Induced Pluripotent Stem Cells to Understand Mucopolysaccharidosis. I: Demonstration of a Migration Defect in Neural Precursors

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Construction of Plasmid and Lentiviral Vector
2.2. Lentiviral Vector Transduction and Rescue
2.3. IDUA Activity Measurement Assay
2.4. Establishment and Characterization of MPS1-H iPSC Line
2.5. hiPSC-Derived NSCs
2.6. NSCs Culture
2.7. NSC Differentiation
2.8. Neurospheres Differentiation Protocol
2.9. Immunofluorescent Analysis
2.10. Scratch Test
2.11. RNA Sequencing
3. Results
3.1. Restoration of IDUAα Activity of MPS1-H Cells Infected with UBI-IDUA-PGK-BSD Lentivector
3.1.1. IDUAα Activity of rMPS1 Fibroblasts
3.1.2. IDUAα Activity of rMPS1-H iPSCs
3.1.3. IDUAα Activity of rMPS1-H iPSCs-Derived NSCs
3.2. Characterization of Rescue rMPS1-H iPSCs
3.3. IDUA Lentiviral Rescue of MPS1-H NSCs Ameliorates Migration In Vitro
3.4. Alteration of Neurite Outgrowth during In Vitro MPS1-H Neuronal Differentiation
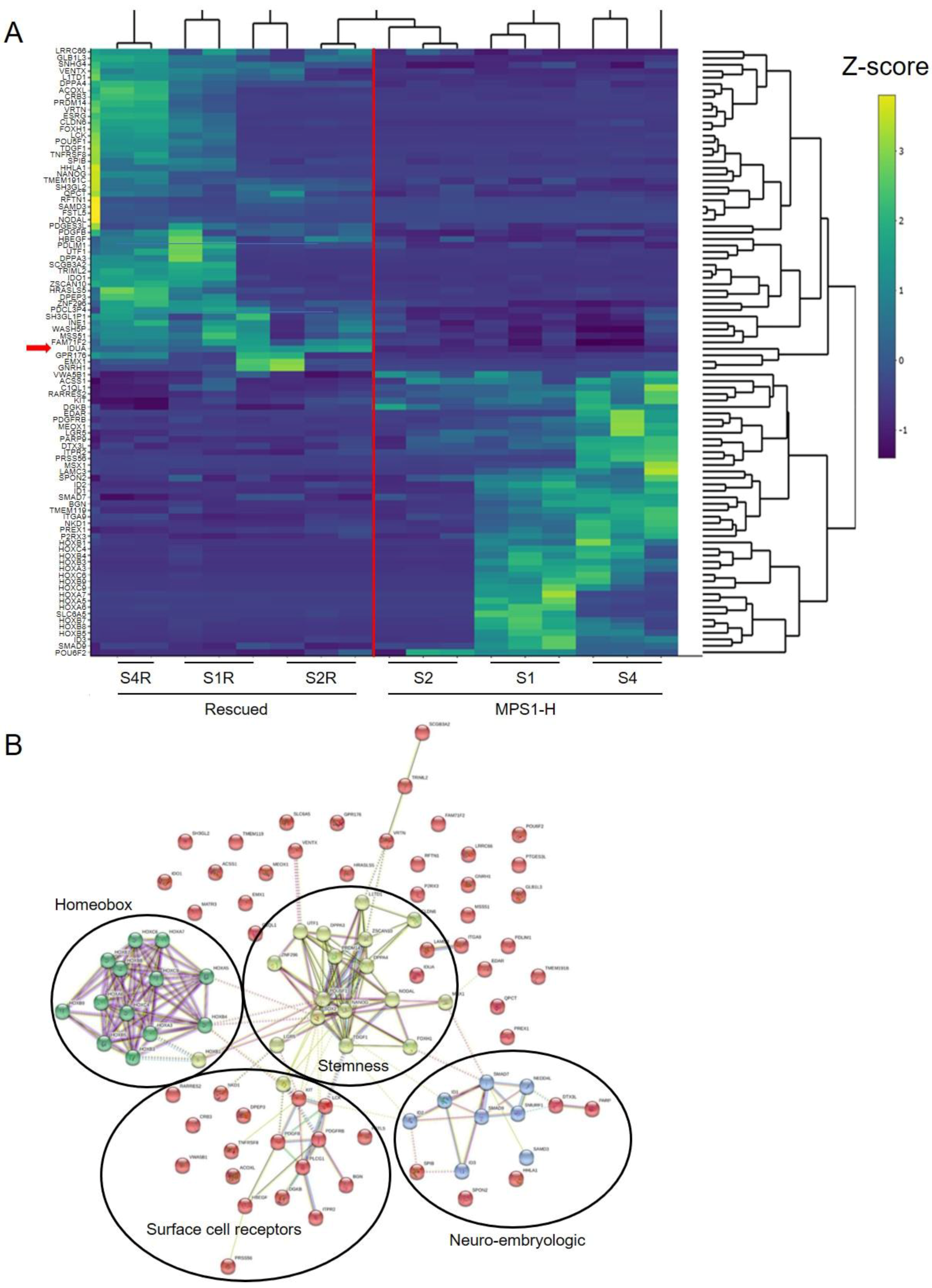
3.5. Transcriptome Analysis of MPS1-H Affected and Rescued iPSCs-Derived Neurospheres
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giugliani, R.; Federhen, A.; Rojas, M.V.M.; Vieira, T.; Artigalás, O.; Pinto, L.L.; Azevedo, A.C.; Acosta, A.; Bonfim, C.; Lourenço, C.M.; et al. Mucopolysaccharidosis I, II, and VI: Brief review and guidelines for treatment. Genet. Mol. Biol. 2010, 33, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F.; Muenzer, J. The Mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill Medical Publishing Division: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Berger, K.I.; Fagondes, S.C.; Giugliani, R.; Hardy, K.A.; Lee, K.S.; McArdle, C.; Scarpa, M.; Tobin, M.J.; Ward, S.A.; Rapoport, D.M. Respiratory and sleep disorders in mucopolysaccharidosis. J. Inherit. Metab. Dis. 2013, 36, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Braunlin, E.A.; Harmatz, P.R.; Scarpa, M.; Furlanetto, B.; Kampmann, C.; Loehr, J.P.; Ponder, K.P.; Roberts, W.C.; Rosenfeld, H.M.; Giugliani, R. Cardiac disease in patients with mucopolysaccharidosis: Presentation, diagnosis and management. J. Inherit. Metab. Dis. 2011, 34, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.G.; Lockman, L.A.; Balthazor, M.; Krivit, W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J. Inherit. Metab. Dis. 1995, 18, 413–429. [Google Scholar] [CrossRef]
- Shapiro, E.G.; Nestrasil, I.; Rudser, K.; Delaney, K.; Kovac, V.; Ahmed, A.; Yund, B.; Orchard, P.J.; Eisengart, J.; Niklason, G.R.; et al. Neurocognition across the spectrum of mucopolysaccharidosis type I: Age, severity, and treatment. Mol. Genet. Metab. 2015, 116, 61–68. [Google Scholar] [CrossRef]
- Gorlin, R.J.; Cohen, M.M., Jr.; Hennekam, R.C.M. Syndromes of the Head and Neck, 4th ed.; Oxford University Press: New York, NY, USA, 2001. [Google Scholar]
- Wraith, J.E.; Rogers, J.G.; Danks, D.M. The mucopolysaccharidoses. J. Paediatr. Child Health 1987, 23, 329–334. [Google Scholar] [CrossRef]
- Bax, M.C.; Colville, G.A. Behaviour in mucopolysaccharide disorders. Arch. Dis. Child. 1995, 73, 77–81. [Google Scholar] [CrossRef]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology 2011, 50, v4–v12. [Google Scholar] [CrossRef]
- Scarpa, M.; Orchard, P.J.; Schulz, A.; Dickson, P.I.; Haskins, M.E.; Escolar, M.L.; Giugliani, R. Treatment of brain disease in the mucopolysaccharidoses. Mol. Genet. Metab. 2017, 122, 25–34. [Google Scholar] [CrossRef]
- Cleary, M.A.; Wraith, J.E. The presenting features of mucopolysaccharidosis type IH (Hurler syndrome). Acta Paediatr. 1995, 84, 337–339. [Google Scholar] [CrossRef]
- Staba, S.L.; Escolar, M.L.; Poe, M.; Kim, Y.; Martin, P.L.; Szabolcs, P.; Allison-Thacker, J.; Wood, S.; Wenger, D.A.; Rubinstein, P.; et al. Cord-Blood Transplants from Unrelated Donors in Patients with Hurler’s Syndrome. N. Engl. J. Med. 2004, 350, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Shapiro, E.G.; Anderson, J.; Henslee-Downey, P.J.; Klemperer, M.R.; Cowan, M.J.; Saunders, E.F.; Dealarcon, P.A.; Twist, C.; Nachman, J.B.; et al. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. The Storage Disease Collaborative Study Group. Blood 1998, 91, 2601–2608. [Google Scholar] [CrossRef] [PubMed]
- Poe, M.D.; Chagnon, S.L.; Escolar, M.L. Early treatment is associated with improved cognition in Hurler syndrome. Ann. Neurol. 2014, 76, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.; Jones, S.A.; Escolar, M.L. Developmental and behavioral aspects of mucopolysaccharidoses with brain manifestations—Neurological signs and symptoms. Mol. Genet. Metab. 2017, 122, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kiely, B.T.; Kohler, J.L.; Coletti, H.Y.; Poe, M.D.; Escolar, M.L. Early disease progression of Hurler syndrome. Orphanet J. Rare Dis. 2017, 12, 1–10. [Google Scholar] [CrossRef]
- Pontz, B.; Albini, A.; Mensing, H.; Cantz, M.; Müller, P. Pattern of collagen synthesis and chemotactic response of fibroblasts derived from mucopolysaccharidosis patients. Exp. Cell Res. 1984, 155, 457–466. [Google Scholar] [CrossRef]
- Lemonnier, T.; Blanchard, S.; Toli, D.; Roy, E.; Bigou, S.; Froissart, R.; Rouvet, I.; Vitry, S.; Heard, J.M.; Bohl, D. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 3653–3666. [Google Scholar] [CrossRef]
- Bruyère, J.; Roy, E.; Ausseil, J.; Lemonnier, T.; Teyre, G.; Bohl, D.; Etienne-Manneville, S.; Lortat-Jacob, H.; Heard, J.M.; Vitry, S. Heparan Sulfate Saccharides Modify Focal Adhesions: Implication in Mucopolysaccharidosis Neuropathophysiology. J. Mol. Biol. 2015, 427, 775–791. [Google Scholar] [CrossRef]
- McGlynn, R.; Dobrenis, K.; Walkley, S.U. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp. Neurol. 2004, 480, 415–426. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A. Mucopolysaccharidosis I: Management and Treatment Guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in Mouse Models of Mucopolysaccharidosis Type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef] [PubMed]
- Constantopoulos, G.; Dekaban, A.S. Neurochemistry of the Mucopolysaccharidoses: Brain Lipids and Lysosomal Enzymes in Patients with Four Types of Mucopolysaccharidosis and in Normal Controls. J. Neurochem. 1978, 30, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Walkley, S.U. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin. Cell Dev. Biol. 2004, 15, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A. The mucopolysaccharidoses: A success of molecular medicine. Expert Rev. Mol. Med. 2008, 10, e1. [Google Scholar] [CrossRef] [PubMed]
- Langford-Smith, A.W.W.; Langford-Smith, K.J.; Jones, S.A.; Wynn, R.; Wraith, J.E.; Wilkinson, F.L.; Bigger, B.W. Female Mucopolysaccharidosis IIIA Mice Exhibit Hyperactivity and a Reduced Sense of Danger in the Open Field Test. PLoS ONE 2011, 6, e25717. [Google Scholar] [CrossRef] [PubMed]
- Langford-Smith, A.W.W.; Malinowska, M.; Langford-Smith, K.J.; Węgrzyn, G.; Jones, S.; Wynn, R.; Wraith, J.E.; Wilkinson, F.L.; Bigger, B.W. Hyperactive behaviour in the mouse model of mucopolysaccharidosis IIIB in the open field and home cage environments. Genes Brain Behav. 2011, 10, 673–682. [Google Scholar] [CrossRef]
- Canal, M.M.; Wilkinson, F.L.; Cooper, J.D.; Wraith, J.E.; Wynn, R.; Bigger, B.W. Circadian rhythm and suprachiasmatic nucleus alterations in the mouse model of mucopolysaccharidosis IIIB. Behav. Brain Res. 2010, 209, 212–220. [Google Scholar] [CrossRef]
- Lito, S.; Burda, P.; Baumgartner, M.; Sloan-Béna, F.; Táncos, Z.; Kobolák, J.; Dinnyés, A.; Krause, K.-H.; Marteyn, A. Generation of human induced pluripotent stem cell line UNIGEi001-A from a 2-years old patient with Mucopolysaccharidosis type IH disease. Stem Cell Res. 2019, 41, 101604. [Google Scholar] [CrossRef]
- Rouhani, F.; Kumasaka, N.; De Brito, M.C.; Bradley, A.; Vallier, L.; Gaffney, D. Genetic Background Drives Transcriptional Variation in Human Induced Pluripotent Stem Cells. PLoS Genet. 2014, 10, e1004432. [Google Scholar] [CrossRef]
- Salmon, P. Generation of Human Cell Lines Using Lentiviral-Mediated Genetic Engineering. Methods Mol. Biol. 2012, 945, 417–448. [Google Scholar] [CrossRef]
- Minami, R.; Watanabe, Y.; Kudoh, T.; Oyanagi, K.; Nakao, T. Fluorometric measurement of alpha-L-iduronidase activity using 4-methylumbelliferyl-alpha-L-iduronide. Tohoku J. Exp. Med. 1980, 130, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Rahimi-Balaei, M.; Bergen, H.; Kong, J.; Marzban, H. Neuronal Migration During Development of the Cerebellum. Front. Cell. Neurosci. 2018, 12, 484. [Google Scholar] [CrossRef]
- Sterneckert, J.L.; Reinhardt, P.; Schöler, H.R. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014, 15, 625–639. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Barberi, T.; Klivenyi, P.; Calingasan, N.Y.; Lee, H.; Kawamata, H.; Loonam, K.; Perrier, A.L.; Bruses, J.; Rubio, M.E.; Topf, N.; et al. Neural subtype specification of fertilization and nuclear transfer embryonic stem cells and application in parkinsonian mice. Nat. Biotechnol. 2003, 21, 1200–1207. [Google Scholar] [CrossRef]
- Harding, S.E.; Ali, N.N.; Brito-Martins, M.; Gorelik, J. The human embryonic stem cell-derived cardiomyocyte as a pharmacological model. Pharmacol. Ther. 2007, 113, 341–353. [Google Scholar] [CrossRef]
- Lavon, N.; Yanuka, O.; Benvenisty, N. Differentiation and isolation of hepatic-like cells from human embryonic stem cells. Differentiation 2004, 72, 230–238. [Google Scholar] [CrossRef]
- Perrier, A.L.; Tabar, V.; Barberi, T.; Rubio, M.E.; Bruses, J.; Topf, N.; Harrison, N.L.; Studer, L. Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2004, 101, 12543–12548. [Google Scholar] [CrossRef]
- Gauthier, M.; Marteyn, A.; Denis, J.A.; Cailleret, M.; Giraud-Triboult, K.; Aubert, S.; Lecuyer, C.; Marie, J.; Furling, D.; Vernet, R.; et al. A defective Krab-domain zinc-finger transcription factor contributes to altered myogenesis in myotonic dystrophy type 1. Hum. Mol. Genet. 2013, 22, 5188–5198. [Google Scholar] [CrossRef] [PubMed]
- Mateizel, I.; De Temmerman, N.; Ullmann, U.; Cauffman, G.; Sermon, K.; Van De Velde, H.; De Rycke, M.; Degreef, E.; Devroey, P.; Liebaers, I.; et al. Derivation of human embryonic stem cell lines from embryos obtained after IVF and after PGD for monogenic disorders. Hum. Reprod. 2005, 21, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Urbach, A.; Schuldiner, M.; Benvenisty, N. Modeling for Lesch-Nyhan Disease by Gene Targeting in Human Embryonic Stem Cells. Stem Cells 2004, 22, 635–641. [Google Scholar] [CrossRef]
- Verlinsky, Y.; Strelchenko, N.; Kukharenko, V.; Rechitsky, S.; Galat, V.; Kuliev, A. Human embryonic stem cell lines with genetic disorders. Reprod. Biomed. Online 2005, 10, 105–110. [Google Scholar] [CrossRef]
- Evans, T. Embryonic stem cells as a model for cardiac development and disease. Drug Discov. Today Dis. Model. 2008, 5, 147–155. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ebert, A.D.; Yu, J.; Rose, F.F., Jr.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hechedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Chestkov, I.V.; Vasilieva, E.A.; Illarioshkin, S.N.; Lagarkova, M.A.; Kiselev, S.L. Patient-Specific Induced Pluripotent Stem Cells for SOD1-Associated Amyotrophic Lateral Sclerosis Pathogenesis Studies. Acta Nat. 2014, 6, 54–60. [Google Scholar] [CrossRef]
- Mungenast, A.E.; Siegert, S.; Tsai, L.-H. Modeling Alzheimer’s disease with human induced pluripotent stem (iPS) cells. Mol. Cell. Neurosci. 2016, 73, 13–31. [Google Scholar] [CrossRef]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016, 11, 27. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Yamanaka, S. Recent Stem Cell Advances: Induced Pluripotent Stem Cells for Disease Modeling and Stem Cell-Based Regeneration. Circulation 2010, 122, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Unternaehrer, J.J.; Daley, G.Q. Induced pluripotent stem cells for modelling human diseases. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 2274–2285. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K. Genome editing of human pluripotent stem cells to generate human cellular disease models. Dis. Model. Mech. 2013, 6, 896–904. [Google Scholar] [CrossRef]
- Ding, Q.; Lee, Y.-K.; Schaefer, E.A.K.; Peters, D.T.; Veres, A.; Kim, K.; Kuperwasser, N.; Motola, D.L.; Meissner, T.B.; Hendriks, W.T.; et al. A TALEN Genome-Editing System for Generating Human Stem Cell-Based Disease Models. Cell Stem Cell 2013, 12, 238–251. [Google Scholar] [CrossRef]
- Horii, T.; Tamura, D.; Morita, S.; Kimura, M.; Hatada, I. Generation of an ICF Syndrome Model by Efficient Genome Editing of Human Induced Pluripotent Stem Cells Using the CRISPR System. Int. J. Mol. Sci. 2013, 14, 19774–19781. [Google Scholar] [CrossRef]
- Tolar, J.; Park, I.-H.; Xia, L.; Lees, C.J.; Peacock, B.; Webber, B.; McElmurry, R.T.; Eide, C.R.; Orchard, P.J.; Kyba, M.; et al. Hematopoietic differentiation of induced pluripotent stem cells from patients with mucopolysaccharidosis type I (Hurler syndrome). Blood 2011, 117, 839–847. [Google Scholar] [CrossRef]
- Maeda, N.; Ishii, M.; Nishimura, K.; Kamimura, K. Functions of Chondroitin Sulfate and Heparan Sulfate in the Developing Brain. Neurochem. Res. 2010, 36, 1228–1240. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan Sulfate Proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef]
- De Wit, J.; Verhaagen, J. Proteoglycans as Modulators of Axon Guidance Cue Function. Biol. Mammary Gland 2007, 600, 73–89. [Google Scholar] [CrossRef]
- Xian, X.; Gopal, S.; Couchman, J.R. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res. 2010, 339, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Marteyn, A.; Maury, Y.; Gauthier, M.M.; Lecuyer, C.; Vernet, R.; Denis, J.A.; Pietu, G.; Peschanski, M.; Martinat, C. Mutant Human Embryonic Stem Cells Reveal Neurite and Synapse Formation Defects in Type 1 Myotonic Dystrophy. Cell Stem Cell 2011, 8, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Bonneh-Barkay, D.; Wiley, C.A. Brain Extracellular Matrix in Neurodegeneration. Brain Pathol. 2009, 19, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L.; De Bruyn, K.; Enserink, J.; Kuiperij, B.; Rangarajan, S.; Rehmann, H.; Riedl, J.; De Rooij, J.; Van Mansfeld, F.; Zwartkruis, F. The role of Rap1 in integrin-mediated cell adhesion. Biochem. Soc. Trans. 2003, 31, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upregulated Genes | Downregulated Genes | ||||
|---|---|---|---|---|---|
| Gene Name | Log FC | False Discovery Rate (FDR) | Gene Name | Log FC | False Discovery Rate (FDR) |
| HOXA3 | 8.498 | 0.014 | LINC01405 | −5.762 | 0.038 |
| HOXA7 | 7.852 | 0.032 | ZFP42 | −5.879 | 0.061 |
| HOXB9 | 7.789 | 0.030 | ACOXL | −6.305 | 0.039 |
| HOXA5 | 7.530 | 0.032 | JAKMIP2-AS1 | −6.335 | 0.034 |
| EDAR | 7.514 | 0.036 | ZSCAN10 | −6.423 | 0.034 |
| SOST | 7.251 | 0.058 | PRDM14 | −6.602 | 0.034 |
| HOXC9 | 7.192 | 0.030 | LINC00428 | −6.659 | 0.034 |
| HOXC6 | 6.280 | 0.039 | VRTN | −6.694 | 0.039 |
| HOXA6 | 6.054 | 0.046 | DPPA3 | −6.734 | 0.032 |
| GDF7 | 5.721 | 0.079 | FOXH1 | −6.858 | 0.032 |
| HOXB3 | 5.710 | 0.046 | LINC00678 | −7.005 | 0.032 |
| SLC6A5 | 5.664 | 0.042 | NODAL | −7.012 | 0.039 |
| HOXB5 | 5.618 | 0.034 | TDGF1 | −7.073 | 0.036 |
| PRSS56 | 5.535 | 0.039 | HHLA1 | −7.299 | 0.027 |
| HOXB4 | 5.501 | 0.046 | LINC01108 | −7.349 | 0.030 |
| HOXB8 | 5.455 | 0.046 | TRIML2 | −7.479 | 0.030 |
| HOXB7 | 5.296 | 0.036 | SCGB3A2 | −7.745 | 0.030 |
| HOXC4 | 5.226 | 0.039 | LOC101929194 | −7.835 | 0.078 |
| ATOH1 | 5.170 | 0.085 | POU5F1 | −7.861 | 0.032 |
| HOXB1 | 5.048 | 0.042 | ESRG | −8.122 | 0.032 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lito, S.; Sidibe, A.; Ilmjarv, S.; Burda, P.; Baumgartner, M.; Wehrle-Haller, B.; Krause, K.-H.; Marteyn, A. Induced Pluripotent Stem Cells to Understand Mucopolysaccharidosis. I: Demonstration of a Migration Defect in Neural Precursors. Cells 2020, 9, 2593. https://doi.org/10.3390/cells9122593
Lito S, Sidibe A, Ilmjarv S, Burda P, Baumgartner M, Wehrle-Haller B, Krause K-H, Marteyn A. Induced Pluripotent Stem Cells to Understand Mucopolysaccharidosis. I: Demonstration of a Migration Defect in Neural Precursors. Cells. 2020; 9(12):2593. https://doi.org/10.3390/cells9122593
Chicago/Turabian StyleLito, Silvin, Adama Sidibe, Sten Ilmjarv, Patricie Burda, Matthias Baumgartner, Bernhard Wehrle-Haller, Karl-Heinz Krause, and Antoine Marteyn. 2020. "Induced Pluripotent Stem Cells to Understand Mucopolysaccharidosis. I: Demonstration of a Migration Defect in Neural Precursors" Cells 9, no. 12: 2593. https://doi.org/10.3390/cells9122593
APA StyleLito, S., Sidibe, A., Ilmjarv, S., Burda, P., Baumgartner, M., Wehrle-Haller, B., Krause, K.-H., & Marteyn, A. (2020). Induced Pluripotent Stem Cells to Understand Mucopolysaccharidosis. I: Demonstration of a Migration Defect in Neural Precursors. Cells, 9(12), 2593. https://doi.org/10.3390/cells9122593