Painting of Fourth and the X-Linked 1.688 Satellite in D. melanogaster Is Involved in Chromosome-Wide Gene Regulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Fly Strains
2.2. Preparation of RNA Library and Sequencing
2.3. Raw Data Processing
2.4. Genome Read Counting
2.5. Sample Similarity Heatmaps
2.6. Differential Expression
2.7. Characterisation of 1.688 Transcripts between CG1840 and Sicily
2.8. Functional Enrichment Analysis
3. Results
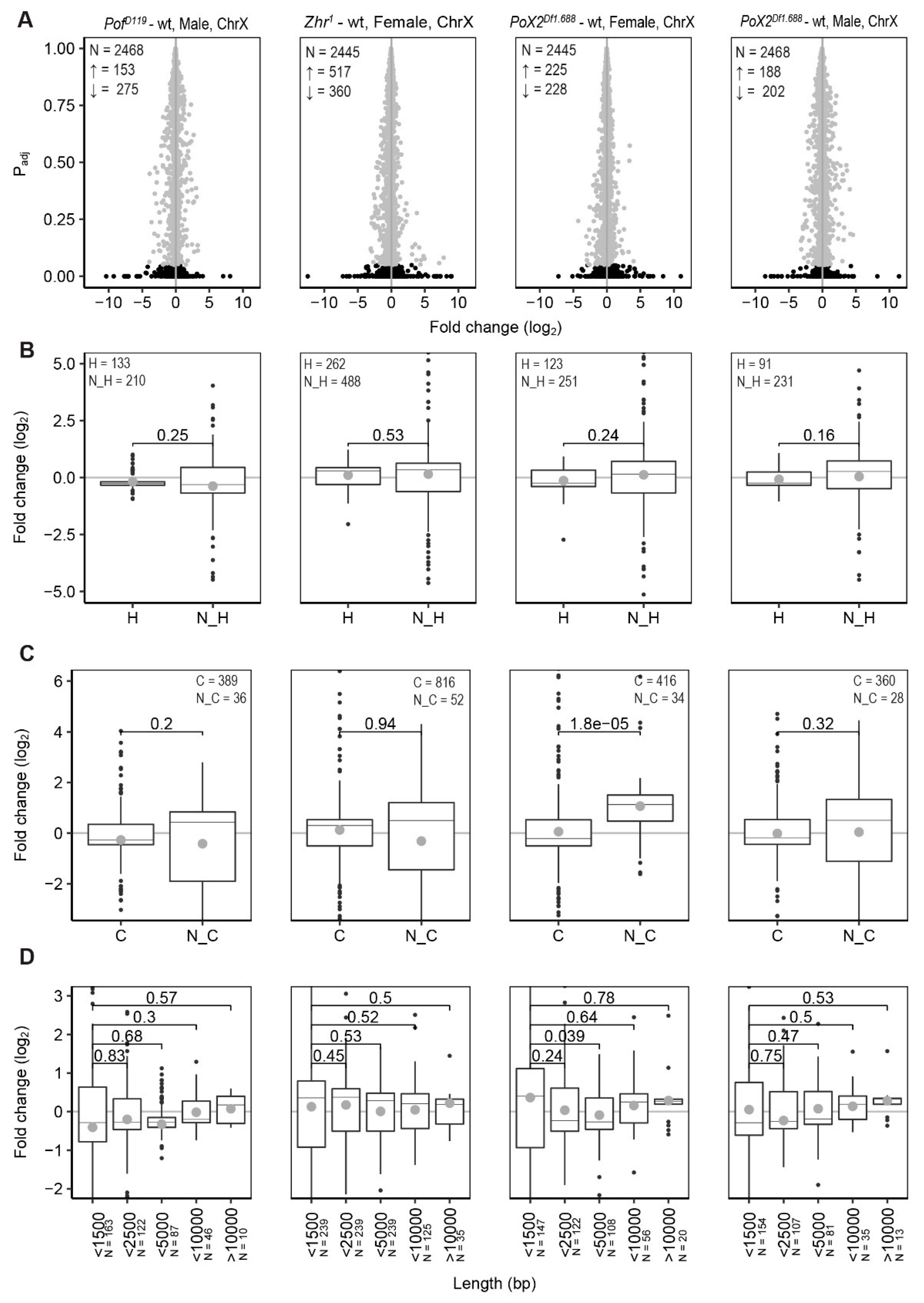
3.1. PofD119, Zhr1, and PoX2Df1.688 Mutants Show Chromosome-Specific Differential Expression
3.2. POF Stimulates Expression on the 4th Chromosome Preferentially on Short, Non-Coding, and Differentially Expressed Genes
3.3. Relationship between X-Linked Transcriptional Alterations and MSL Complex Mediated Dosage-Compensation
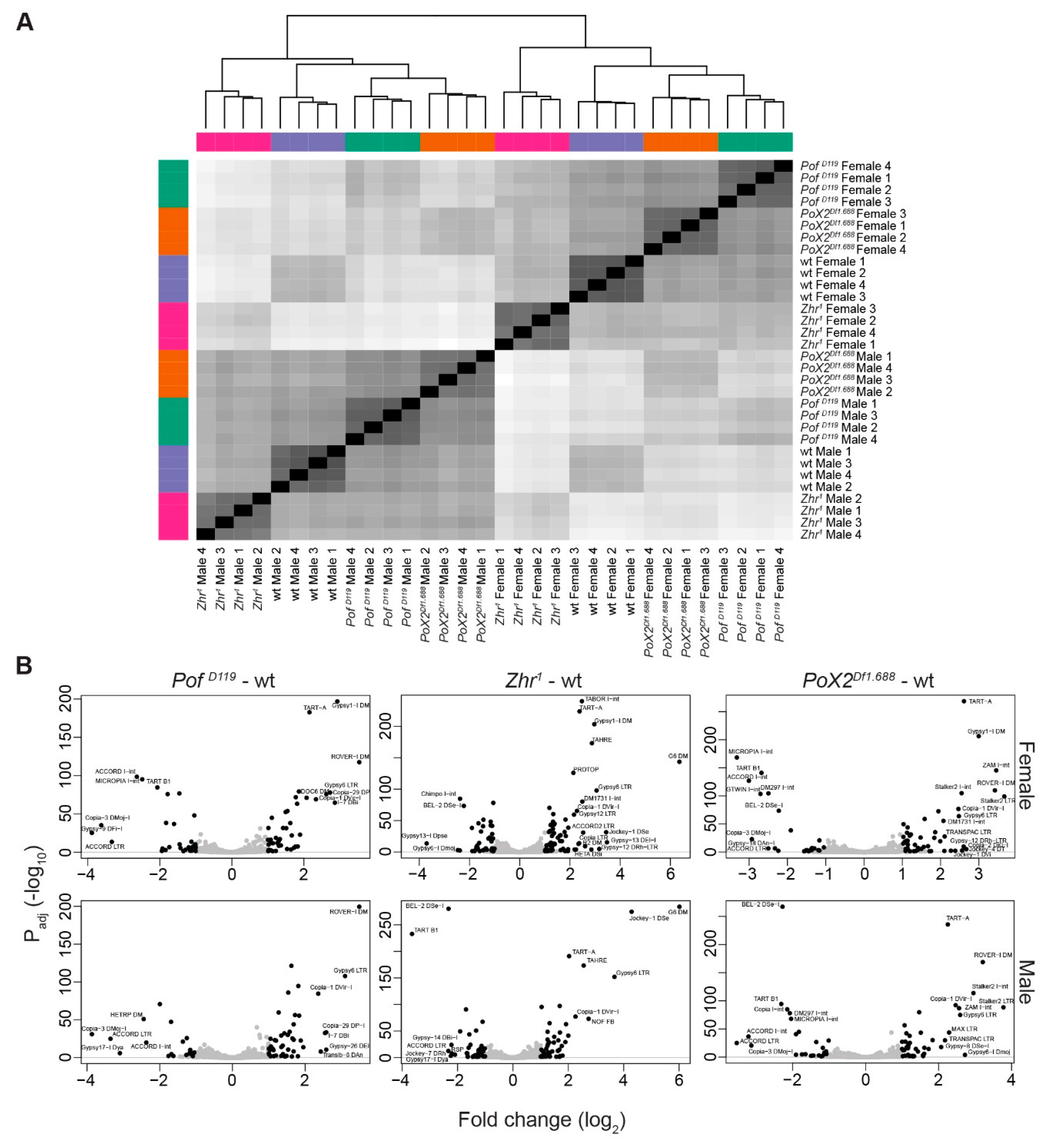
3.4. The Zhr1 Mutant Reduces the Expression of 1.688 Satellites in Trans
3.5. Expression Analyses of X-Linked 1.688 Satellites
3.6. Transposon de-Repression in Zhr1 Mutant
3.7. Female-Biased Genes Related to Eggshell Formation Show Increased Expression in Zhr1 Mutant Females
4. Discussion
4.1. Chromosome Specific Differential Expression in Male PofD119 and Female Zhr1
4.2. Loss of the Pericentromeric 1.688 Satellite Region on the X-Chromosome Reduces Satellite Expression in Cis and Trans, and Induces Transposon de-Repression
4.3. The 1.688 Satellite Modulates Chorion Family Expression in Female D. melanogaster
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Larsson, J.; Meller, V.H. Dosage compensation, the origin and the afterlife of sex chromosomes. Chromosome Res. 2006, 14, 417–431. [Google Scholar] [CrossRef]
- Stenberg, P.; Larsson, J. Buffering and the evolution of chromosome-wide gene regulation. Chromosoma 2011, 120, 213–225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mank, J.E. Sex chromosome dosage compensation: Definitely not for everyone. Trends Genet. 2013, 29, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Prestel, M.; Feller, C.; Becker, P.B. Dosage compensation and the global re-balancing of aneuploid genomes. Genome Biol. 2010, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, J.C.; Kelly, W.G.; Panning, B. Chromatin remodeling in dosage compensation. Annu. Rev. Genet. 2005, 39, 615–651. [Google Scholar] [CrossRef]
- Hochman, B. The fourth chromosome of Drosophila melanogaster. In The Genetics and Biology of Drosophila; Ashburner, M., Novitski, E., Eds.; Academic Press: London, UK, 1976; Volume 1B, pp. 903–928. [Google Scholar]
- Ashburner, M.; Golic, K.G.; Hawley, R.S. Drosophila a Laboratory Handbook; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2005. [Google Scholar]
- Vicoso, B.; Bachtrog, D. Reversal of an ancient sex chromosome to an autosome in Drosophila. Nature 2013, 499, 332–335. [Google Scholar] [CrossRef]
- Vicoso, B.; Bachtrog, D. Numerous transitions of sex chromosomes in Diptera. PLoS Biol. 2015, 13, e1002078. [Google Scholar] [CrossRef]
- Lundberg, L.E.; Figueiredo, M.L.; Stenberg, P.; Larsson, J. Buffering and proteolysis are induced by segmental monosomy in Drosophila melanogaster. Nucleic Acids Res. 2012, 40, 5926–5937. [Google Scholar] [CrossRef]
- Stenberg, P.; Lundberg, L.E.; Johansson, A.M.; Rydén, P.; Svensson, M.J.; Larsson, J. Buffering of segmental and chromosomal aneuploidies in Drosoph. Melanogaster. PLoS Genet. 2009, 5, e100302. [Google Scholar] [CrossRef]
- Zhang, Y.; Oliver, B. An evolutionary consequence of dosage compensation on Drosophila melanogaster female X-chromatin structure? Bmc Genom. 2010, 11, 6. [Google Scholar] [CrossRef]
- Kuroda, M.I.; Hilfiker, A.; Lucchesi, J.C. Dosage compensation in Drosophila-a model for the coordinate regulation of transcription. Genetics 2016, 204, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, M.L.; Kim, M.; Philip, P.; Allgardsson, A.; Stenberg, P.; Larsson, J. Non-coding roX RNAs prevent the binding of the MSL-complex to heterochromatic regions. PLoS Genet. 2014, 10, e1004865. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Faucillion, M.L.; Larsson, J. RNA-on-X 1 and 2 in Drosophila melanogaster fulfill separate functions in dosage compensation. PLoS Genet. 2018, 14, e1007842. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.M.; Stenberg, P.; Allgardsson, A.; Larsson, J. POF regulates the expression of genes on the fourth chromosome in Drosophila melanogaster by binding to nascent RNA. Mol. Cell. Biol. 2012, 32, 2121–2134. [Google Scholar] [CrossRef]
- Johansson, A.M.; Stenberg, P.; Bernhardsson, C.; Larsson, J. Painting of fourth and chromosome-wide regulation of the 4th chromosome in Drosophila melanogaster. Embo J. 2007, 26, 2307–2316. [Google Scholar] [CrossRef]
- Larsson, J.; Chen, J.D.; Rasheva, V.; Rasmuson Lestander, A.; Pirrotta, V. Painting of fourth, a chromosome-specific protein in Drosophila. Proc. Natl. Acad. Sci. USA 2001, 98, 6273–6278. [Google Scholar] [CrossRef]
- Johansson, A.M.; Stenberg, P.; Pettersson, F.; Larsson, J. POF and HP1 bind expressed exons, suggesting a balancing mechanism for gene regulation. PLoS Genet. 2007, 3, e209. [Google Scholar] [CrossRef]
- Figueiredo, M.L.; Philip, P.; Stenberg, P.; Larsson, J. HP1a recruitment to promoters is independent of H3K9 methylation in Drosophila melanogaster. PLoS Genet. 2012, 8, e1003061. [Google Scholar] [CrossRef]
- Lundberg, L.E.; Kim, M.; Johansson, A.M.; Faucillion, M.L.; Josupeit, R.; Larsson, J. Targeting of Painting of fourth to roX1 and roX2 proximal sites suggests evolutionary links between dosage compensation and the regulation of the fourth chromosome in Drosophila melanogaster. G3 2013, 3, 1325–1334. [Google Scholar] [CrossRef]
- Bone, J.R.; Kuroda, M.I. Dosage compensation regulatory proteins and the evolution of sex chromosomes in Drosophila. Genetics 1996, 144, 705–713. [Google Scholar]
- Marin, I.; Franke, A.; Bashaw, G.J.; Baker, B.S. The dosage compensation system of Drosophila is co-opted by newly evolved X chromosomes. Nature 1996, 383, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Ellison, C.; Bachtrog, D. Contingency in the convergent evolution of a regulatory network: Dosage compensation in Drosophila. PLoS Biol. 2019, 17, e3000094. [Google Scholar] [CrossRef] [PubMed]
- Ellison, C.E.; Bachtrog, D. Dosage compensation via transposable element mediated rewiring of a regulatory network. Science 2013, 342, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J.; Svensson, M.J.; Stenberg, P.; Mäkitalo, M. Painting of fourth in genus Drosophila suggests autosome-specific gene regulation. Proc. Natl. Acad. Sci. USA 2004, 101, 9728–9733. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ekhteraei-Tousi, S.; Lewerentz, J.; Larsson, J. The X-linked 1.688 satellite in Drosophila melanogaster promotes specific targeting by Painting of Fourth. Genetics 2018, 208, 623–632. [Google Scholar] [CrossRef]
- Alekseyenko, A.A.; Ellison, C.E.; Gorchakov, A.A.; Zhou, Q.; Kaiser, V.B.; Toda, N.; Walton, Z.; Peng, S.; Park, P.J.; Bachtrog, D.; et al. Conservation and de novo acquisition of dosage compensation on newly evolved sex chromosomes in Drosophila. Genes Dev. 2013, 27, 853–858. [Google Scholar] [CrossRef]
- Pardue, M.L.; Lowenhaupt, K.; Rich, A.; Nordheim, A. (dC-dA)n.(dG-dT)n sequences have evolutionarily conserved chromosomal locations in Drosophila with implications for roles in chromosome structure and function. Embo J. 1987, 6, 1781–1789. [Google Scholar] [CrossRef]
- Waring, G.L.; Pollack, J.C. Cloning and characterization of a dispersed, multicopy, X chromosome sequence in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1987, 84, 2843–2847. [Google Scholar] [CrossRef]
- Gallach, M. Recurrent turnover of chromosome-specific satellites in Drosophila. Genome Biol. Evol. 2014, 6, 1279–1286. [Google Scholar] [CrossRef]
- Gallach, M. 1.688 g/cm3 satellite-related repeats: A missing link to dosage compensation and speciation. Mol. Ecol. 2015, 24, 4340–4347. [Google Scholar] [CrossRef]
- DiBartolomeis, S.M.; Tartof, K.D.; Jackson, F.R. A superfamily of Drosophila satellite related (SR) DNA repeats restricted to the X chromosome euchromatin. Nucleic Acids Res. 1992, 20, 1113–1116. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, G.C.; Kuttler, H.; Moreira-Filho, O.; Heslop-Harrison, J.S. The 1.688 repetitive DNA of Drosophila: Concerted evolution at different genomic scales and association with genes. Mol. Biol. Evol. 2012, 29, 7–11. [Google Scholar] [CrossRef]
- Joshi, S.S.; Meller, V.H. Satellite repeats identify X chromatin for dosage compensation in Drosophila melanogaster males. Curr. Biol. 2017, 27, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Menon, D.U.; Coarfa, C.; Xiao, W.; Gunaratne, P.H.; Meller, V.H. siRNAs from an X-linked satellite repeat promote X-chromosome recognition in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2014, 111, 16460–16465. [Google Scholar] [CrossRef] [PubMed]
- Lohe, A.R.; Hilliker, A.J.; Roberts, P.A. Mapping simple repeated DNA sequences in heterochromatin of Drosophila melanogaster. Genetics 1993, 134, 1149–1174. [Google Scholar] [CrossRef]
- Hsieh, T.; Brutlag, D. Sequence and sequence variation within the 1.688 g/cm3 satellite DNA of Drosophila melanogaster. J. Mol. Biol. 1979, 135, 465–481. [Google Scholar] [CrossRef]
- Rosic, S.; Kohler, F.; Erhardt, S. Repetitive centromeric satellite RNA is essential for kinetochore formation and cell division. J. Cell Biol. 2014, 207, 335–349. [Google Scholar] [CrossRef]
- Rosic, S.; Erhardt, S. No longer a nuisance: Long non-coding RNAs join CENP-A in epigenetic centromere regulation. Cell. Mol. Life Sci. 2016, 73, 1387–1398. [Google Scholar] [CrossRef]
- Ferree, P.M.; Barbash, D.A. Species-specific heterochromatin prevents mitotic chromosome segregation to cause hybrid lethality in Drosophila. PLoS Biol. 2009, 7, e1000234. [Google Scholar] [CrossRef]
- Sawamura, K.; Yamamoto, M.T.; Watanabe, T.K. Hybrid lethal systems in the Drosophila melanogaster species complex. II. The Zygotic hybrid rescue (Zhr) gene of D. melanogaster. Genetics 1993, 133, 307–313. [Google Scholar]
- Sawamura, K.; Yamamoto, M.T. Cytogenetical localization of Zygotic hybrid rescue (Zhr), a Drosophila melanogaster gene that rescues interspecific hybrids from embryonic lethality. Mol. Gen. Genet. 1993, 239, 441–449. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kopylova, E.; Noe, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Tempel, S. Using and understanding RepeatMasker. Methods Mol. Biol. 2012, 859, 29–51. [Google Scholar] [CrossRef]
- Nishimura, D. RepeatMasker. Biotech. Softw. Internet Rep. 2000, 1, 36–39. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- R_Core_Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 18 November 2019).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Warnes, G.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Mächler, M.; Magnusson, A.; Möller, S. gplots: Various R Programming Tools for Plotting Data; The Comprehensive R Archive Network; Institute for Statistics and Mathematics, Vienna University of Economics and Business: Vienna, Austria, 2005; Volume 2. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Lundberg, L.E.; Stenberg, P.; Larsson, J. HP1a, Su(var)3-9, SETDB1 and POF stimulate or repress gene expression depending on genomic position, gene length and expression pattern in Drosophila melanogaster. Nucleic Acids Res. 2013, 41, 4481–4494. [Google Scholar] [CrossRef]
- Kind, J.; Vaquerizas, J.M.; Gebhardt, P.; Gentzel, M.; Luscombe, N.M.; Bertone, P.; Akhtar, A. Genome-wide analysis reveals MOF as a key regulator of dosage compensation and gene expression in Drosophila. Cell 2008, 133, 813–828. [Google Scholar] [CrossRef]
- Philip, P.; Stenberg, P. Male X-linked genes in Drosophila melanogaster are compensated independently of the Male-Specific Lethal complex. Epigenetics Chromatin 2013, 6, 35. [Google Scholar] [CrossRef]
- Soruco, M.M.; Larschan, E. A new player in X identification: The CLAMP protein is a key factor in Drosophila dosage compensation. Chromosome Res. 2014, 22, 505–515. [Google Scholar] [CrossRef]
- Kuzu, G.; Kaye, E.G.; Chery, J.; Siggers, T.; Yang, L.; Dobson, J.R.; Boor, S.; Bliss, J.; Liu, W.; Jogl, G.; et al. Expansion of GA dinucleotide repeats increases the density of CLAMP binding sites on the X-chromosome to promote Drosophila dosage compensation. PLoS Genet. 2016, 12, e1006120. [Google Scholar] [CrossRef]
- Soruco, M.M.; Chery, J.; Bishop, E.P.; Siggers, T.; Tolstorukov, M.Y.; Leydon, A.R.; Sugden, A.U.; Goebel, K.; Feng, J.; Xia, P.; et al. The CLAMP protein links the MSL complex to the X chromosome during Drosophila dosage compensation. Genes Dev. 2013, 27, 1551–1556. [Google Scholar] [CrossRef]
- Albig, C.; Tikhonova, E.; Krause, S.; Maksimenko, O.; Regnard, C.; Becker, P.B. Factor cooperation for chromosome discrimination in Drosophila. Nucleic Acids Res. 2019, 47, 1706–1724. [Google Scholar] [CrossRef]
- Usakin, L.; Abad, J.; Vagin, V.V.; de Pablos, B.; Villasante, A.; Gvozdev, V.A. Transcription of the 1.688 satellite DNA family is under the control of RNA interference machinery in Drosophila melanogaster ovaries. Genetics 2007, 176, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Palomeque, T.; Lorite, P. Satellite DNA in insects: A review. Heredity 2008, 100, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, V.; Bernardi, F.; Romani, P.; Duchi, S.; Gargiulo, G. Building up the Drosophila eggshell: First of all the eggshell genes must be transcribed. Dev. Dyn. 2008, 237, 2061–2072. [Google Scholar] [CrossRef]
- Tootle, T.L.; Williams, D.; Hubb, A.; Frederick, R.; Spradling, A. Drosophila eggshell production: Identification of new genes and coordination by Pxt. PLoS ONE 2011, 6, e19943. [Google Scholar] [CrossRef]
- Prayitno, K.; Schauer, T.; Regnard, C.; Becker, P.B. Progressive dosage compensation during Drosophila embryogenesis is reflected by gene arrangement. EMBO Rep. 2019, 20, e48138. [Google Scholar] [CrossRef]
- Krassovsky, K.; Henikoff, S. Distinct chromatin features characterize different classes of repeat sequences in Drosophila melanogaster. BMC Genom. 2014, 15, 105. [Google Scholar] [CrossRef]
- Haldane, J.B.S. Sex ratio and unisexual sterility in hybrid animals. J. Genet. 1922, 12, 101–109. [Google Scholar] [CrossRef]
- Chatterjee, R.n.; Kuthe, S.; Chatterjee, P. Eggshells of Drosophila melanogaster and D. simulans: Ultrastructure, Measurement and Analyses. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 2015, 87. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ekhteraei-Tousi, S.; Lewerentz, J.; Larsson, J. Painting of Fourth and the X-Linked 1.688 Satellite in D. melanogaster Is Involved in Chromosome-Wide Gene Regulation. Cells 2020, 9, 323. https://doi.org/10.3390/cells9020323
Ekhteraei-Tousi S, Lewerentz J, Larsson J. Painting of Fourth and the X-Linked 1.688 Satellite in D. melanogaster Is Involved in Chromosome-Wide Gene Regulation. Cells. 2020; 9(2):323. https://doi.org/10.3390/cells9020323
Chicago/Turabian StyleEkhteraei-Tousi, Samaneh, Jacob Lewerentz, and Jan Larsson. 2020. "Painting of Fourth and the X-Linked 1.688 Satellite in D. melanogaster Is Involved in Chromosome-Wide Gene Regulation" Cells 9, no. 2: 323. https://doi.org/10.3390/cells9020323
APA StyleEkhteraei-Tousi, S., Lewerentz, J., & Larsson, J. (2020). Painting of Fourth and the X-Linked 1.688 Satellite in D. melanogaster Is Involved in Chromosome-Wide Gene Regulation. Cells, 9(2), 323. https://doi.org/10.3390/cells9020323