of the PI3K/AKT/mTOR pathway in Ewing sarcoma cells. Nucleic Acids Res. 2017, 45, 12270–12284. [Google Scholar] [CrossRef]
- Savola, S.; Klami, A.; Tripathi, A.; Niini, T.; Serra, M.; Picci, P.; Kaski, S.; Zambelli, D.; Scotlandi, K.; Knuutila, S. Combined use of expression and CGH arrays pinpoints novel candidate genes in Ewing sarcoma family of tumors. BMC Cancer 2009, 9, 17. [Google Scholar] [CrossRef]
- Available online: http://cbio.mskcc.org/Public/sarcoma_array_data/ (accessed on 1 December 2011).
- Smith, C.W.; Valcárcel, J. Alternative pre-mRNA splicing: The logic of combinatorial control. Trends Biochem. Sci. 2000, 25, 381–388. [Google Scholar] [CrossRef]
- Cáceres, J.F.; Kornblihtt, A.R. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends Genet. 2002, 18, 186–193. [Google Scholar] [CrossRef]
- Chakraborty, P.; Huang, J.T.J.; Hiom, K. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018, 9, 4346. [Google Scholar] [CrossRef]
- Aktaş, T.; Avşar Ilık, İ.; Maticzka, D.; Bhardwaj, V.; Pessoa Rodrigues, C.; Mittler, G.; Manke, T.; Backofen, R.; Akhtar, A. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature 2017, 544, 115–119. [Google Scholar] [CrossRef]
- Balamuth, N.J.; Womer, R.B. Ewing’s sarcoma. Lancet Oncol. 2010, 11, 184–192. [Google Scholar] [CrossRef]
- Trincado, J.L.; Sebestyén, E.; Pagés, A.; Eyras, E. The prognostic potential of alternative transcript isoforms across human tumors. Genome Med. 2016, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Peng, Z.G.; He, R.Q.; Luo, B.; Ma, J.; Hu, X.H.; Dang, Y.W.; Chen, G.; Pan, S.L. Prognostic index of aberrant mRNA splicing profiling acts as a predictive indicator for hepatocellular carcinoma based on TCGA SpliceSeq data. Int. J. Oncol. 2019, 55, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Selvanathan, S.P.; Graham, G.T.; Grego, A.R.; Baker, T.M.; Hogg, J.R.; Simpson, M.; Batish, M.; Crompton, B.; Stegmaier, K.; Tomazou, E.M.; et al. EWS-FLI1 modulated alternative splicing of ARID1A reveals novel oncogenic function through the BAF complex. Nucleic Acids Res. 2019, 47, 9619–9636. [Google Scholar] [CrossRef] [PubMed]
- Selvanathan, S.P.; Graham, G.T.; Erkizan, H.V.; Dirksen, U.; Natarajan, T.G.; Dakic, A.; Yu, S.; Liu, X.; Paulsen, M.T.; Ljungman, M.E.; et al. Oncogenic fusion protein EWS-FLI1 is a network hub that regulates alternative splicing. Proc. Natl. Acad. Sci. USA 2015, 112, E1307–E1316. [Google Scholar] [CrossRef]
- Sand, L.G.; Szuhai, K.; Hogendoorn, P.C. Sequencing Overview of Ewing Sarcoma: A Journey across Genomic, Epigenomic and Transcriptomic Landscapes. Int. J. Mol. Sci. 2015, 16, 16176–16215. [Google Scholar] [CrossRef]
- Paronetto, M.P.; Bernardis, I.; Volpe, E.; Bechara, E.; Sebestyén, E.; Eyras, E.; Valcárcel, J. Regulation of FAS exon definition and apoptosis by the Ewing sarcoma protein. Cell Rep. 2014, 7, 1211–1226. [Google Scholar] [CrossRef]
- Sanchez, G.; Bittencourt, D.; Laud, K.; Barbier, J.; Delattre, O.; Auboeuf, D.; Dutertre, M. Alteration of cyclin D1 transcript elongation by a mutated transcription factor up-regulates the oncogenic D1b splice isoform in cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 6004–6009. [Google Scholar] [CrossRef]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Obeng, E.A.; Stewart, C.; Abdel-Wahab, O. Altered RNA Processing in Cancer Pathogenesis and Therapy. Cancer Discov. 2019, 9, 1493–1510. [Google Scholar] [CrossRef]
- Anczuków, O.; Krainer, A.R. Splicing-factor alterations in cancers. RNA 2016, 22, 1285–1301. [Google Scholar] [CrossRef]
- Swanson, M.S.; Dreyfuss, G. RNA binding specificity of hnRNP proteins: A subset bind to the 3′ end of introns. EMBO J. 1988, 7, 3519–3529. [Google Scholar] [CrossRef] [PubMed]
- Datar, K.V.; Dreyfuss, G.; Swanson, M.S. The human hnRNP M proteins: Identification of a methionine/arginine-rich repeat motif in ribonucleoproteins. Nucleic Acids Res. 1993, 21, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Hovhannisyan, R.H.; Carstens, R.P. Heterogeneous ribonucleoprotein m is a splicing regulatory protein that can enhance or silence splicing of alternatively spliced exons. J. Biol. Chem. 2007, 282, 36265–36274. [Google Scholar] [CrossRef] [PubMed]
- Huelga, S.C.; Vu, A.Q.; Arnold, J.D.; Liang, T.Y.; Liu, P.P.; Yan, B.Y.; Donohue, J.P.; Shiue, L.; Hoon, S.; Brenner, S.; et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012, 1, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gao, X.D.; Lee, J.H.; Huang, H.; Tan, H.; Ahn, J.; Reinke, L.M.; Peter, M.E.; Feng, Y.; Gius, D.; et al. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev. 2014, 28, 1191–1203. [Google Scholar] [CrossRef]
- Zahler, A.M.; Lane, W.S.; Stolk, J.A.; Roth, M.B. SR proteins: A conserved family of pre-mRNA splicing factors. Genes Dev. 1992, 6, 837–847. [Google Scholar] [CrossRef]
- Huang, Y.; Steitz, J.A. Splicing factors SRp20 and 9G8 promote the nucleocytoplasmic export of mRNA. Mol. Cell 2001, 7, 899–905. [Google Scholar] [CrossRef]
- Lou, H.; Neugebauer, K.M.; Gagel, R.F.; Berget, S.M. Regulation of alternative polyadenylation by U1 snRNPs and SRp20. Mol. Cell Biol. 1998, 18, 4977–4985. [Google Scholar] [CrossRef]
- Kim, J.; Park, R.Y.; Chen, J.K.; Jeong, S.; Ohn, T. Splicing factor SRSF3 represses the translation of programmed cell death 4 mRNA by associating with the 5′-UTR region. Cell Death Differ. 2014, 21, 481–490. [Google Scholar] [CrossRef]
- Bedard, K.M.; Daijogo, S.; Semler, B.L. A nucleo-cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J. 2007, 26, 459–467. [Google Scholar] [CrossRef]
- Auyeung, V.C.; Ulitsky, I.; McGeary, S.E.; Bartel, D.P. Beyond secondary structure: Primary-sequence determinants license pri-miRNA hairpins for processing. Cell 2013, 152, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Loomis, R.J.; Naoe, Y.; Parker, J.B.; Savic, V.; Bozovsky, M.R.; Macfarlan, T.; Manley, J.L.; Chakravarti, D. Chromatin binding of SRp20 and ASF/SF2 and dissociation from mitotic chromosomes is modulated by histone H3 serine 10 phosphorylation. Mol. Cell 2009, 33, 450–461. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Arslan, A.D.; Pool, M.D.; Ho, T.T.; Darcy, K.M.; Coon, J.S.; Beck, W.T. Knockdown of splicing factor SRp20 causes apoptosis in ovarian cancer cells and its expression is associated with malignancy of epithelial ovarian cancer. Oncogene 2011, 30, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, M.; Jia, R.; Yang, Y.; Zhu, J.; Zheng, Z.M. A genome landscape of SRSF3-regulated splicing events and gene expression in human osteosarcoma U2OS cells. Nucleic Acids Res. 2016, 44, 1854–1870. [Google Scholar] [CrossRef]
- Jia, R.; Li, C.; McCoy, J.P.; Deng, C.X.; Zheng, Z.M. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int. J. Biol. Sci. 2010, 6, 806–826. [Google Scholar] [CrossRef] [PubMed]
- Hurt, J.A.; Robertson, A.D.; Burge, C.B. Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res. 2013, 23, 1636–1650. [Google Scholar] [CrossRef]
- Ratnadiwakara, M.; Archer, S.K.; Dent, C.I.; Ruiz De Los Mozos, I.; Beilharz, T.H.; Knaupp, A.S.; Nefzger, C.M.; Polo, J.M.; Anko, M.L. SRSF3 promotes pluripotency through. Elife 2018, 7. [Google Scholar] [CrossRef]
- Santos, G.C.; Zielenska, M.; Prasad, M.; Squire, J.A. Chromosome 6p amplification and cancer progression. J. Clin. Pathol. 2007, 60, 1–7. [Google Scholar] [CrossRef]
- Sun, Y.; Yan, L.; Guo, J.; Shao, J.; Jia, R. Downregulation of SRSF3 by antisense oligonucleotides sensitizes oral squamous cell carcinoma and breast cancer cells to paclitaxel treatment. Cancer Chemother Pharm. 2019, 84, 1133–1143. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palombo, R.; Verdile, V.; Paronetto, M.P. Poison-Exon Inclusion in DHX9 Reduces Its Expression and Sensitizes Ewing Sarcoma Cells to Chemotherapeutic Treatment. Cells 2020, 9, 328. https://doi.org/10.3390/cells9020328
Palombo R, Verdile V, Paronetto MP. Poison-Exon Inclusion in DHX9 Reduces Its Expression and Sensitizes Ewing Sarcoma Cells to Chemotherapeutic Treatment. Cells. 2020; 9(2):328. https://doi.org/10.3390/cells9020328
Chicago/Turabian StylePalombo, Ramona, Veronica Verdile, and Maria Paola Paronetto. 2020. "Poison-Exon Inclusion in DHX9 Reduces Its Expression and Sensitizes Ewing Sarcoma Cells to Chemotherapeutic Treatment" Cells 9, no. 2: 328. https://doi.org/10.3390/cells9020328
APA StylePalombo, R., Verdile, V., & Paronetto, M. P. (2020). Poison-Exon Inclusion in DHX9 Reduces Its Expression and Sensitizes Ewing Sarcoma Cells to Chemotherapeutic Treatment. Cells, 9(2), 328. https://doi.org/10.3390/cells9020328