Comprehensive Structural and Molecular Comparison of Spike Proteins of SARS-CoV-2, SARS-CoV and MERS-CoV, and Their Interactions with ACE2

 , ,
, ,  ,
,  ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
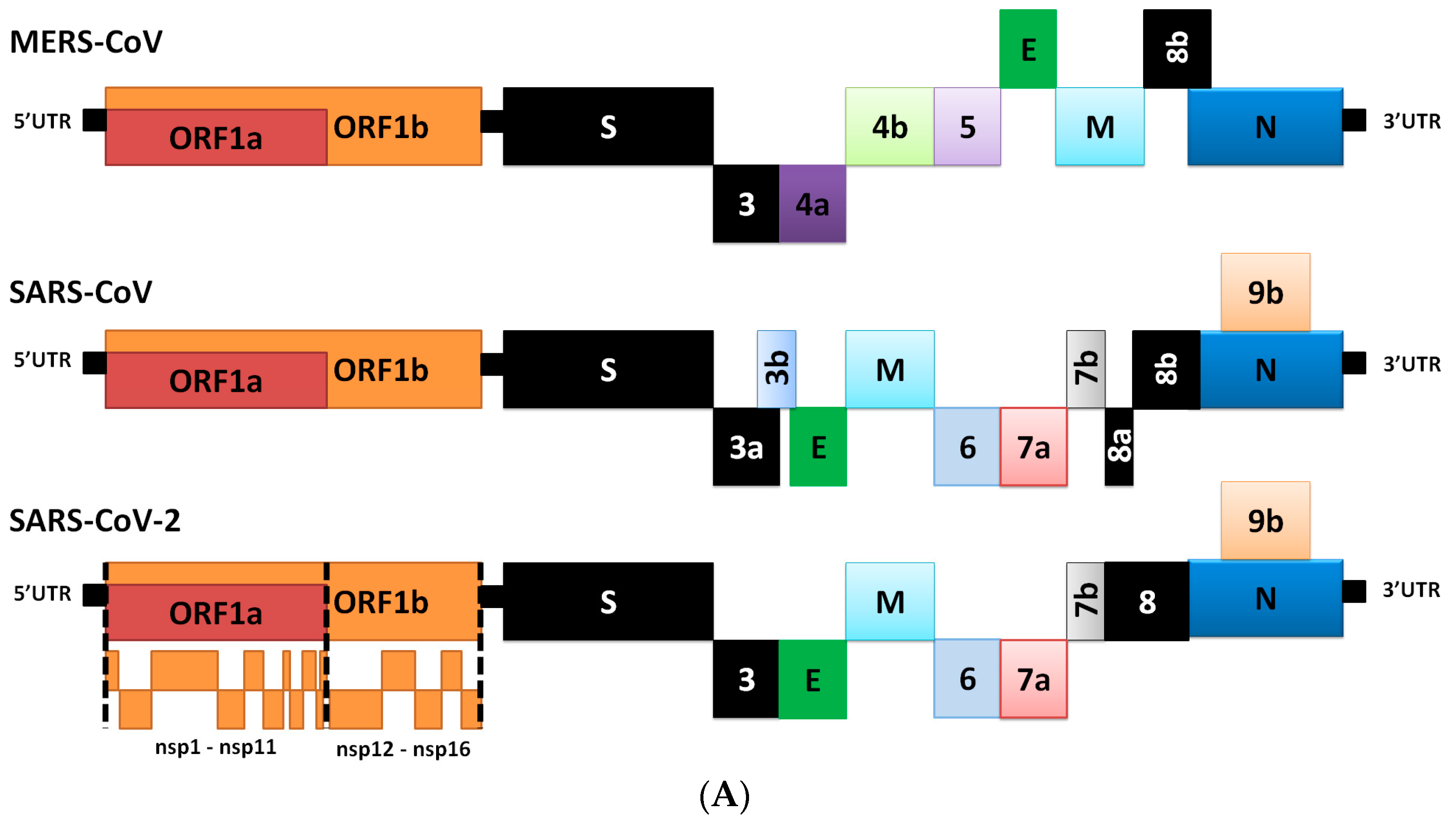
2. Genome Comparison of Coronaviruses
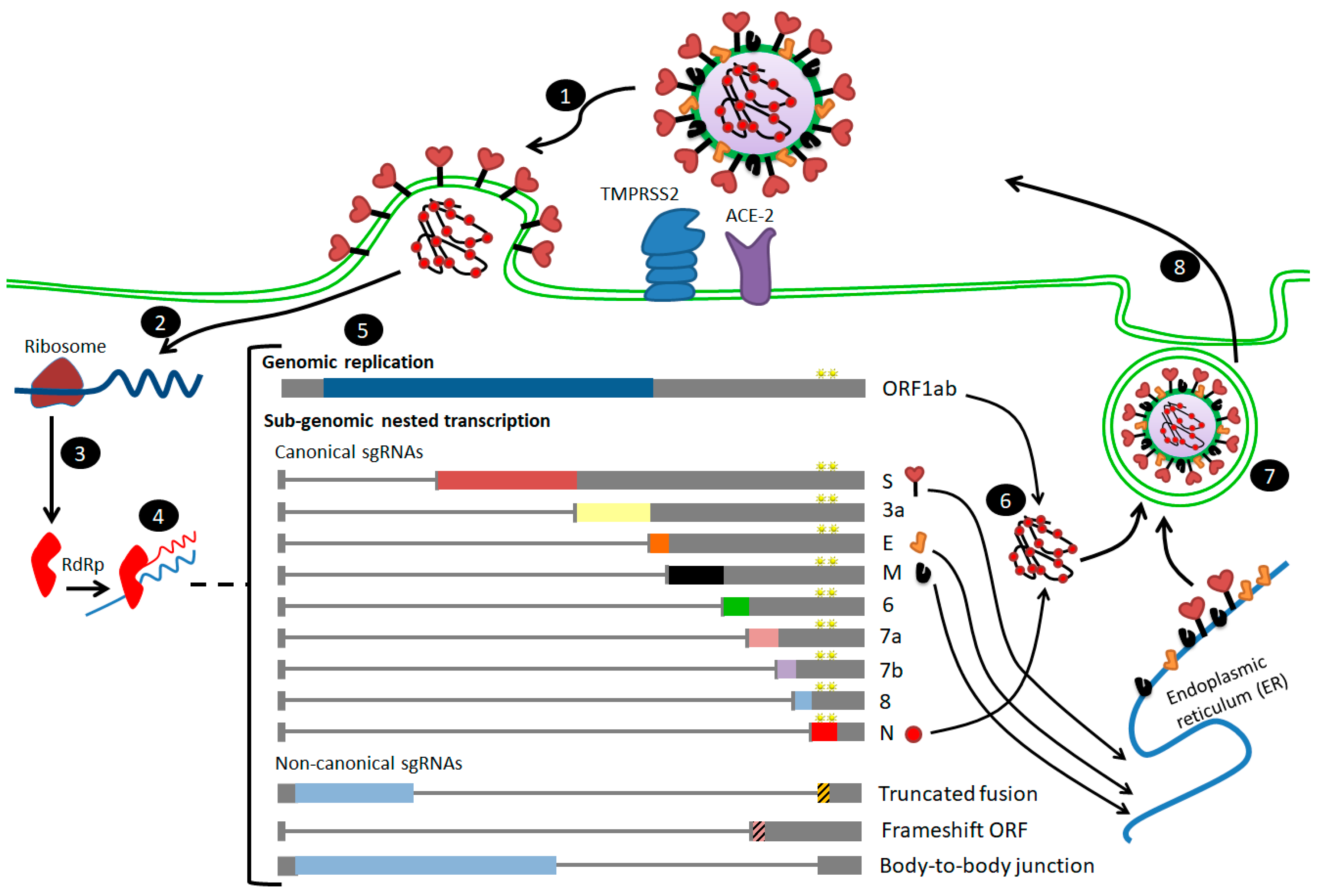
3. Differences in Transcriptome
4. Role of S Protein in Pathogenesis
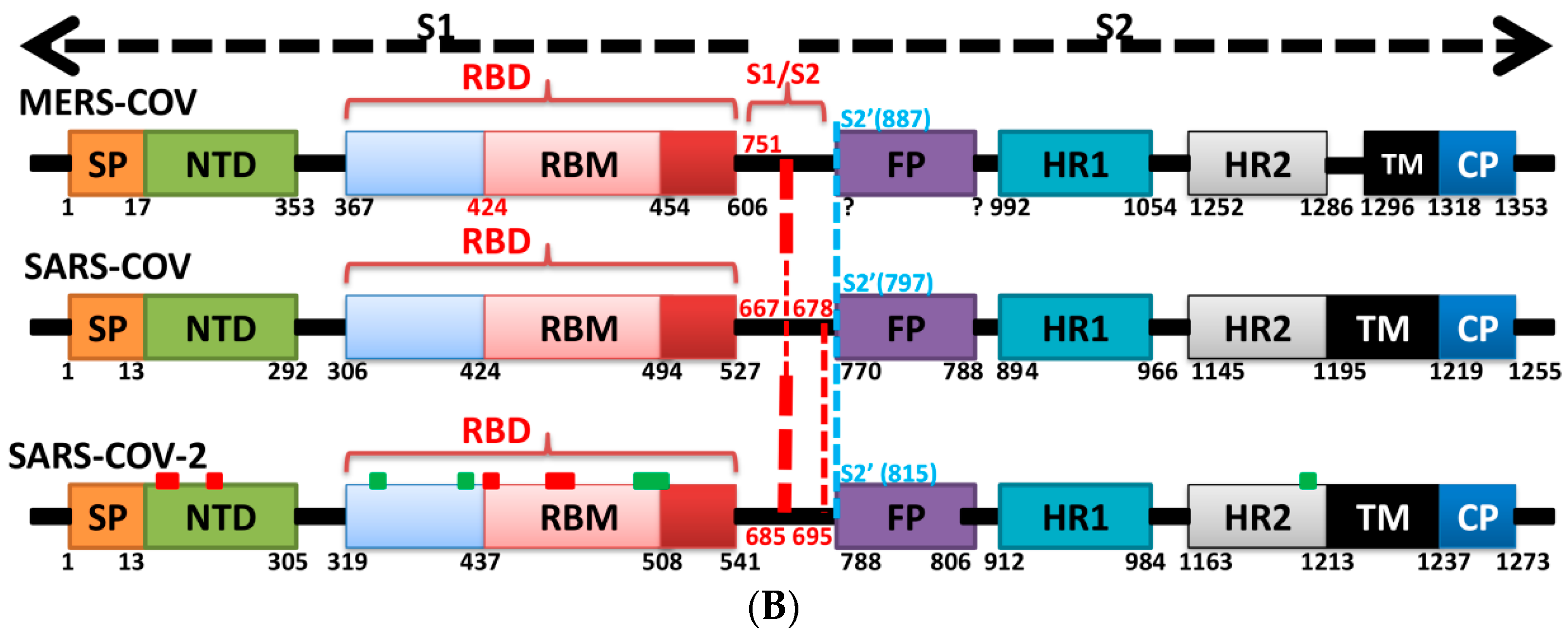
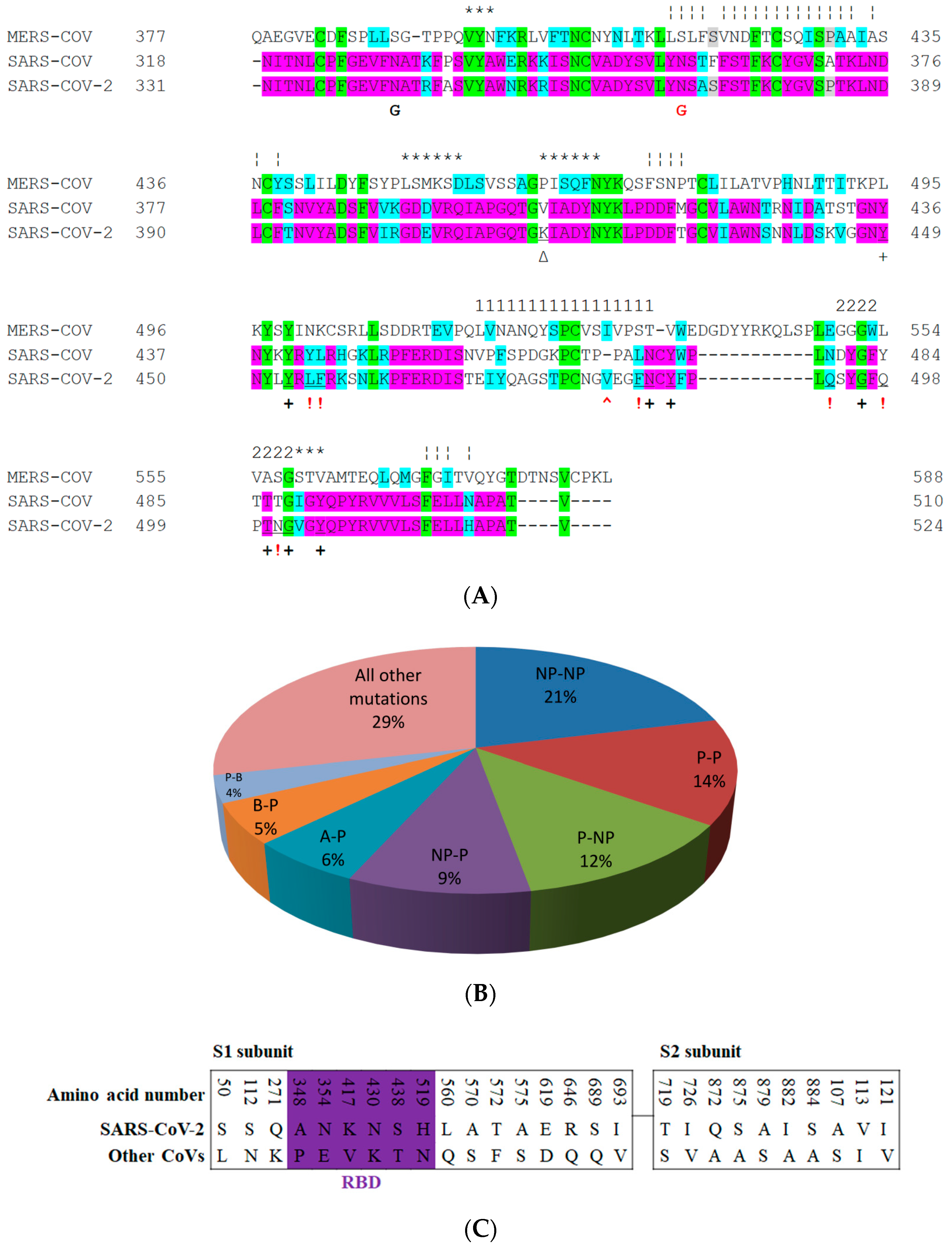
5. The Primary, Secondary and Tertiary Structures of SARS-CoV-2 S Protein, and Differences from Other Coronaviruses
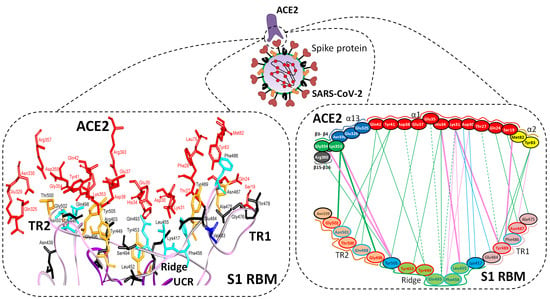
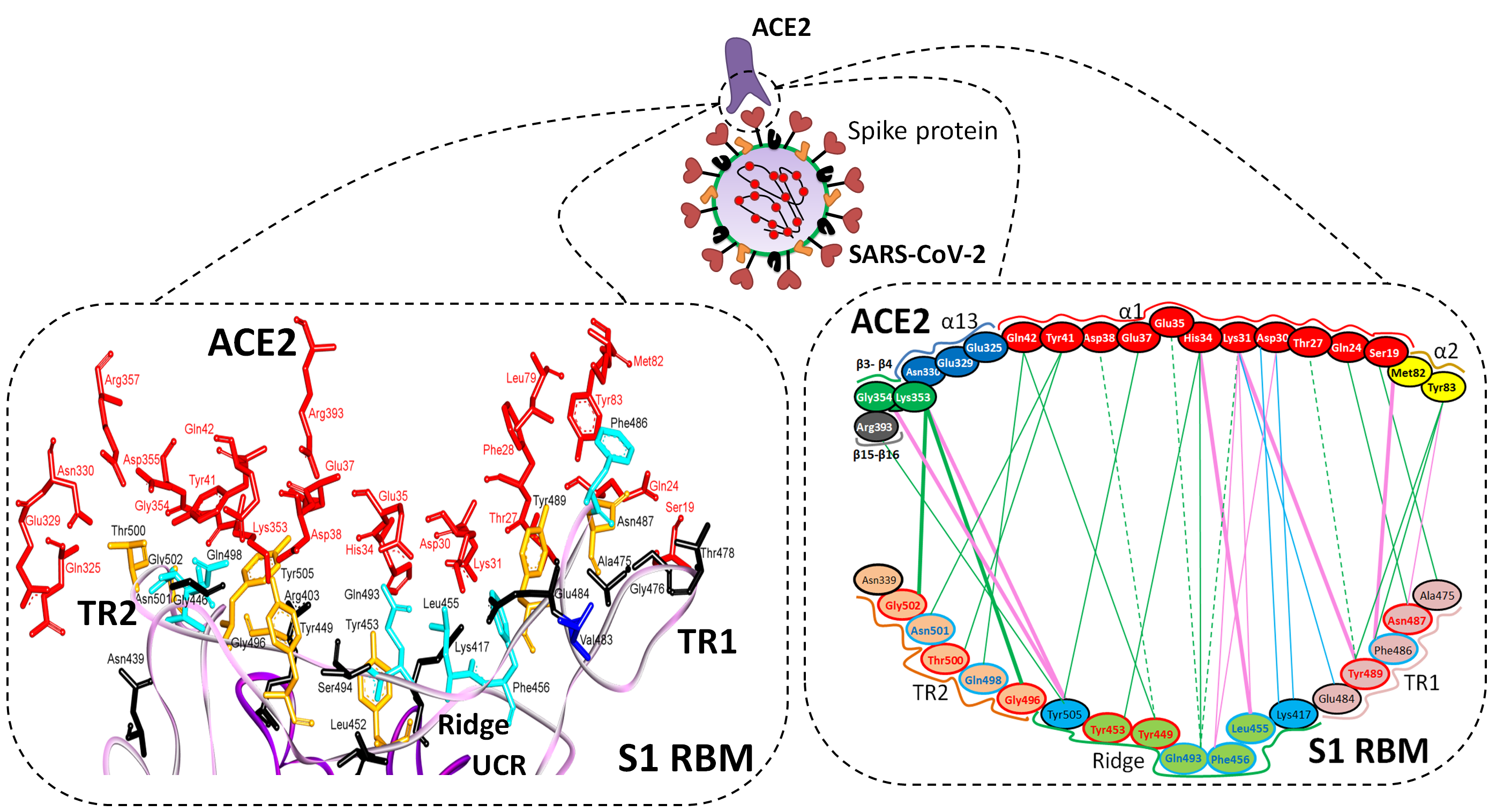
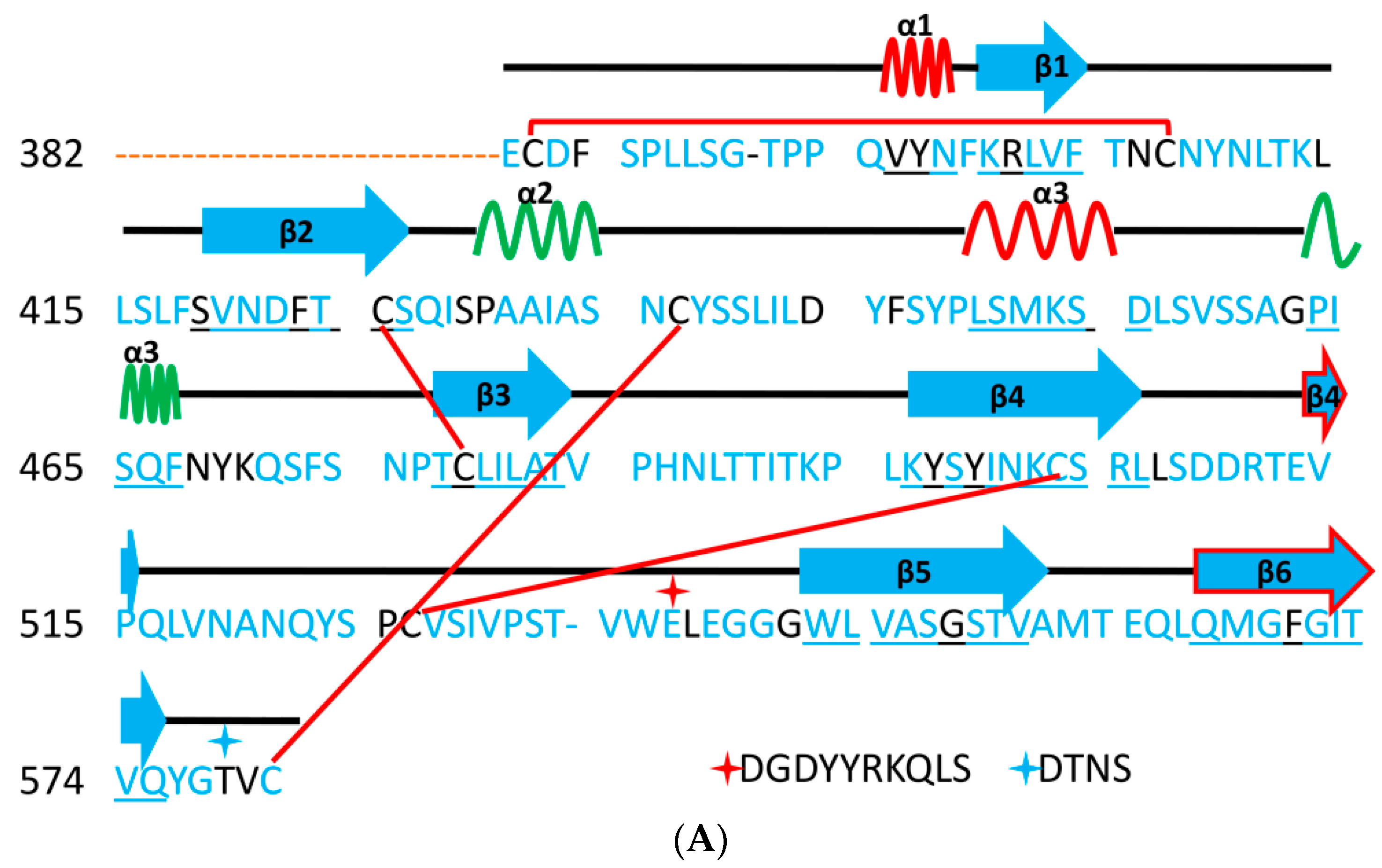
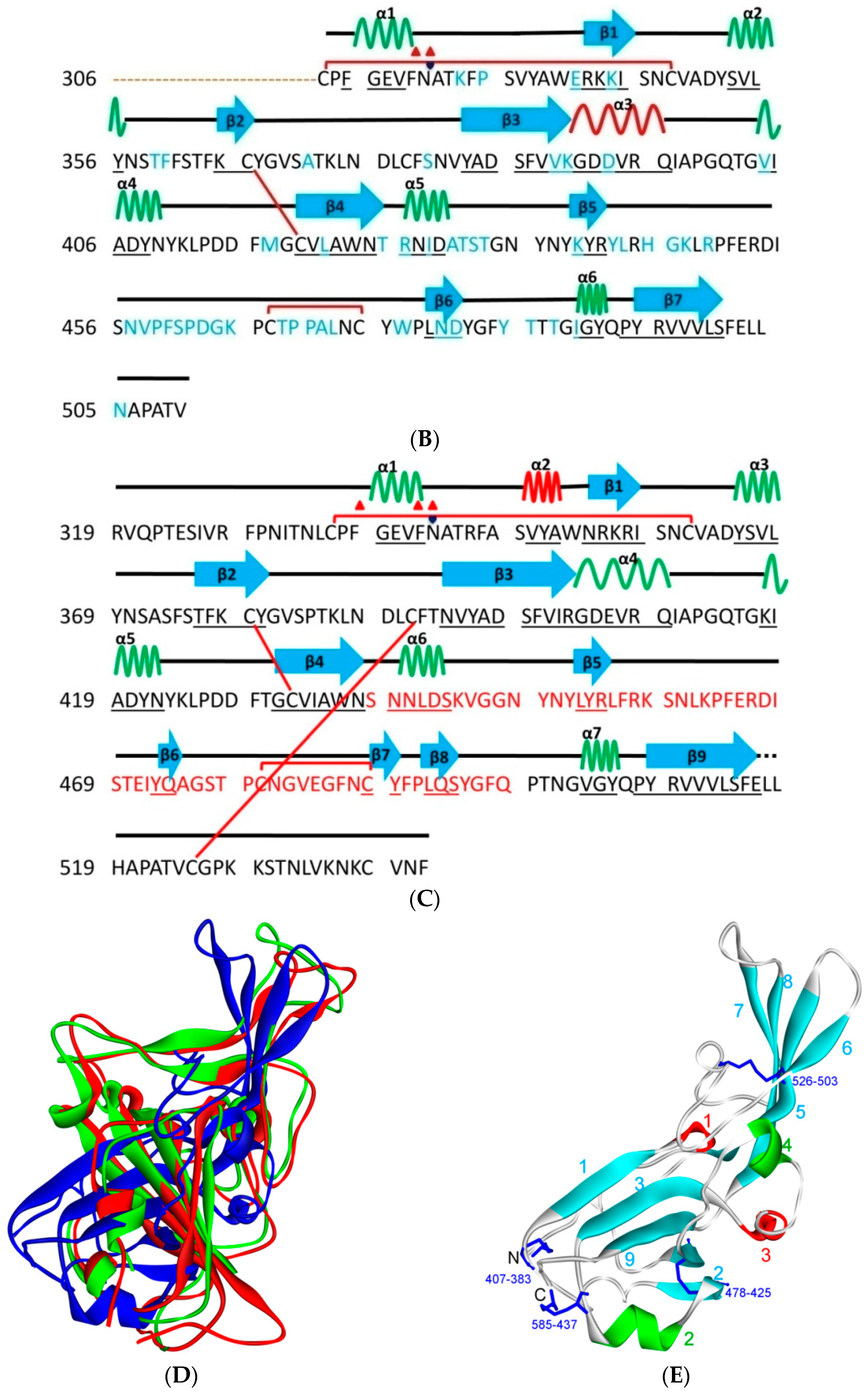
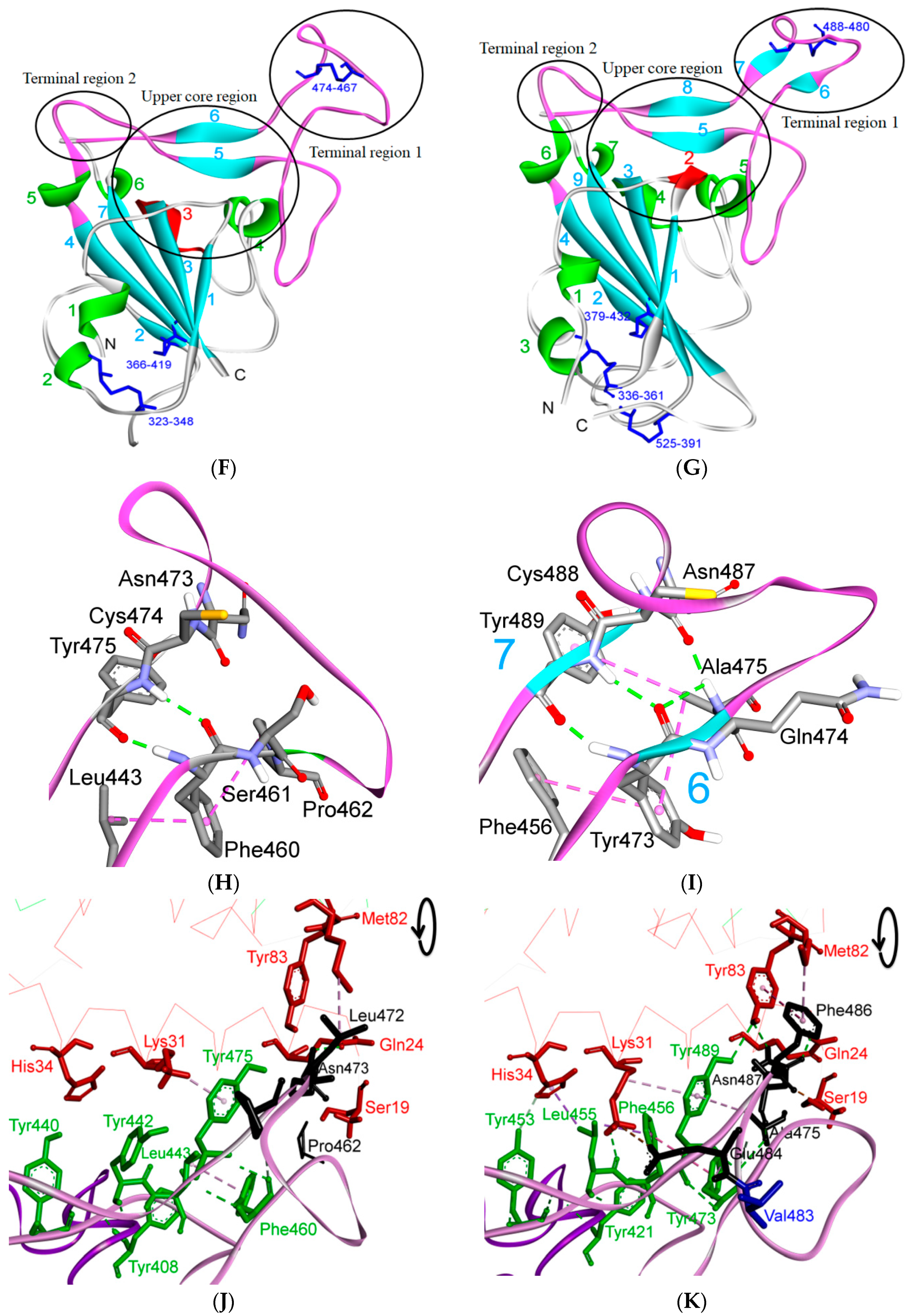
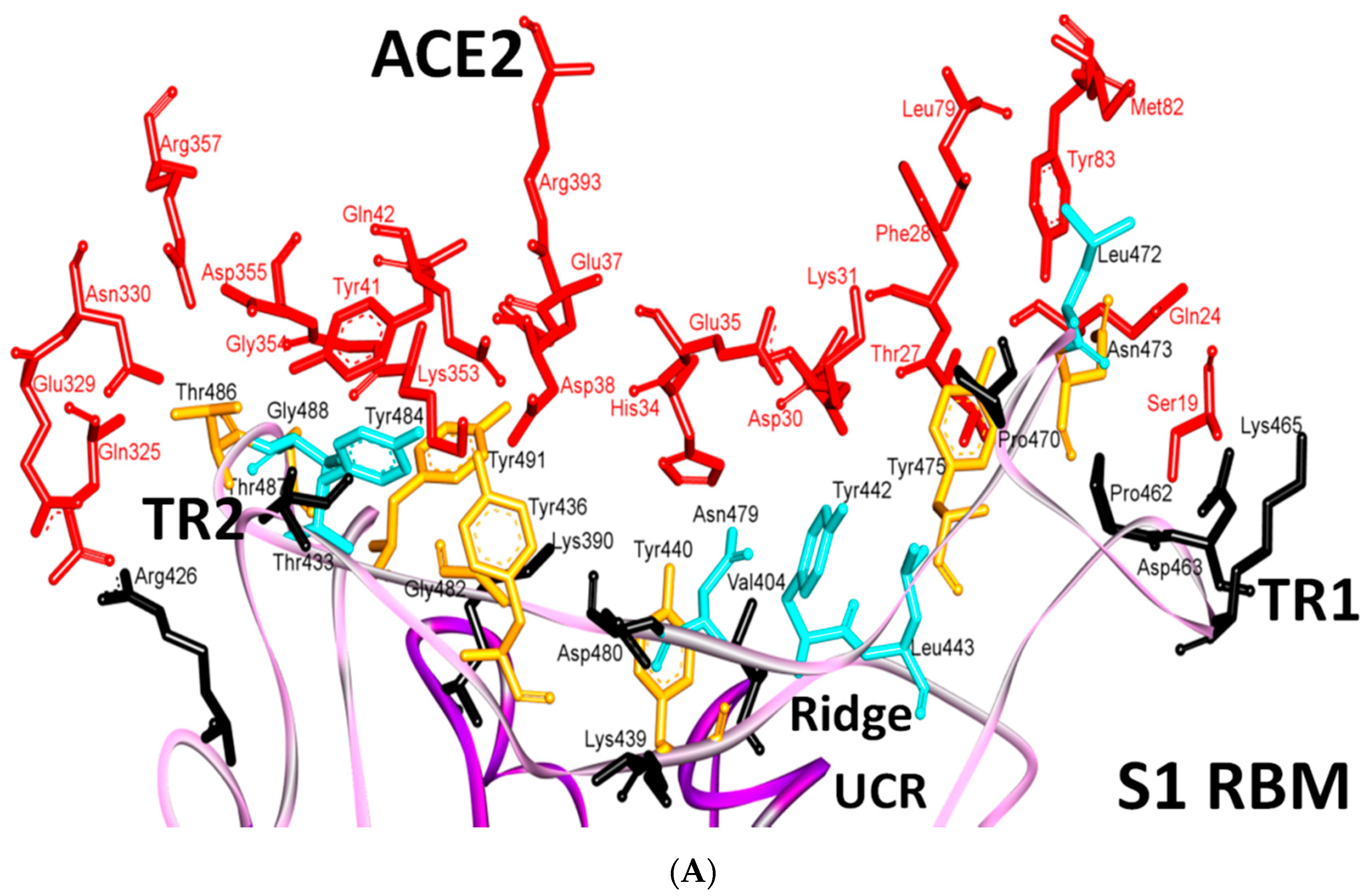
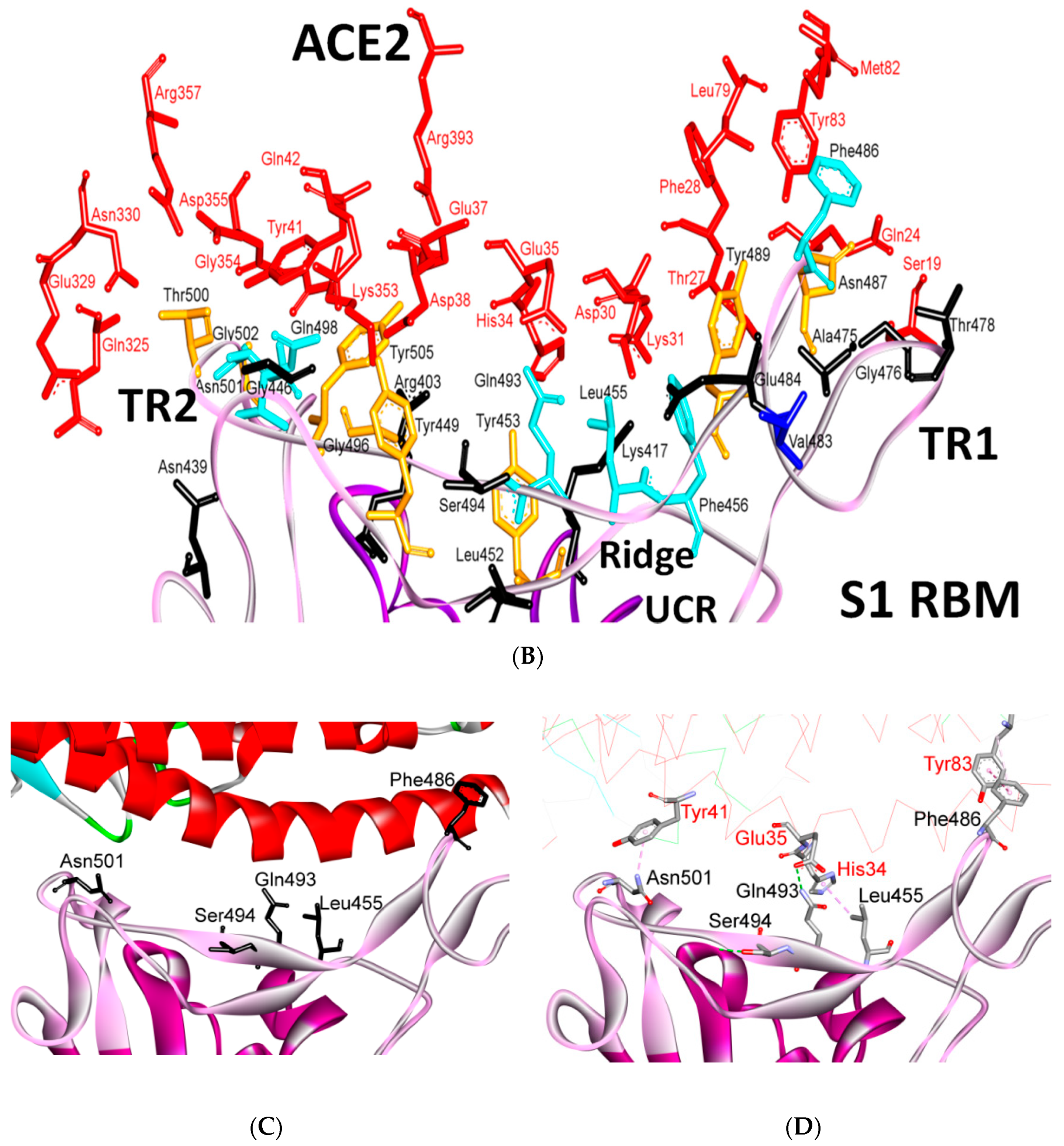
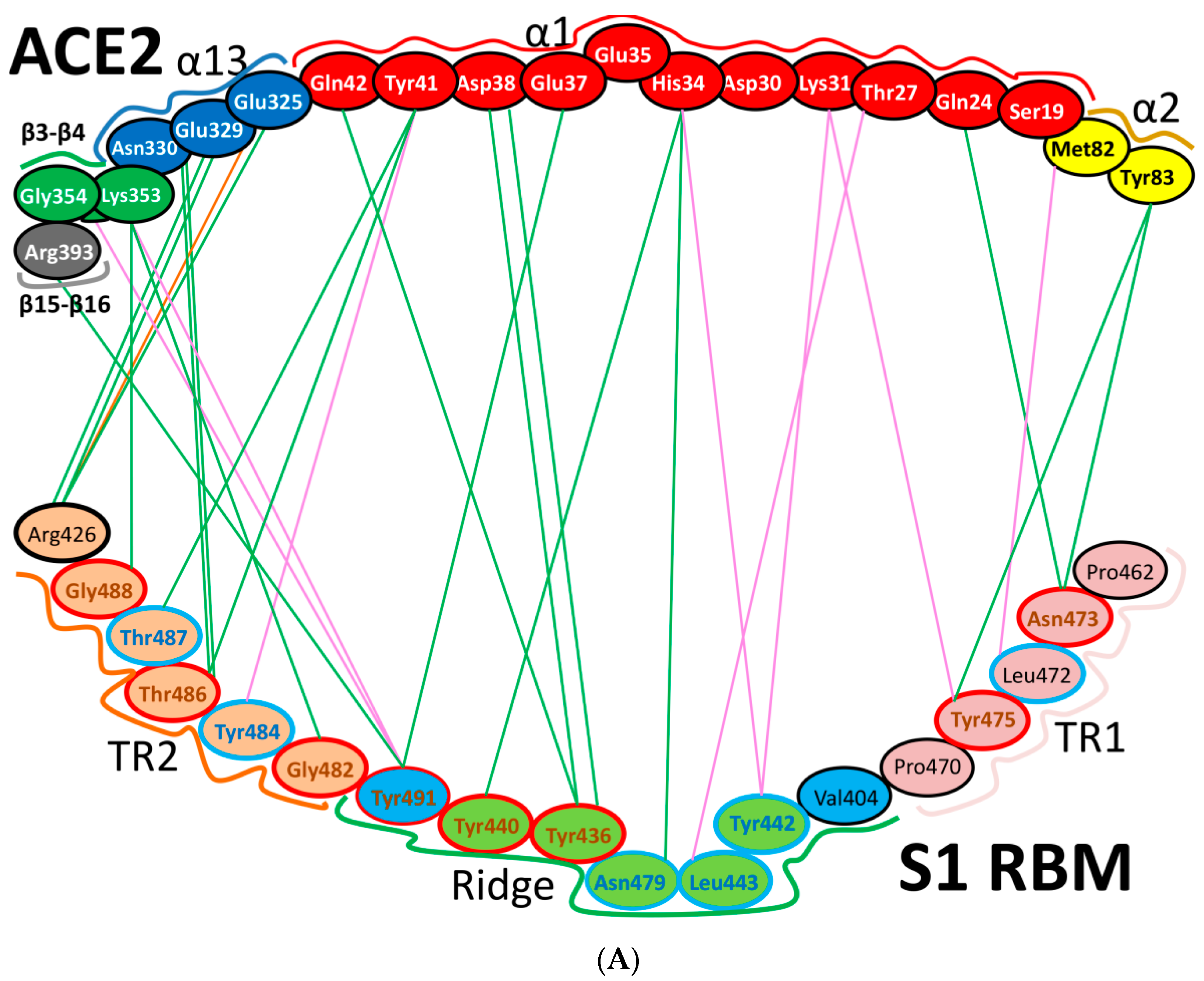
5.1. Terminal Region 1
5.2. Terminal Region 2
5.3. Middle Ridge of RBM and Upper Core Region of RBD
5.4. Other Regions of S Protein
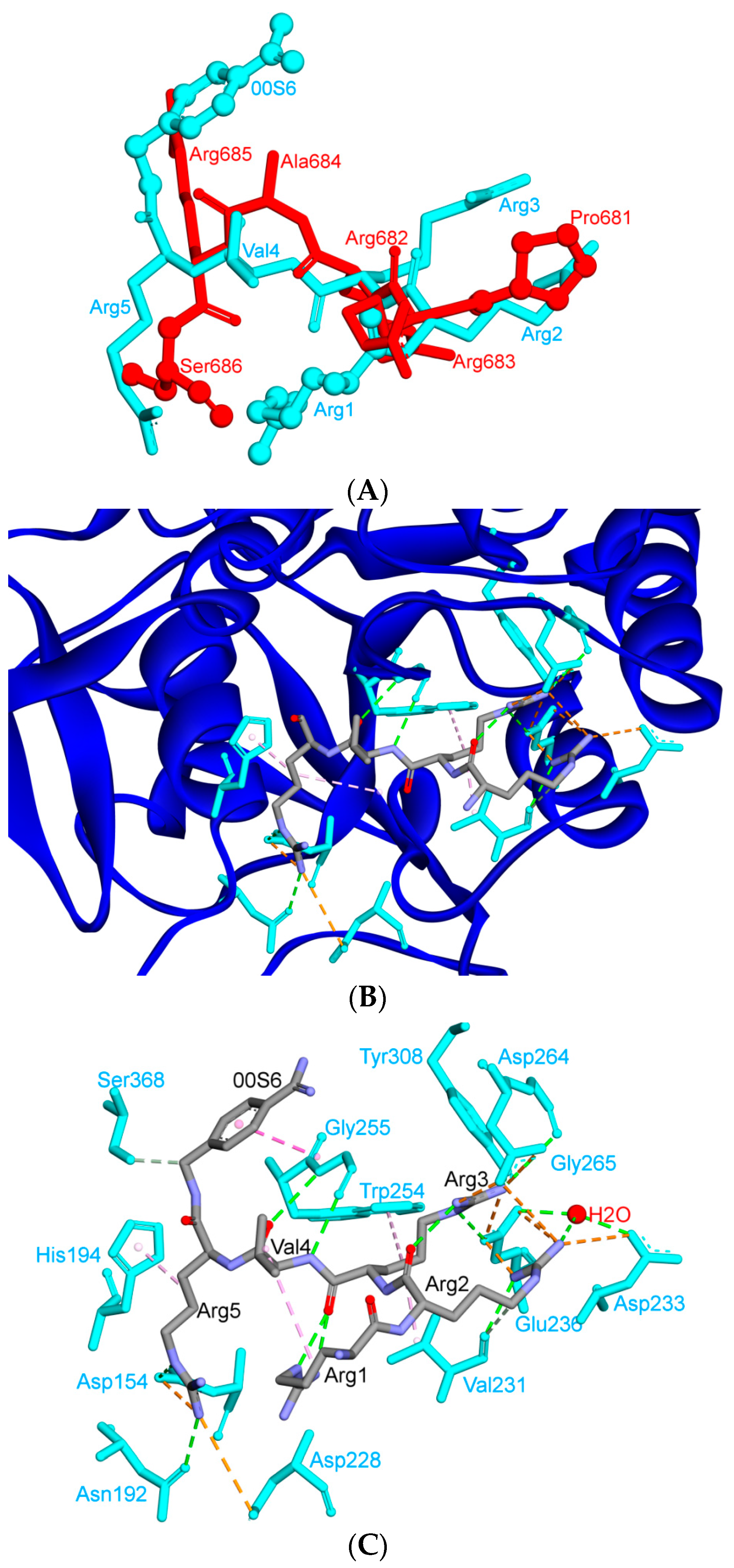
6. Is ACE2 the Receptor for S Protein?
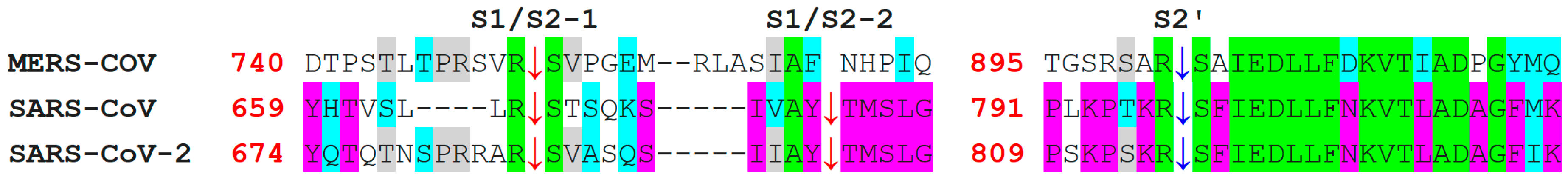
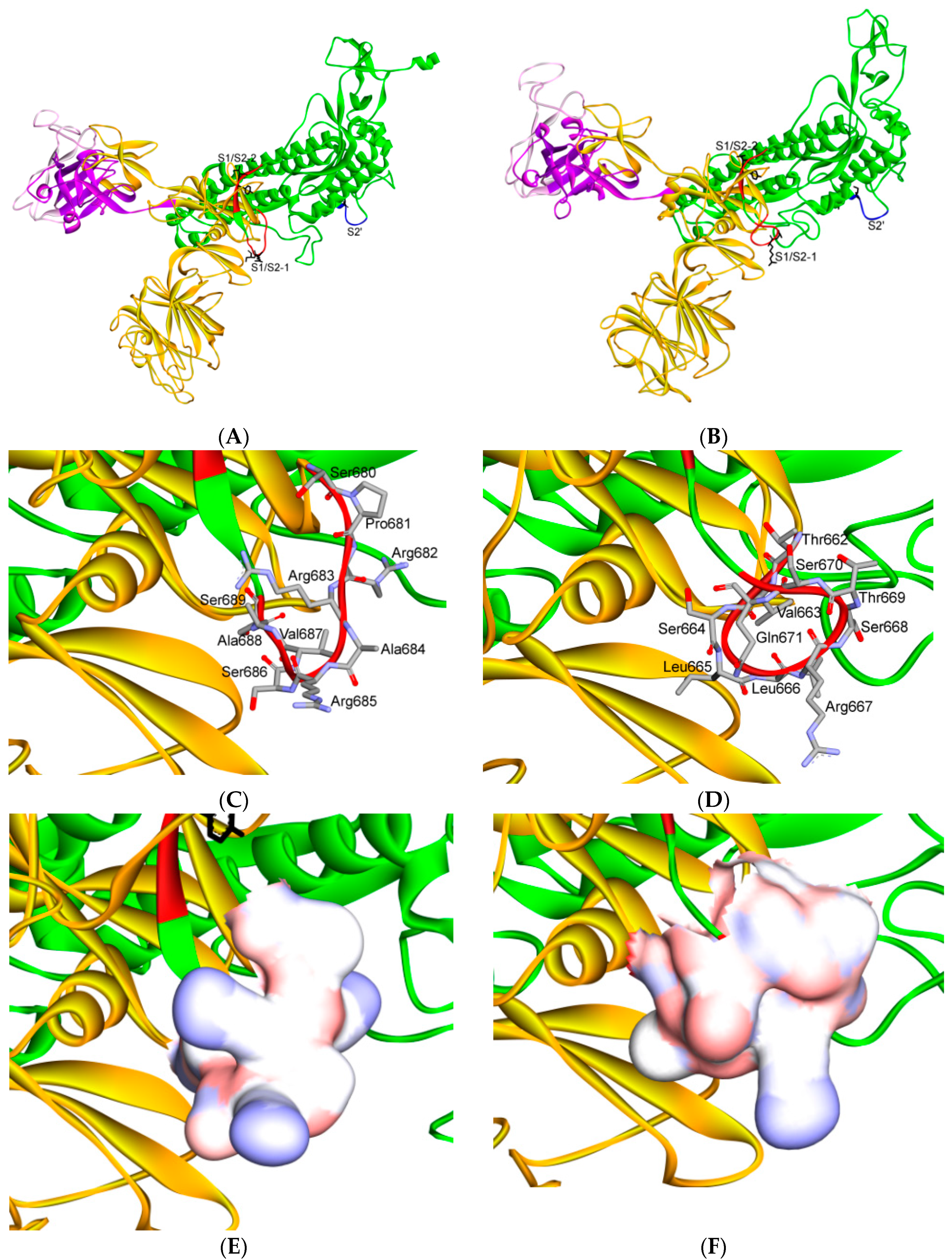
7. Differences in Furin-Like Protease Recognition Pattern
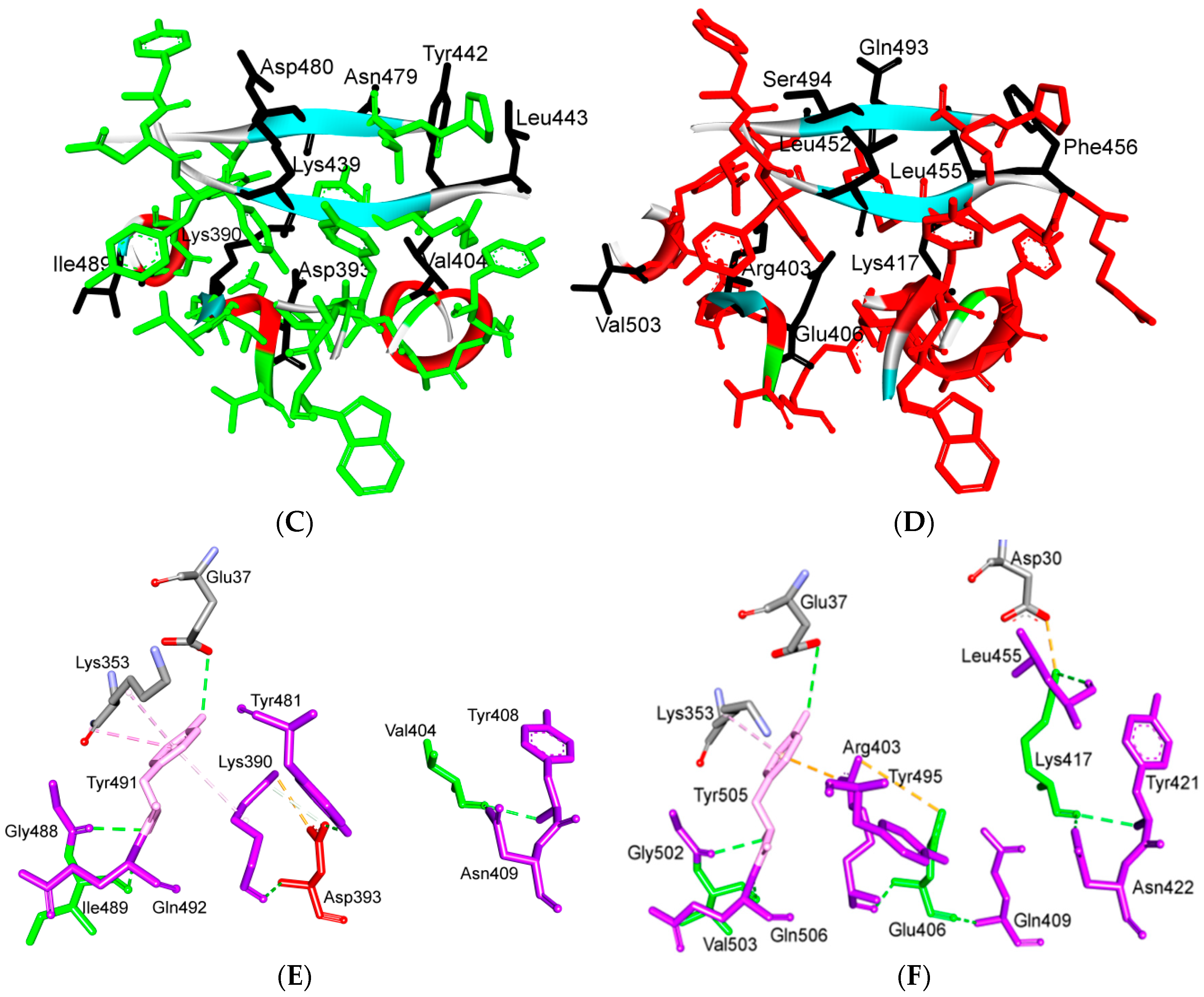
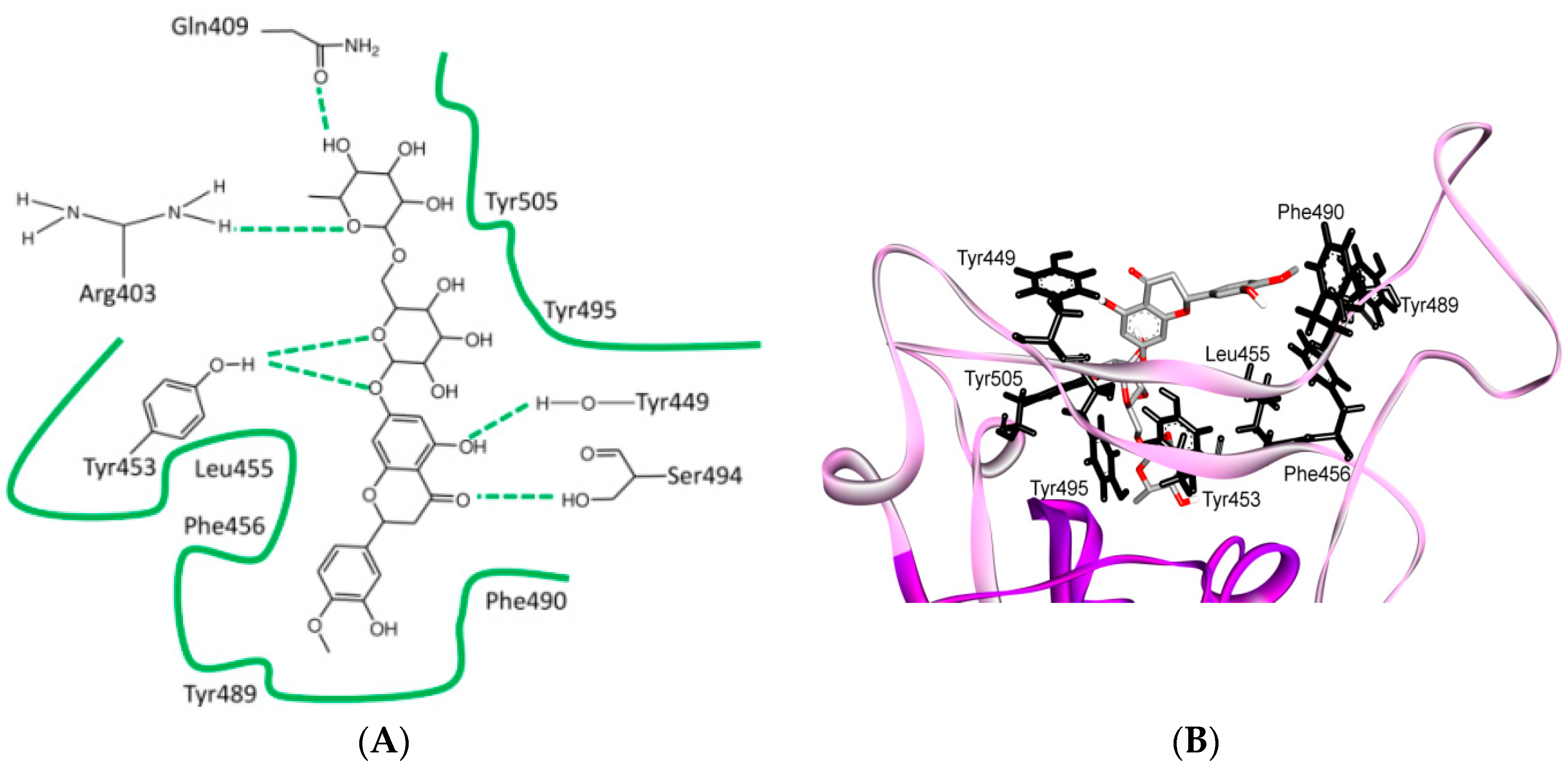
8. Effect of the S Protein and ACE2 Differences on the Binding Contacts, Affinity and Virulence
9. S Protein and ACE2 Differences Determine the Risk of Being an Intermediate Host
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by the novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020, 94, 94. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef] [PubMed]
- The, L.I.D. Challenges of coronavirus disease 2019. Lancet Infect. Dis. 2020, 20, 261. [Google Scholar]
- Huang, J.; Song, W.; Huang, H.; Sun, Q. Pharmacological therapeutics targeting RNA-dependent RNA polymerase, proteinase and spike protein: From mechanistic studies to clinical trials for COVID-19. J. Clin. Med. 2020, 9, 1131. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses: Methods and Protocols; Maier, H.J., Bickerton, E., Britton, P., Eds.; Springer: New York, NY, USA, 2015; pp. 1–23. [Google Scholar]
- Turner, A. Chapter 25-ACE2 cell biology, regulation, and physiological functions. In The Protective Arm of the Renin Angiotensin System (RAS): Functional Aspects; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Han, Y.; Král, P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef]
- Deng, X.; Cao, M.; Zhang, J.; Hu, K.; Yin, Z.; Zhou, Z.; Xiao, X.; Yang, Y.; Sheng, W.; Wu, Y. Hyaluronic acid-chitosan nanoparticles for co-delivery of MiR-34a and doxorubicin in therapy against triple negative breast cancer. Biomaterials 2014, 35, 4333–4344. [Google Scholar] [CrossRef]
- Magrone, T.; Magrone, M.; Jirillo, E. Focus on Receptors for Coronaviruses with Special Reference to Angiotensin-converting Enzyme 2 as a Potential Drug Target—A Perspective. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 807–811. [Google Scholar] [CrossRef]
- Batlle, D.; Wysocki, J.; Satchell, K. Soluble angiotensin-converting enzyme 2: A potential approach for coronavirus infection therapy? Clin. Sci. 2020, 134, 543–545. [Google Scholar] [CrossRef]
- Robson, B. COVID-19 Coronavirus spike protein analysis for synthetic vaccines, a peptidomimetic antagonist, and therapeutic drugs, and analysis of a proposed achilles’ heel conserved region to minimize probability of escape mutations and drug resistance. Comput. Biol. Med. 2020, 121, 103749. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef]
- Snijder, E.; Decroly, E.; Ziebuhr, J. The nonstructural proteins directing coronavirus RNA synthesis and processing. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2016; Volume 96, pp. 59–126. [Google Scholar]
- Sola, I.; Almazan, F.; Zuniga, S.; Enjuanes, L. Continuous and discontinuous RNA synthesis in coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef]
- Guruprasad, L. Evolutionary relationships and sequence-structure determinants in human SARS coronavirus-2 spike proteins for host receptor recognition. Proteins 2020, 88, 1387–1393. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Saltalamacchia, A.; Magistrato, A. Is the Rigidity of SARS-CoV-2 Spike Receptor-Binding Motif the Hallmark for Its Enhanced Infectivity? Insights from All-Atom Simulations. J. Phys. Chem. Lett. 2020, 11, 4785–4790. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Virus. Res. 2016, 96, 59–126. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-Y.; Ke, T.-Y.; Liao, W.-Y.; Chang, N.-Y. Regulation of coronaviral poly (A) tail length during infection. PLoS ONE 2013, 8, e70548. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zheng, C. Replication and transmission mechanisms of highly pathogenic human coronaviruses. Zhejiang Da Xue Xue Bao Yi Xue ban J. Zhejiang Univ. Med Sci. 2020, 49, 324–339. [Google Scholar]
- Tvarogová, J.; Madhugiri, R.; Bylapudi, G.; Ferguson, L.J.; Karl, N.; Ziebuhr, J. Identification and characterization of a human coronavirus 229E nonstructural protein 8-associated RNA 3′-terminal adenylyltransferase activity. J. Virol. 2019, 93, 93. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Malik, Y.A. Properties of Coronavirus and SARS-CoV-2. Malays. J. Pathol. 2020, 42, 3–11. [Google Scholar] [PubMed]
- Al-Hatamleh, M.A.I.; Hatmal, M.M.; Sattar, K.; Ahmad, S.; Mustafa, M.Z.; Bittencourt, M.D.C.; Mohamud, R. Antiviral and Immunomodulatory Effects of Phytochemicals from Honey against COVID-19: Potential Mechanisms of Action and Future Directions. Molecules 2020, 25, 5017. [Google Scholar] [CrossRef] [PubMed]
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-CoV: A comparative overview. Infez. Med. 2020, 28, 174–184. [Google Scholar]
- Ji, H.-L.; Zhao, R.; Matalon, S.; Matthay, M.A. Elevated plasmin (ogen) as a common risk factor for COVID-19 susceptibility. Physiol. Rev. 2020, 100, 1065–1075. [Google Scholar] [CrossRef]
- Kumar, S.; Maurya, V.K.; Prasad, A.K.; Bhatt, M.L.; Saxena, S.K. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV). Virusdisease 2020, 31, 13–21. [Google Scholar] [CrossRef]
- Yin, C. Genotyping coronavirus SARS-CoV-2: Methods and implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef]
- Xia, S.; Liu, Q.; Wang, Q.; Sun, Z.; Su, S.; Du, L.; Ying, T.; Lu, L.; Jiang, S. Middle East respiratory syndrome coronavirus (MERS-CoV) entry inhibitors targeting spike protein. Virus Res. 2014, 194, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining ‘host jump’of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic analysis and structural modeling of SARS-CoV-2 spike protein reveals an evolutionary distinct and proteolytically-sensitive activation loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef] [PubMed]
- Hulswit, R.; De Haan, C.; Bosch, B.-J. Coronavirus spike protein and tropism changes. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2016; Volume 96, pp. 29–57. [Google Scholar]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Chang, T.-J.; Yang, D.-M.; Wang, M.-L.; Liang, K.-H.; Tsai, P.-H.; Chiou, S.-H.; Lin, T.-H.; Wang, C.-T. Genomic analysis and comparative multiple sequences of SARS-CoV2. J. Chin. Med. Assoc. 2020, 83, 537–543. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997, 2, 173–181. [Google Scholar] [CrossRef]
- Rowland, R.R.; Yoo, D. Nucleolar-cytoplasmic shuttling of PRRSV nucleocapsid protein: A simple case of molecular mimicry or the complex regulation by nuclear import, nucleolar localization and nuclear export signal sequences. Virus Res. 2003, 95, 23–33. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiao, X.; Wei, X.; Li, J.; Yang, J.; Tan, H.; Zhu, J.; Zhang, Q.; Wu, J.; Liu, L. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 2020, 92, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Lu, Y.; Jin, X.; Zhang, L. Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem. Biophys. Res. Commun. 2020, 526, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O. How large B-factors can be in protein crystal structures. BMC Bioinform. 2018, 19, 61. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Joshi, M.; Degani, M.S. Tackling SARS-CoV-2: Proposed targets and repurposed drugs. Future Med. Chem. 2020, 12, 1579–1601. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Dahanayake, J.N.; Mitchell-Koch, K.R. How does solvation layer mobility affect protein structural dynamics? Front. Mol. Biosci. 2018, 5, 65. [Google Scholar] [CrossRef]
- Kay, B.K.; Williamson, M.P.; Sudol, M. The importance of being proline: The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000, 14, 231–241. [Google Scholar] [CrossRef]
- Sudol, M.; Bedford, M.T. Competitive Binding of Proline-Rich Sequences by SH3, WW, and Other Functionally Related Protein Domains. In Proteomics and Protein-Protein Interactions: Biology, Chemistry, Bioinformatics, and Drug Design; Waksman, G., Ed.; Springer: Boston, MA, USA, 2005; pp. 185–201. [Google Scholar]
- Lise, S.; Jones, D. Sequence patterns associated with disordered regions in proteins. PROTEINS Struct. Funct. Bioinform. 2005, 58, 144–150. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Kimzey, A.L.; Masselon, C.D.; Bruce, J.E.; Garner, E.C.; Brown, C.J.; Dunker, A.K.; Smith, R.D.; Ackerman, E.J. Identification of intrinsic order and disorder in the DNA repair protein XPA. Protein Sci. 2001, 10, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Bright, J.N.; Sansom, M.S. The flexing/twirling helix: Exploring the flexibility about molecular hinges formed by proline and glycine motifs in transmembrane helices. J. Phys. Chem. B 2003, 107, 627–636. [Google Scholar] [CrossRef]
- Chowdhury, R.; Maranas, C.D. Biophysical characterization of the SARS-CoV-2 spike protein binding with the ACE2 receptor explains increased COVID-19 pathogenesis. bioRxiv 2020. preprint. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.-C.D.; So, R.T.; Lv, H.; Mok, C.K.; Wilson, I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Di Scala, C.; Chahinian, H.; Yahi, N. Structural and molecular modeling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int. J. Antimicrob. Agents 2020, 55, 105960. [Google Scholar] [CrossRef] [PubMed]
- Brufsky, A. Distinct Viral Clades of SARS-CoV-2: Implications for Modeling of Viral Spread. J. Med. Virol. 2020, 92, 1386–1390. [Google Scholar] [CrossRef]
- Reva, B.A.; Finkelstein, A.V.; Skolnick, J. What is the probability of a chance prediction of a protein structure with an rmsd of 6 A? Fold. Des. 1998, 3, 141–147. [Google Scholar] [CrossRef]
- Hatmal, M.M.; Taha, M.O. Combining Stochastic Deformation/Relaxation and Intermolecular Contacts Analysis for Extracting Pharmacophores from Ligand-Receptor Complexes. J. Chem. Inf. Model. 2018, 58, 879–893. [Google Scholar] [CrossRef]
- Maiorov, V.N.; Crippen, G.M. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef]
- Kannan, S.; Ali, P.S.S.; Sheeza, A.; Hemalatha, K. COVID-19 (Novel Coronavirus 2019)-recent trends. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2006–2011. [Google Scholar] [PubMed]
- Kuster, G.M.; Pfister, O.; Burkard, T.; Zhou, Q.; Twerenbold, R.; Haaf, P.; Widmer, A.F.; Osswald, S. SARS-CoV2: Should inhibitors of the renin–angiotensin system be withdrawn in patients with COVID-19? Eur. Heart J. 2020, 41, 1801–1803. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Korteweg, C. Pathology and pathogenesis of severe acute respiratory syndrome. Am. J. Pathol. 2007, 170, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Delmas, B.; Laude, H. Assembly of coronavirus spike protein into trimers and its role in epitope expression. J. Virol. 1990, 64, 5367–5375. [Google Scholar] [CrossRef]
- Kim, E.; Erdos, G.; Huang, S.; Kenniston, T.W.; Balmert, S.C.; Carey, C.D.; Raj, V.S.; Epperly, M.W.; Klimstra, W.B.; Haagmans, B.L. Microneedle array delivered recombinant coronavirus vaccines: Immunogenicity and rapid translational development. EBioMedicine 2020, 55, 102743. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; Van Der Zee, R.; De Haan, C.A.M.; Rottier, P.J.M. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 2020. preprint. [Google Scholar] [CrossRef]
- Dhama, K.; Sharun, K.; Tiwari, R.; Dadar, M.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W. COVID-19, an emerging coronavirus infection: Advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. Hum. Vaccines Immunother. 2020, 16, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Dahms, S.O.; Hardes, K.; Steinmetzer, T.; Than, M.E. X-ray structures of the proprotein convertase furin bound with substrate analogue inhibitors reveal substrate specificity determinants beyond the s4 pocket. Biochemistry 2018, 57, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Elshuber, S.; Mandl, C.W. Resuscitating mutations in a furin cleavage-deficient mutant of the flavivirus tick-borne encephalitis virus. J. Virol. 2005, 79, 11813–11823. [Google Scholar] [CrossRef] [PubMed]
- Shiryaev, S.A.; Chernov, A.V.; Golubkov, V.S.; Thomsen, E.R.; Chudin, E.; Chee, M.S.; Kozlov, I.A.; Strongin, A.Y.; Cieplak, P. High-resolution analysis and functional mapping of cleavage sites and substrate proteins of furin in the human proteome. PLoS ONE 2013, 8, e54290. [Google Scholar] [CrossRef]
- Follis, K.E.; York, J.; Nunberg, J.H. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell–cell fusion but does not affect virion entry. Virology 2006, 350, 358–369. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Remacle, A.G.; Ratnikov, B.I.; Nelson, N.A.; Savinov, A.Y.; Wei, G.; Bottini, M.; Rega, M.F.; Parent, A.; Desjardins, R. Targeting host cell furin proprotein convertases as a therapeutic strategy against bacterial toxins and viral pathogens. J. Biol. Chem. 2007, 282, 20847–20853. [Google Scholar] [CrossRef]
- Duckert, P.; Brunak, S.; Blom, N. Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 2004, 17, 107–112. [Google Scholar] [CrossRef]
- Tian, S.; Huajun, W.; Wu, J. Computational prediction of furin cleavage sites by a hybrid method and understanding mechanism underlying diseases. Sci. Rep. 2012, 2, 261. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 2020, 30, 1346–1351. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Braun, E.; Sauter, D. Furin-mediated protein processing in infectious diseases and cancer. Clin. Transl. Immunol. 2019, 8, e1073. [Google Scholar] [CrossRef] [PubMed]
- Izaguirre, G. The proteolytic regulation of virus cell entry by furin and other proprotein convertases. Viruses 2019, 11, 837. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Prat, A. The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Decha, P.; Rungrotmongkol, T.; Intharathep, P.; Malaisree, M.; Aruksakunwong, O.; Laohpongspaisan, C.; Parasuk, V.; Sompornpisut, P.; Pianwanit, S.; Kokpol, S. Source of high pathogenicity of an avian influenza virus H5N1: Why H5 is better cleaved by furin. Biophys. J. 2008, 95, 128–134. [Google Scholar] [CrossRef][Green Version]
- Schwab, M. Encyclopedia of Cancer; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Bottomley, K.M.; Bradshaw, D.; Nixon, J.S. Metalloproteinases as Targets for Anti-Inflammatory Drugs; Springer: Basel, Switzerland, 1999; Volume 14. [Google Scholar]
- Nagase, H.; Woessner, J.F. Matrix metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [Google Scholar] [CrossRef]
- Hernandez-Barrantes, S.; Bernardo, M.; Toth, M.; Fridman, R. Regulation of membrane type-matrix metalloproteinases. Semin. Cancer Biol. 2002, 12, 131–138. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Ratnikov, B.I.; Yu, Q.; Baciu, P.C.; Rozanov, D.V.; Strongin, A.Y. Prointegrin maturation follows rapid trafficking and processing of MT1-MMP in Furin-Negative Colon Carcinoma LoVo Cells. Traffic 2004, 5, 627–641. [Google Scholar] [CrossRef]
- Nakai, K.; Kanehisa, M. A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics 1992, 14, 897–911. [Google Scholar] [CrossRef]
- Rowland, R.R.; Kervin, R.; Kuckleburg, C.; Sperlich, A.; Benfield, D.A. The localization of porcine reproductive and respiratory syndrome virus nucleocapsid protein to the nucleolus of infected cells and identification of a potential nucleolar localization signal sequence. Virus. Res. 1999, 64, 1–12. [Google Scholar] [CrossRef]
- Rowland, R.R.; Schneider, P.; Fang, Y.; Wootton, S.; Yoo, D.; Benfield, D.A. Peptide domains involved in the localization of the porcine reproductive and respiratory syndrome virus nucleocapsid protein to the nucleolus. Virology 2003, 316, 135–145. [Google Scholar] [CrossRef]
- Tijms, M.A.; van der Meer, Y.; Snijder, E.J. Nuclear localization of non-structural protein 1 and nucleocapsid protein of equine arteritis virus. J. Gen. Virol. 2002, 83, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.; Wootton, S.K.; Li, G.; Song, C.; Rowland, R.R. Colocalization and interaction of the porcine arterivirus nucleocapsid protein with the small nucleolar RNA-associated protein fibrillarin. J. Virol. 2003, 77, 12173–12183. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, J. The nucleolus—A gateway to viral infection? Arch. Virol. 2002, 147, 1077–1089. [Google Scholar] [CrossRef]
- Hiscox, J.A.; Wurm, T.; Wilson, L.; Britton, P.; Cavanagh, D.; Brooks, G. The coronavirus infectious bronchitis virus nucleoprotein localizes to the nucleolus. J. Virol. 2001, 75, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Rowland, R.R.; Chauhan, V.; Fang, Y.; Pekosz, A.; Kerrigan, M.; Burton, M.D. Intracellular localization of the severe acute respiratory syndrome coronavirus nucleocapsid protein: Absence of nucleolar accumulation during infection and after expression as a recombinant protein in vero cells. J. Virol. 2005, 79, 11507–11512. [Google Scholar] [CrossRef] [PubMed]
- Satarker, S.; Nampoothiri, M. Structural Proteins in Severe Acute Respiratory Syndrome Coronavirus-2. Arch. Med. Res. 2020, 51, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Sorour, M.K.; Kasry, A. Comparing the binding interactions in the receptor binding domains of SARS-CoV-2 and SARS-CoV. J. Phys. Chem. Lett. 2020, 11, 4897–4900. [Google Scholar] [CrossRef]
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI J. 2020, 19, 410. [Google Scholar]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
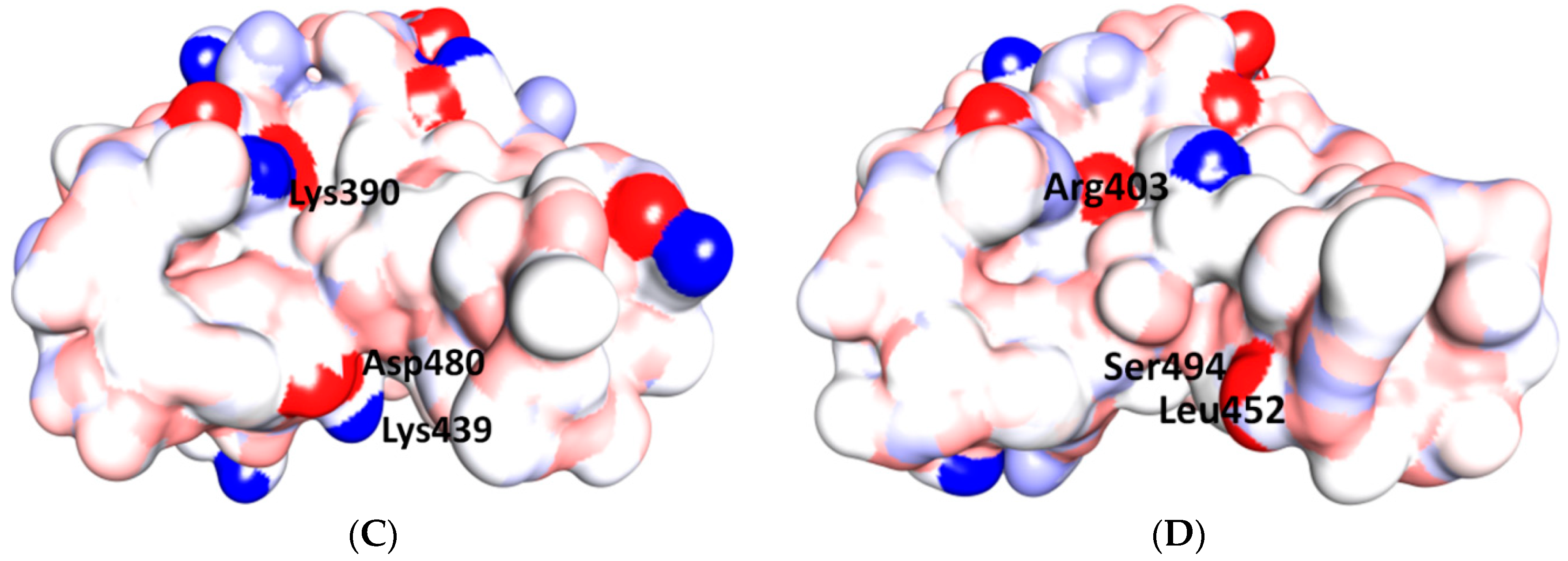{kind=link}
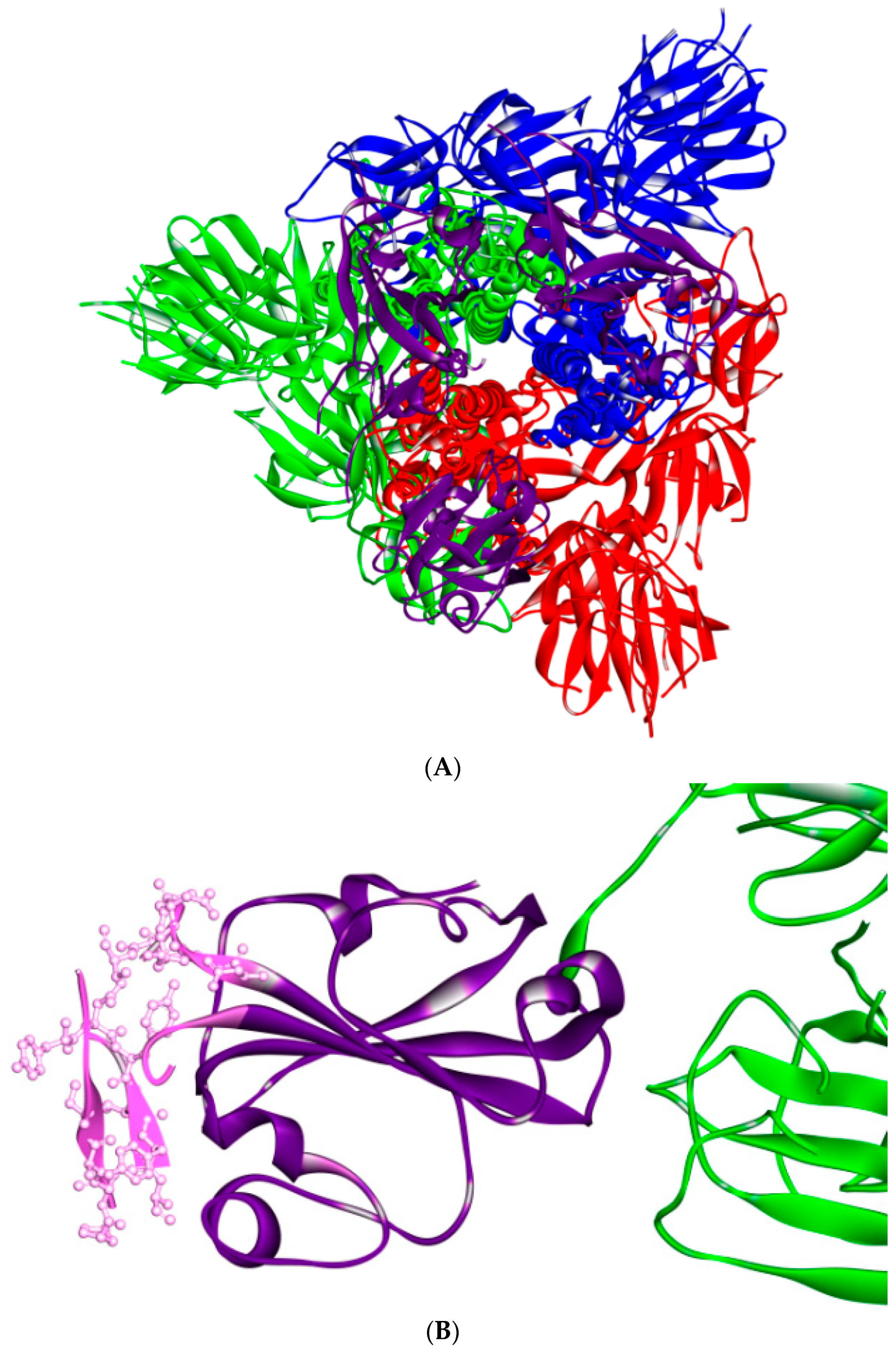{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function |
|---|---|
| nsp1, nsp3 | Inhibition of IFN signaling and blocking of host’s innate immune response by promotion of cellular degradation and blockage of translation of host’s RNA. |
| nsp2 | Binding to prohibition protein. |
| nsp3 | Papain-like protease domain, it promotes cytokine expression and cleavage of viral polyprotein. |
| nsp4, nsp6 | Contribution to structure of DNA methylation valleys (DMVs) as transmembrane scaffold protein (DMVs formation). |
| nsps5 | 3C-like protease domain. |
| nsp7, nsp8 | Processivity clamp for RNA polymerase by arms hexadecameric complex; nsp 8 has adenylyltransferase activity. |
| nsp9 | RNA binding protein phosphatase. |
| nsps12 | RNA-dependent RNA polymerase. |
| nsp10, nsp16, nsp14 | Stimulation of ExoN and 2-O-MT activity. |
| nsp13 | RNA helicase, 5′ triphosphatase. |
| nsp14 | Proofreading of viral genome. |
| S protein | Forms homotrimers that protrude in the viral surface and facilitate binding of envelope viruses to host cells by attraction with ACE2 that is expressed in lower respiratory tract cells. This glycoprotein is cleaved by the host cell furin-like protease into two subunits, S1 and S2. S1 is responsible for the determination of the host virus range and cellular tropism with the receptor binding domain make-up, while S2 mediates virus fusion in transmitting host cells. |
| N protein | Structural component localizes in the endoplasmic reticulum-Golgi region that structurally is bound to the nucleic acid material of the virus. The protein is bound to RNA, so the protein is involved in processes related to the viral genome, the viral replication cycle, and the cellular response of host cells to viral infections. N protein is also heavily phosphorylated and it is suggested that its presence leads to structural changes that enhance the affinity for viral RNA. |
| M protein | The most tightly structured protein, it plays a role in the determination of the shape of the virus envelope. This protein can bind to all other structural proteins. Binding with M protein helps to stabilize nucleocapsids or N proteins and promotes completion of viral assembly by stabilizing N protein-RNA complex, inside the internal virion. |
| E protein | The smallest of the major structural proteins. It plays a role in the production and maturation of this virus. |
| Virus | Position 1 | Position 2 | Position 3 | Position 4 | Position 5 |
|---|---|---|---|---|---|
| SARS-CoV | F > Y, Y, S | F > L > P, L = P, F | N, R > K = N, N | D > G, G > D, D | T >> S, S, N |
| SARS-CoV-2 | L | F | Q | S | N |
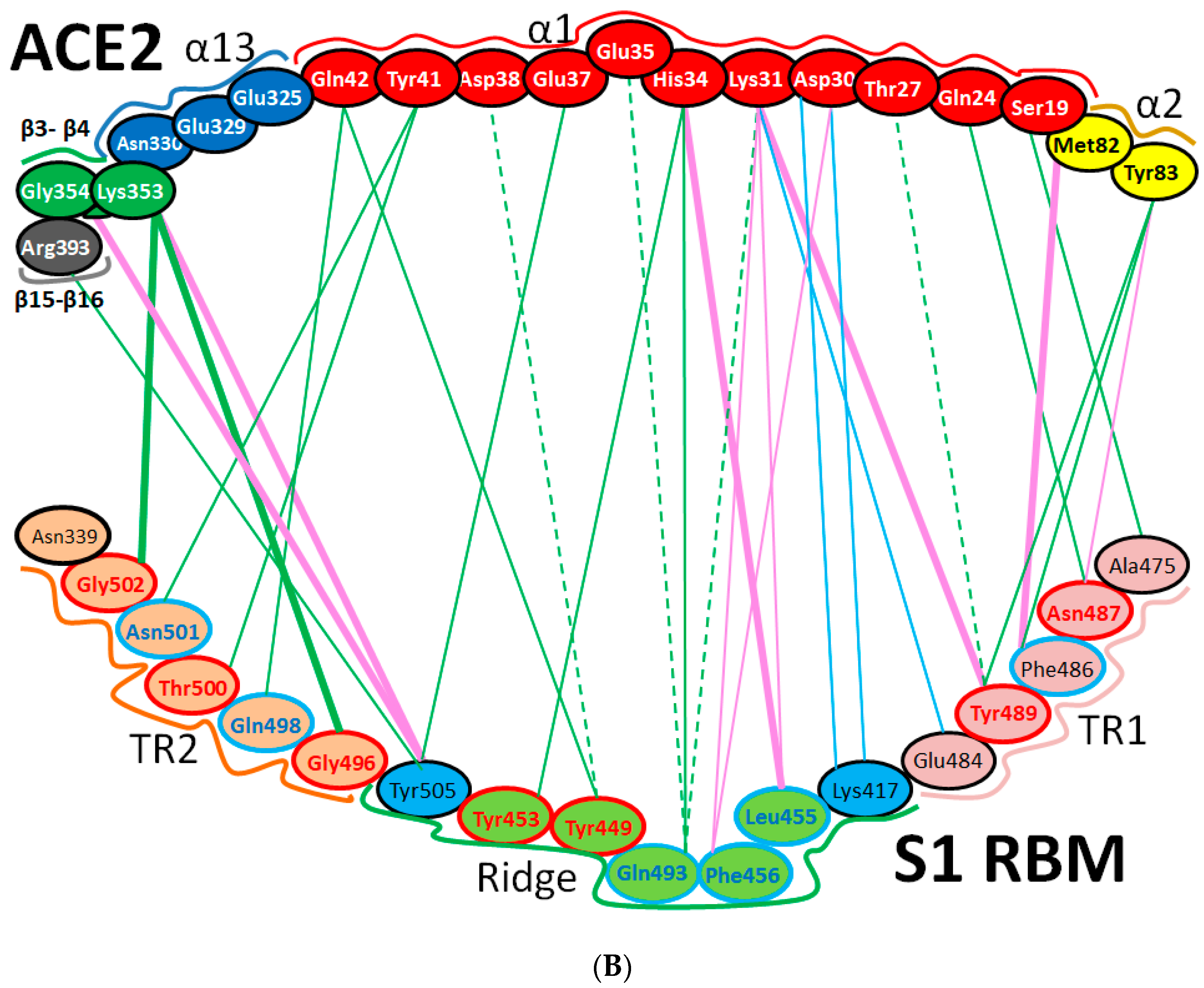
| Region | Number | Amino Acid | Amino Acid ACE2 | Interactions | Details |
|---|---|---|---|---|---|
| TR1 | 1 | A475 | S19 | Strong polar contacts | H: A475(O)-S19(HG): 3.2 (6LZG) |
| P462 | - | No interactions | |||
| 2 | N487 | Q24 | Strong polar contacts | H: N487(ND2)-Q24(HE21): 2.2 (6LZG) H: N487(HD22)-Y83(OH): 2.3 (6LZG) | |
| N473 | Q24,Y83 | Strong polar contacts | H: N473(ND2)-Q24(OE1): 2.5 (2AJF) H: N473(ND2)- Y83(HH): 2.5 (2AJF) | ||
| 3 | T486 | N330 | Hydrogen bonding | H: T486(O)-N330(HD22): 2.3 (2AJF) H: T486(OG1)-Y41(HH): 2.6 (2AJF) H: T486(O)-N330(HD22): 2.3 (2AJF) | |
| F486 | Y83, M82 | Stacking and hydrophobic interactions | ST (Pi-Pi): F486-Y83: 5.2 (6LZG) D (Pi-alkyl): F486-M82: 4.6 (6LZG, 6M17) | ||
| 4 | Y489 | K31 | Hydrophobic and hydrogen interactions | ST (Pi-alkyl): Y489-K31: 4.8 (6LZG, 6M17) H: Y489(HH)-Y83(OH): 3.0 (6LZG) H: Y489(HH)-T27(O): 1.7 (6M17) | |
| Y475 | K31 | Hydrophobic and hydrogen interactions | ST (Pi-alkyl): Y475- K31: 5.2 (2AJF) H: Y475(HH)-Y83(OH): 2.8 (2AJF) | ||
| 5 | E484 | K31 | Strong polar contacts | S: E484(OE2)-K31(NZ): 4.2 (6LZG) | |
| P470 | - | No interactions | - | ||
| Ridge and upper core region | 6 | K417 | D30 | Electrostatic interactions (Salt bridge) | HS: K417(HZ1)-D30(OD1): 2.1 (6LZG) HS: K417(HZ3)-D30(OD2): 3.0 (6LZG) |
| V404 | - | No interactions | - | ||
| 7 | L455 | K31, H34 | Hydrophobic interactions | D (Pi-alkyl): L455-H34: 3.5 (6LZG, 6M17) D: L455(CD2)-K31(CA): 4.3 (6LZG, 6M17) | |
| Y442 | K31, H34 | - | ST: (Pi-Pi): Y442-H34: 6.0 (2AJF) D:Y442(CE1)-K31(CA): 4.1 (2AJF) | ||
| 8 | F456 | D30,K31 | Stacking | ST: (Pi-alkyl): F456-K31: 4.5(6M17) ST: (Pi-alkyl): F456-D30: 4.4 (6M17) | |
| L443 | T27 | Hydrophobic interactions | D: L443(CD2)-T27(CG2): 4.2 (2AJF) | ||
| 9 | Q493 | K31, H34, E35 | Hydrogen bonding | H: Q493(HE22)-E35(OE2): 1.9 (6M17) H: Q493(HE21)-H34(O): 2.9 (6LZG) H: Q493(OE1)-K31(HZ2/HZ3): 3.0 (6M17) | |
| N479 | H34 | - | H: N479(HD21)-H34(ND1): 3.1 (2AJF) | ||
| 10 | Y449 | D38, Q42 | Hydrogen bonding | H: Y449(HH)-D38(OD1): 2.8 ((6M17) H: Y449(OH)-Q42(OE1): 2.5 (6LZG) | |
| Y436 | D38 | Hydrogen bonding | H: Y436(HH)-D38(OD1): 2.8 (2AJF) H: Y436(HH)-D38(OD2): 3.4 (2AJF) H: Y436(HH)-Q42(OE1): 3.4 (2AJF) | ||
| 11 | Y453 | H34 | Polar contacts | H: Y453(HH)-H34(NE2): 3.2 (6M17) | |
| Y440 | H34 | Polar contacts | H: Y440(HH)-H34(NE2): 3.3 (2AJF) | ||
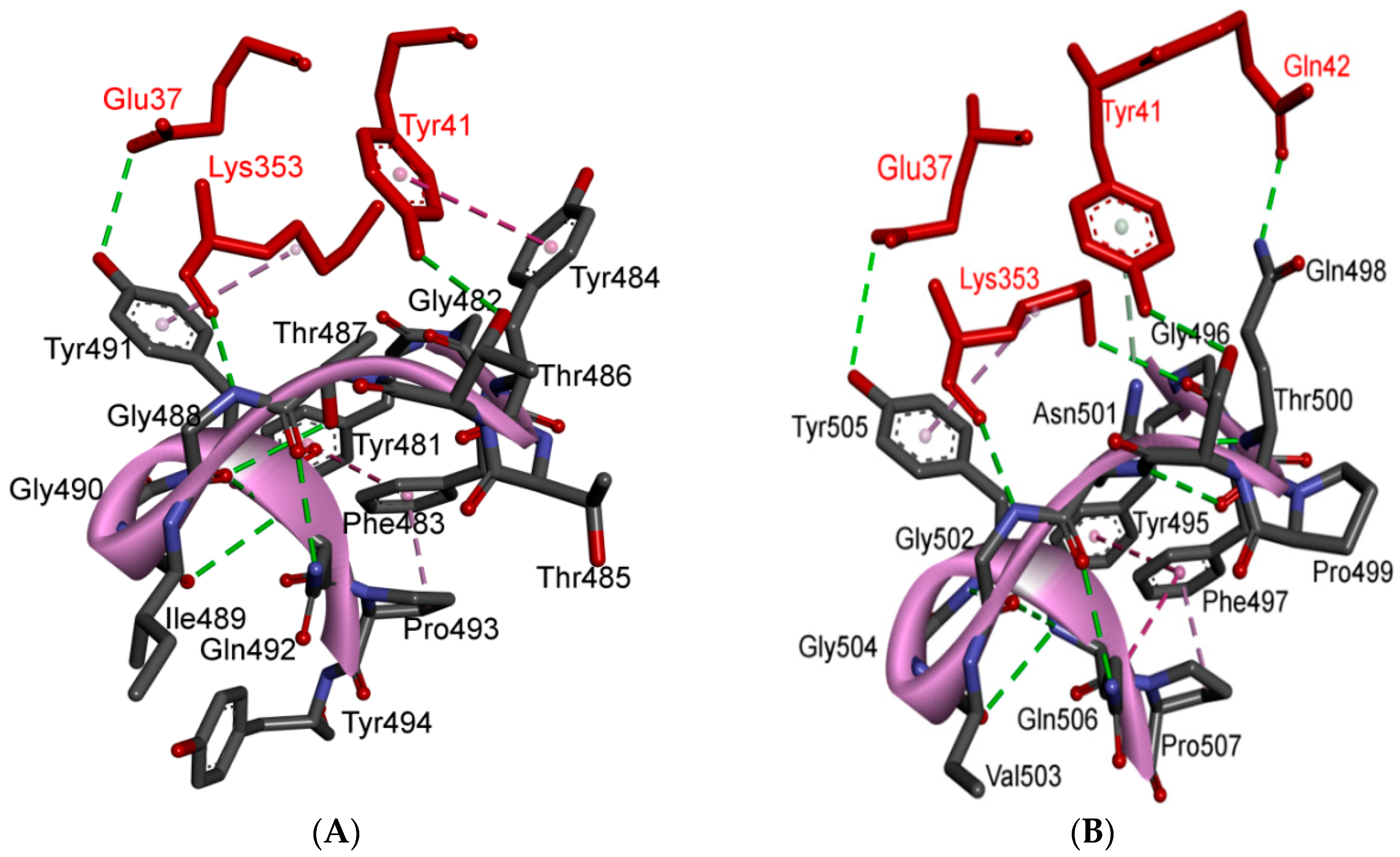
| 12 | Y505 | - | - | H: Y505(HH)-E37(OE1): 2.6 (6LZG, 6M17) H: Y505(OH)-R393(HH22): 3.4 (6LZG, 6M17) ST: (Pi-amid): Y505-K353/Gly354: 4.0 (6LZG, 6M17) ST (Pi-alkyl):Y505-K353: 4.6 (6LZG, 6M17) | |
| Y491 | - | - | H: Y491(HH)-E37(OE1): 2.4 (2AJF) H: Y491(OH)-R393(HH22): 3.4 (2AJF) ST: (Pi-amid): Y491-K353/Gly354: 4.1 (2AJF) ST: (Pi-alkyl): Y491-K353: 4.3 (2AJF) | ||
| TR2 | 13 | G496 | K353 * | Hydrogen bonding | H: G496(O)-K353(HZ2/HZ3): 2.3 (6LZG, 6M17) |
| G482 | K353 * | Hydrogen bonding | H: G482(O)-K353(HZ2/HZ3): 3.9 (2AJF) | ||
| 14 | Q498 | Q48 | Hydrogen bonding | H: Q498(HE22)-Q42(OE1): 2.2 (6LZG) | |
| Y484 | Y41 | Stacking | ST (Pi-Pi): Y484-Y41: 5.3 (2AJF) | ||
| 15 | T500 | Y41 | Hydrogen bonding | H: T500(OG1)-Y41(HH): 2.8 (6LZG, 6M17) | |
| T486 | N330 | Hydrogen bonding | H: T486(O)-N330(HD22): 2.3 (2AJF) H: T486(OG1)-Y41(HH): 2.6 (2AJF) H: T486(O)-N330(HD22): 2.3 (2AJF) | ||
| 16 | N501 | Y41 | Hydrogen bonding | H: N501(HD21)- Y41(OH): 2.3 (6LZG) | |
| T487 | Y41 | Hydrogen bonding | H: T487(HG1)- Y41(HH): 4.0 (2AJF) | ||
| 17 | G502 | K353 * | Hydrogen bonding | H: G502(HN)-K353(O): 1.7 (6LZG, 6M17) | |
| G488 | K353 * | Hydrogen bonding | H: G488(HN)-K353(O): 1.5 (2AJF) | ||
| 18 | N439 | - | No interactions | - | |
| R426 | E329, Q325 | - | S: R426(NH2)-E329(OE2): 3.0 (2AJF) H: R426(HH21)-Q325(OE1): 3.5 (2AJF) H: R426(HH12)-E329(OE2): 3.1 (2AJF) H: R426(HH22)-E329(OE2): 2.0 (2AJF) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatmal, M.M.; Alshaer, W.; Al-Hatamleh, M.A.I.; Hatmal, M.; Smadi, O.; Taha, M.O.; Oweida, A.J.; Boer, J.C.; Mohamud, R.; Plebanski, M. Comprehensive Structural and Molecular Comparison of Spike Proteins of SARS-CoV-2, SARS-CoV and MERS-CoV, and Their Interactions with ACE2. Cells 2020, 9, 2638. https://doi.org/10.3390/cells9122638
Hatmal MM, Alshaer W, Al-Hatamleh MAI, Hatmal M, Smadi O, Taha MO, Oweida AJ, Boer JC, Mohamud R, Plebanski M. Comprehensive Structural and Molecular Comparison of Spike Proteins of SARS-CoV-2, SARS-CoV and MERS-CoV, and Their Interactions with ACE2. Cells. 2020; 9(12):2638. https://doi.org/10.3390/cells9122638
Chicago/Turabian StyleHatmal, Ma’mon M., Walhan Alshaer, Mohammad A. I. Al-Hatamleh, Malik Hatmal, Othman Smadi, Mutasem O. Taha, Ayman J. Oweida, Jennifer C. Boer, Rohimah Mohamud, and Magdalena Plebanski. 2020. "Comprehensive Structural and Molecular Comparison of Spike Proteins of SARS-CoV-2, SARS-CoV and MERS-CoV, and Their Interactions with ACE2" Cells 9, no. 12: 2638. https://doi.org/10.3390/cells9122638
APA StyleHatmal, M. M., Alshaer, W., Al-Hatamleh, M. A. I., Hatmal, M., Smadi, O., Taha, M. O., Oweida, A. J., Boer, J. C., Mohamud, R., & Plebanski, M. (2020). Comprehensive Structural and Molecular Comparison of Spike Proteins of SARS-CoV-2, SARS-CoV and MERS-CoV, and Their Interactions with ACE2. Cells, 9(12), 2638. https://doi.org/10.3390/cells9122638