Single-Nucleus Sequencing of an Entire Mammalian Heart: Cell Type Composition and Velocity

 ,
,  ,
,  ,
,  and
and 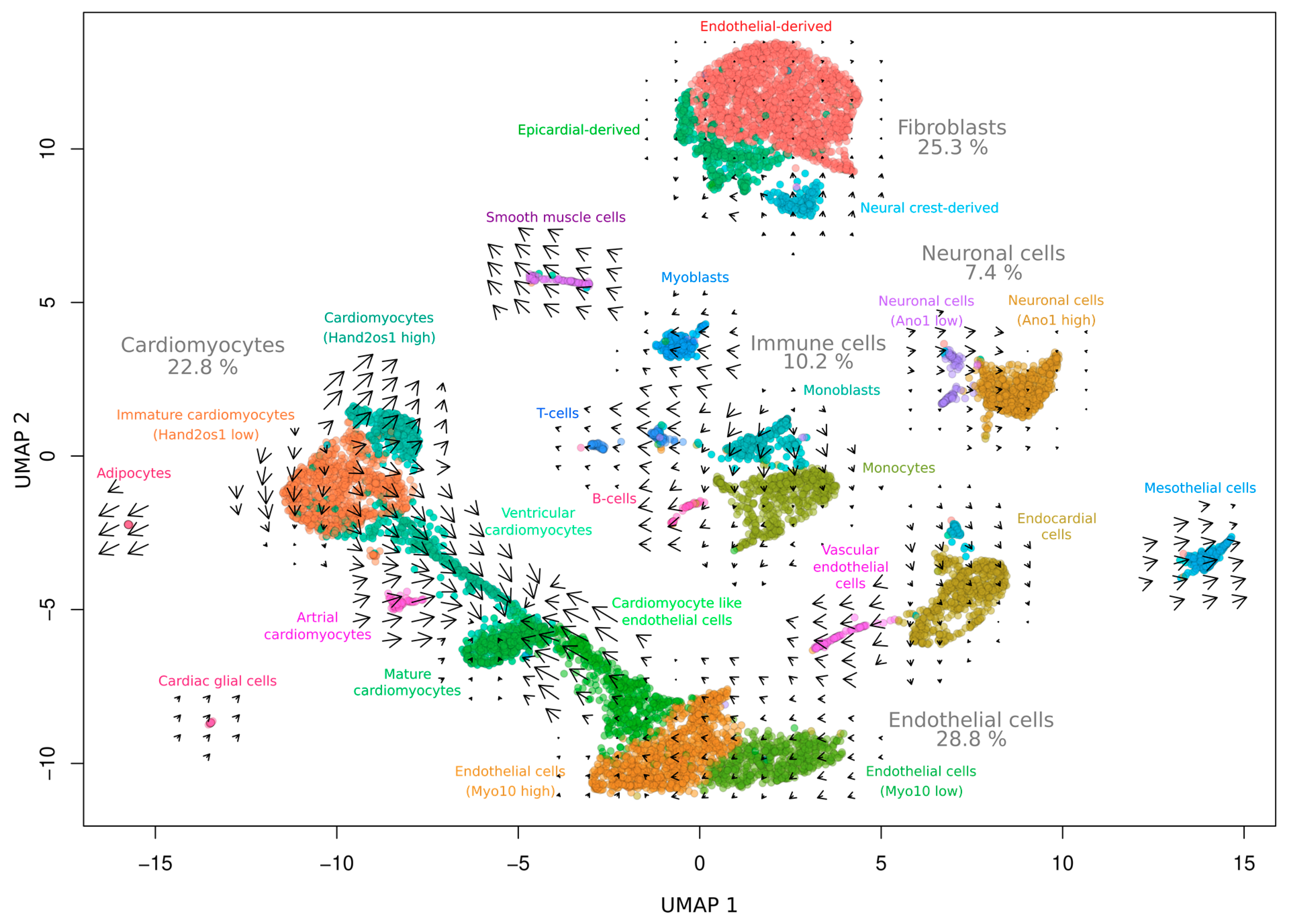{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of Nuclei
2.2. Computational Data Analysis
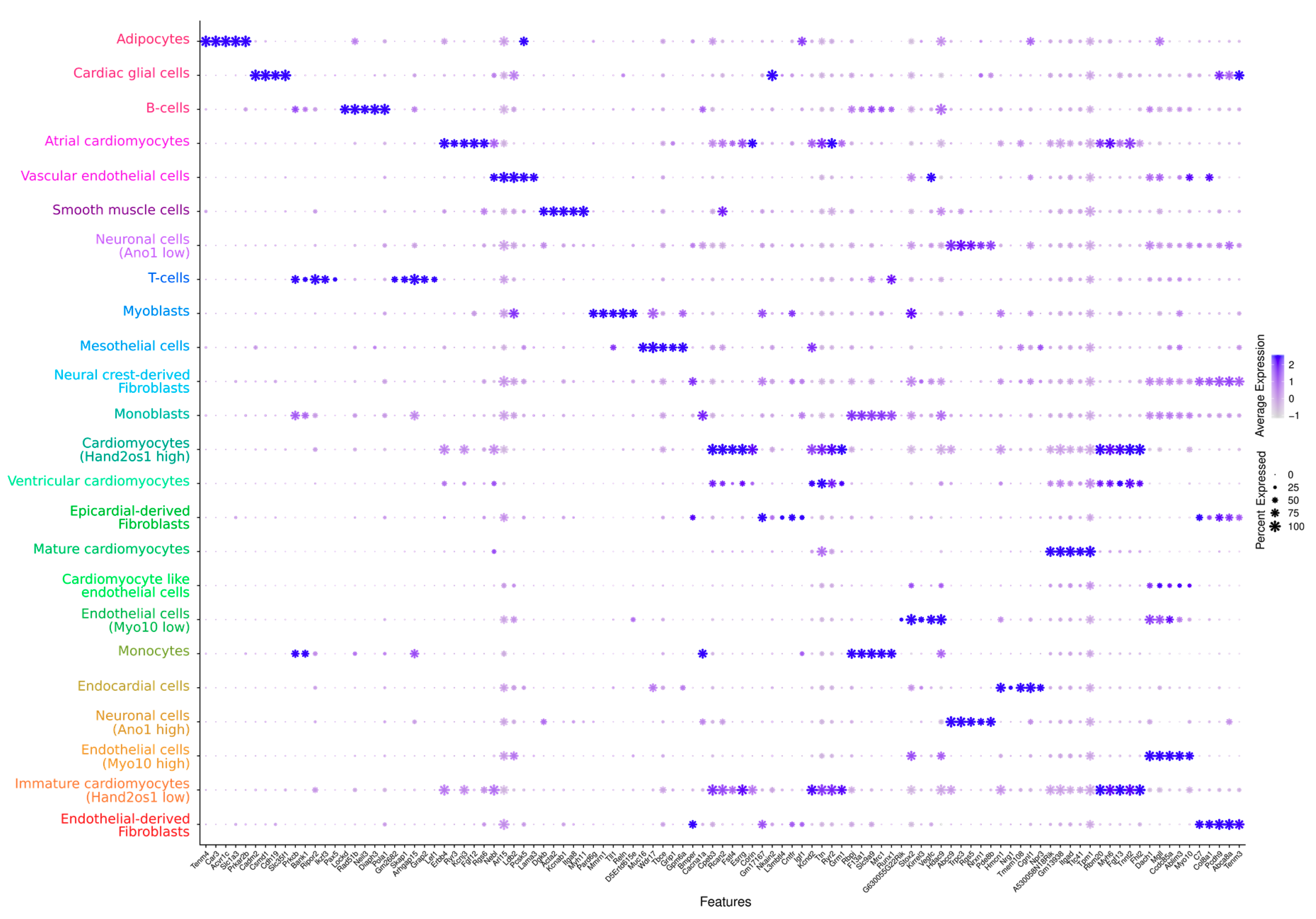
3. Results and Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Schaum, N.; Karkanias, J.; Neff, N.F.; May, A.P.; Quake, S.R.; Wyss-Coray, T.; Darmanis, S.; Batson, J.; Botvinnik, O.; Chen, M.B.; et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nat. Nat. Publ. Group 2018, 562, 367–372. [Google Scholar]
- Ackers-Johnson, M.; Tan, W.L.W.; Foo, R.S.-Y. Following hearts, one cell at a time: Recent applications of single-cell RNA sequencing to the understanding of heart disease. Nat. Commun. 2018, 9, 4434. [Google Scholar] [CrossRef]
- Bakken, T.E.; Hodge, R.D.; Miller, J.A.; Yao, Z.; Nguyen, T.N.; Aevermann, B.; Barkan, E.; Bertagnolli, D.; Casper, T.; Dee, N.; et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE 2018, 13, e0209648. [Google Scholar] [CrossRef] [PubMed]
- Linscheid, N.; Logantha, S.J.R.J.; Poulsen, P.C.; Zhang, S.; Schrölkamp, M.; Egerod, K.L.; Thompson, J.J.; Kitmitto, A.; Galli, G.; Humphries, M.J.; et al. Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nat. Commun. 2019, 10, 2889. [Google Scholar] [CrossRef]
- Hu, P.; Liu, J.; Zhao, J.; Wilkins, B.J.; Lupino, K.; Wu, H.; Pei, L. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018, 32, 1344–1357. [Google Scholar] [CrossRef] [PubMed]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lönnerberg, P.; Furlan, A.; et al. RNA velocity of single cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Dietl, G.; Langhammer, M.; Renne, U. Model simulations for genetic random drift in the outbred strain Fzt:DU. Arch. Anim. Breed. 2004, 47, 595–604. [Google Scholar] [CrossRef]
- Melsted, P.; Booeshaghi, A.S.; Gao, F.; Beltrame, E.D.V.; Lu, L.; Hjorleifsson, K.E.; Gehring, J.; Pachter, L. Modular and Efficient Pre-Processing of Single-Cell RNA-Seq; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2019; p. 673285. [Google Scholar]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.-R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting cardiac cellular composition. Circ. Res. Lippincott Williams Wilkins 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhang, J.; Liu, Y.; Fan, X.; Ai, S.; Luo, Y.; Li, X.; Jin, H.; Luo, S.; Zheng, H.; et al. The lncRNA Hand2os1/Uph locus orchestrates heart development through regulation of precise expression of Hand2. Development 2019, 146, dev176198. [Google Scholar] [CrossRef] [PubMed]
- Ritter, N.; Ali, T.; Kopitchinski, N.; Schuster, P.; Beisaw, A.; Hendrix, D.A.; Schulz, M.H.; Müller-McNicoll, M.; Dimmeler, S.; Grote, P. The lncRNA Locus Handsdown Regulates Cardiac Gene Programs and Is Essential for Early Mouse Development. Dev. Cell 2019, 50, 644–657.e8. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.M.; Anderson, U.M.; McAnally, J.R.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature 2016, 539, 433–436. [Google Scholar] [CrossRef] [PubMed]
- De Soysa, T.Y.; Ranade, S.S.; Okawa, S.; Ravichandran, S.; Huang, Y.; Salunga, H.T.; Schricker, A.; Del Sol, A.; Gifford, C.A.; Srivastava, D. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 2019, 572, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Borello, U.; De Angelis, L.; Latronico, M.; Sirabella, D.; Coletta, M.; Galli, R.; Balconi, G.; Follenzi, A.; Frati, G.; et al. Cardiomyocytes induce endothelial cells to trans-differentiate into cardiac muscle: Implications for myocardium regeneration. Proc. Natl. Acad. Sci. USA 2001, 98, 10733–10738. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolfien, M.; Galow, A.-M.; Müller, P.; Bartsch, M.; Brunner, R.M.; Goldammer, T.; Wolkenhauer, O.; Hoeflich, A.; David, R. Single-Nucleus Sequencing of an Entire Mammalian Heart: Cell Type Composition and Velocity. Cells 2020, 9, 318. https://doi.org/10.3390/cells9020318
Wolfien M, Galow A-M, Müller P, Bartsch M, Brunner RM, Goldammer T, Wolkenhauer O, Hoeflich A, David R. Single-Nucleus Sequencing of an Entire Mammalian Heart: Cell Type Composition and Velocity. Cells. 2020; 9(2):318. https://doi.org/10.3390/cells9020318
Chicago/Turabian StyleWolfien, Markus, Anne-Marie Galow, Paula Müller, Madeleine Bartsch, Ronald M. Brunner, Tom Goldammer, Olaf Wolkenhauer, Andreas Hoeflich, and Robert David. 2020. "Single-Nucleus Sequencing of an Entire Mammalian Heart: Cell Type Composition and Velocity" Cells 9, no. 2: 318. https://doi.org/10.3390/cells9020318
APA StyleWolfien, M., Galow, A.-M., Müller, P., Bartsch, M., Brunner, R. M., Goldammer, T., Wolkenhauer, O., Hoeflich, A., & David, R. (2020). Single-Nucleus Sequencing of an Entire Mammalian Heart: Cell Type Composition and Velocity. Cells, 9(2), 318. https://doi.org/10.3390/cells9020318