Proteomic Characterization of Synaptosomes from Human Substantia Nigra Indicates Altered Mitochondrial Translation in Parkinson’s Disease

 ,
,  , and
, and 
Abstract
1. Introduction
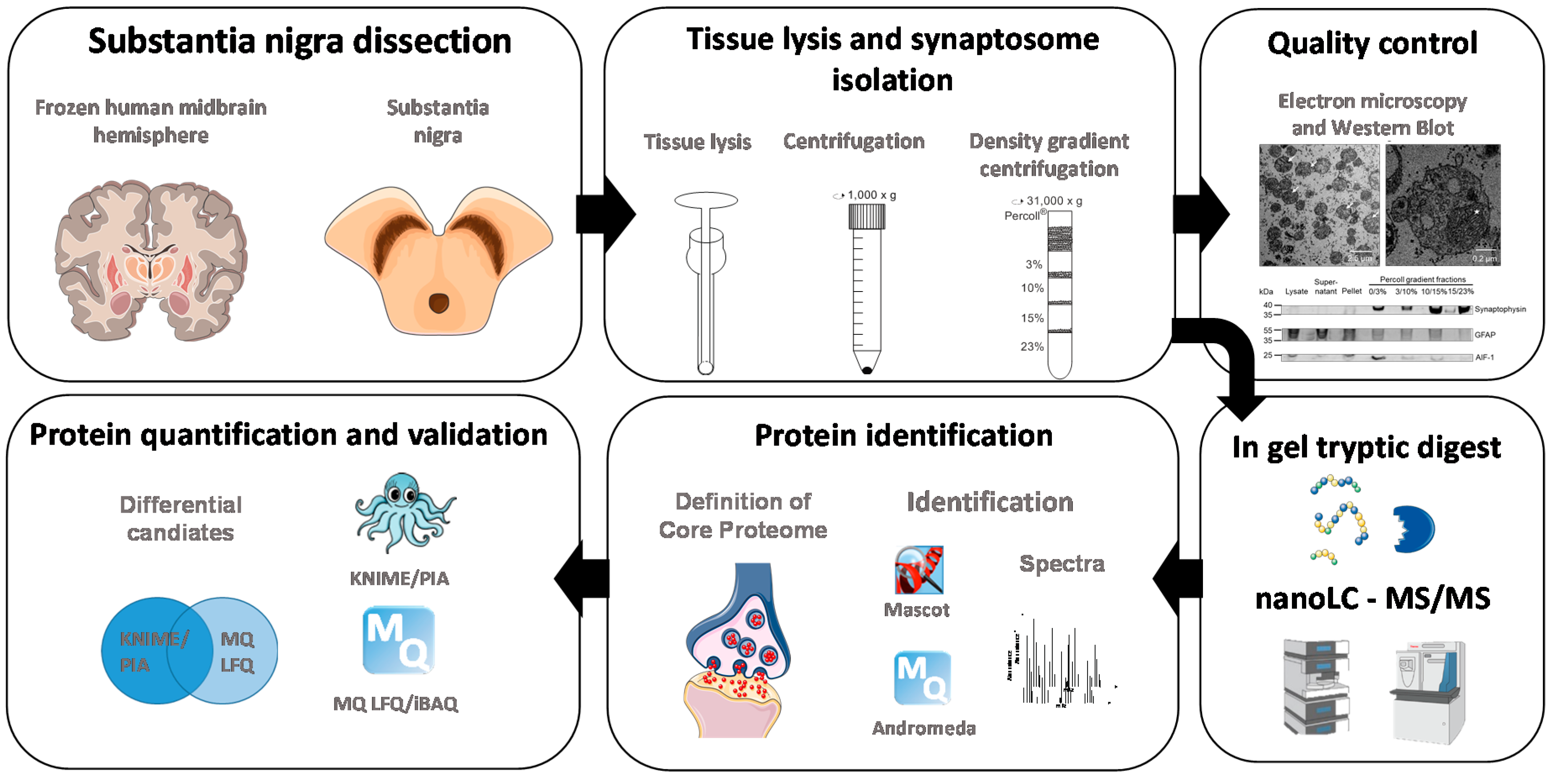
2. Materials and Methods
2.1. Ethical Statement
2.2. Study Cohort for Subsequent Proteomic Analyses
2.3. Tissue and Sample Preparation for LC-MS/MS Experiments
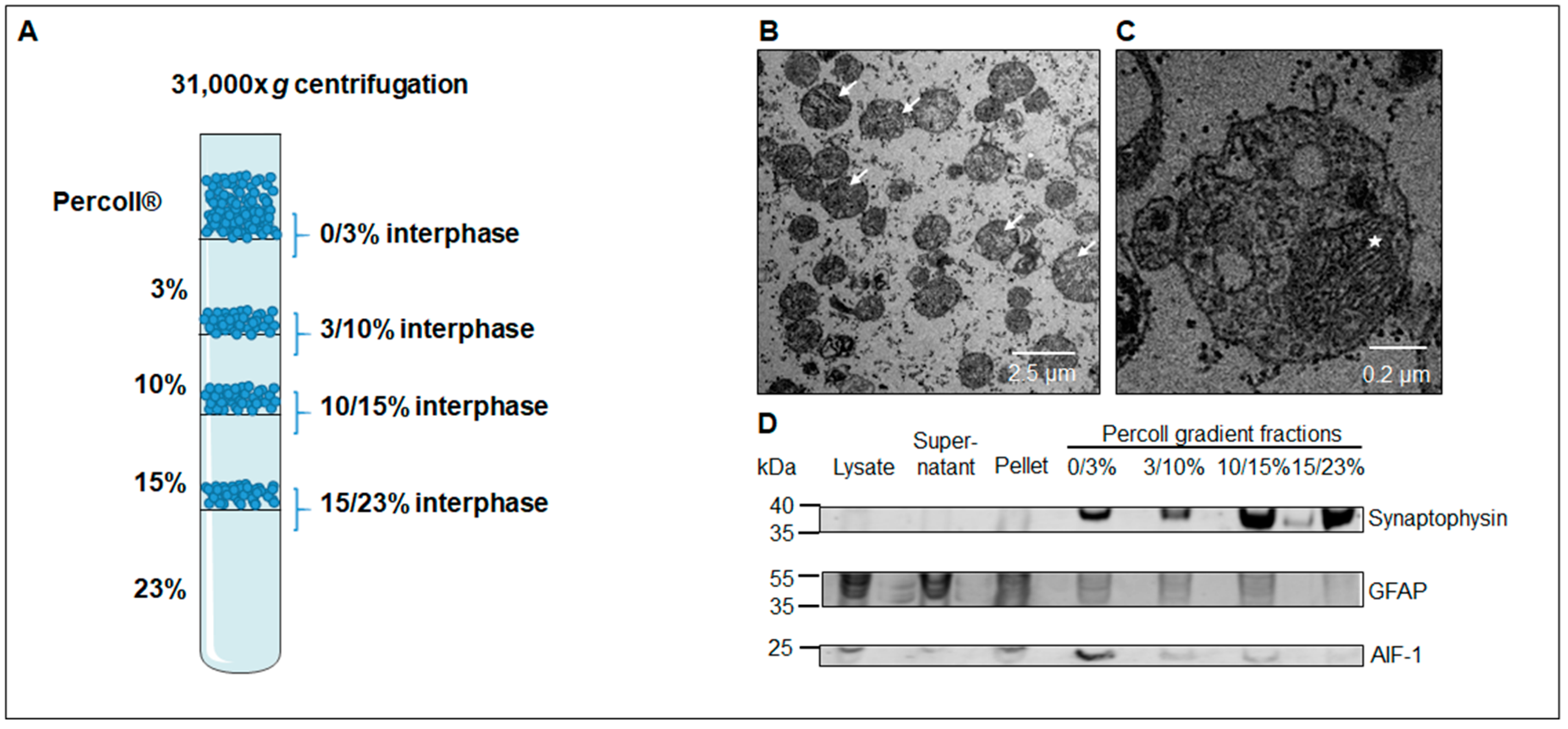
2.4. Electron Microscopy
2.5. Mass Spectrometric Analysis
2.6. Data Analysis
2.7. Analysis of Protein Annotations
2.8. Western Blotting
3. Results
3.1. Western Blot Analysis Identifies a Sub-Fraction Suitable for In-Depth Proteomic Analysis
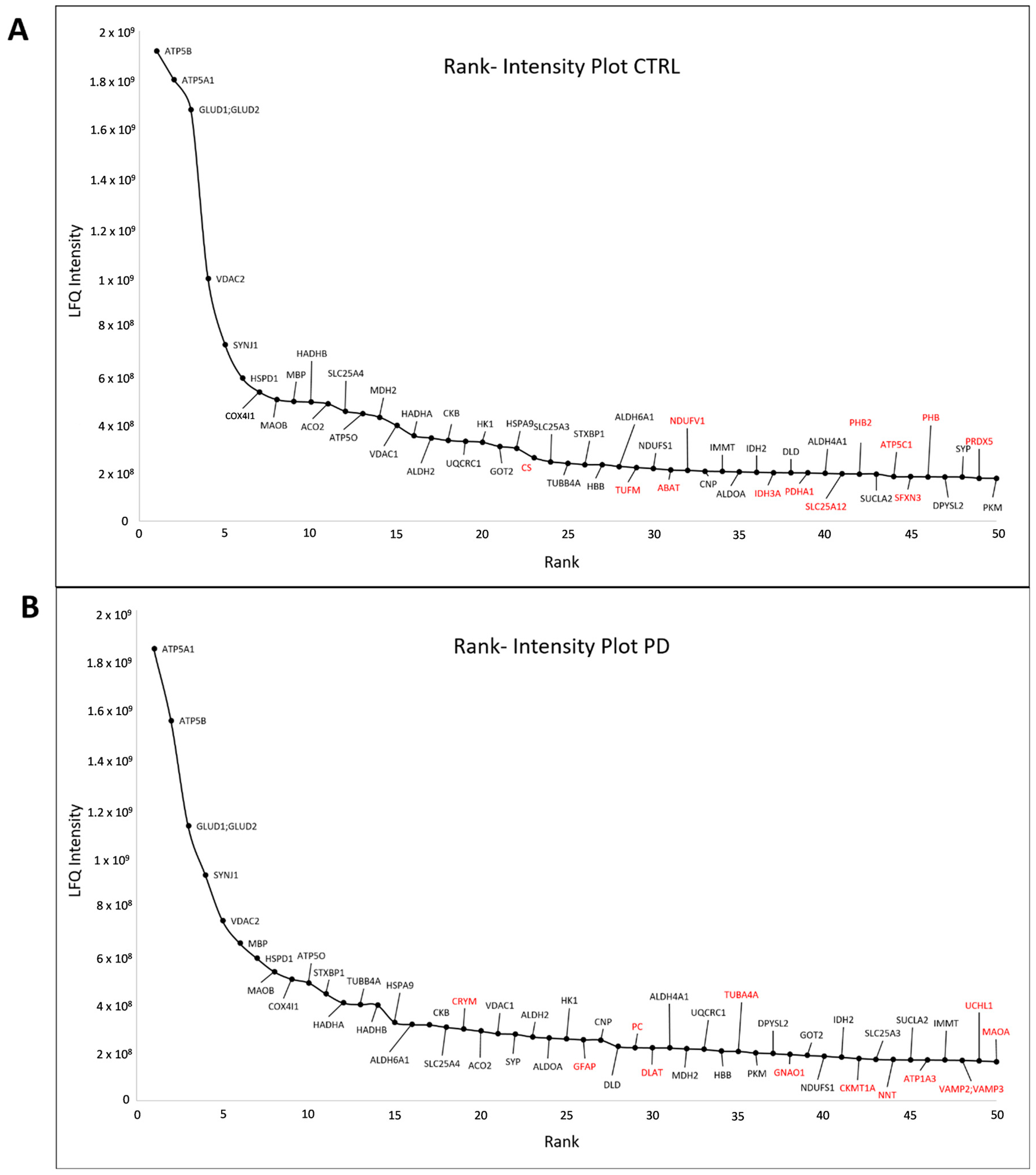
3.2. Core Proteome of Synapses Isolated from the Substantia Nigra
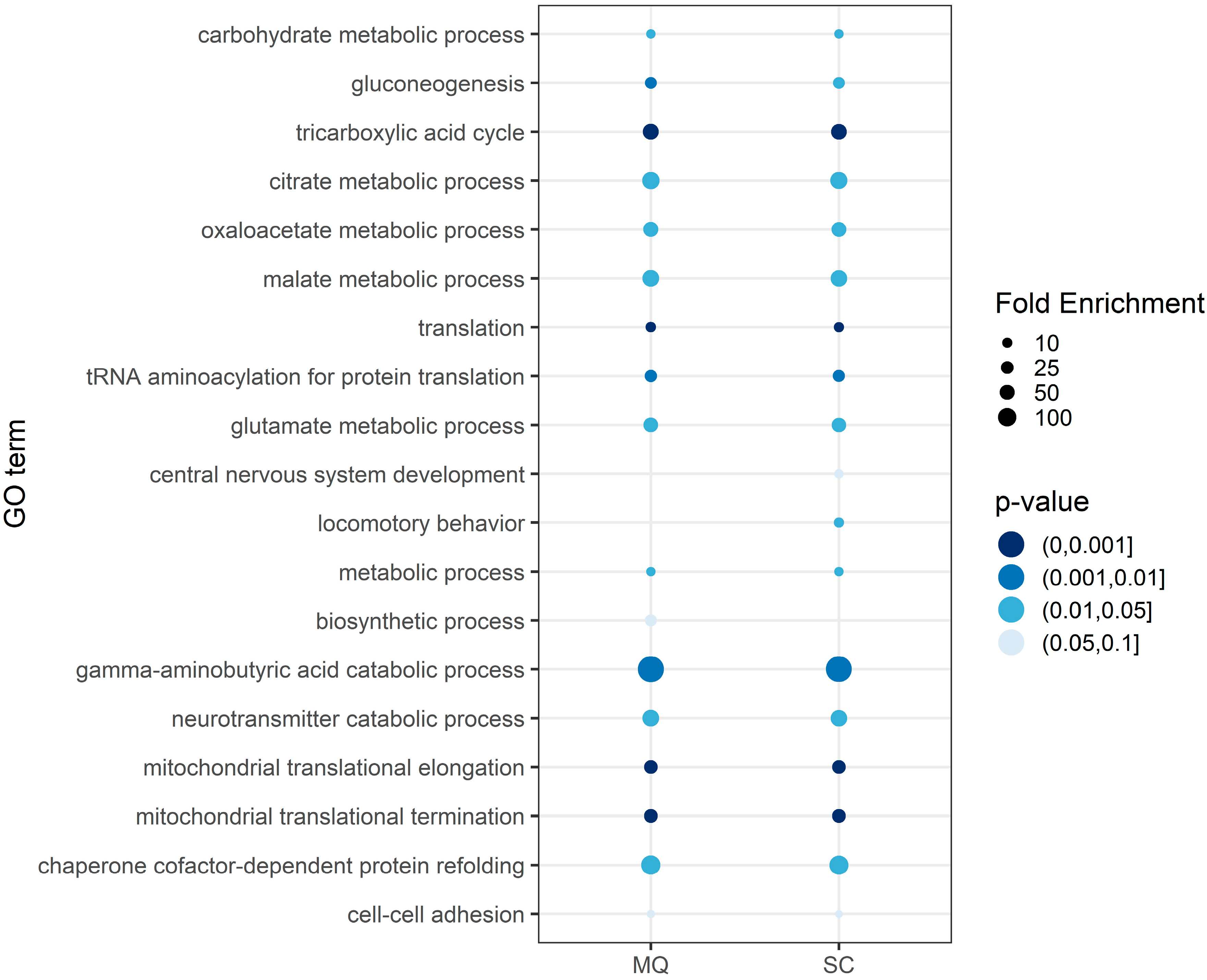
3.3. Functional Annotation of the Core Proteome
3.4. Proteins Altered between Parkinson’s Disease Patients and Control Subjects
3.5. Confirmation of Altered Mitochondrial Translation in PD Synaptosomes
4. Discussion
4.1. Isolation of Synaptosomes from and Definition of Synaptosome Core Proteome in the Substantia Nigra
4.2. Identification of PD-Related Changes
4.3. Changed Mitochondrial Translation and mtDNA Synthesis in PD
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Sian, J.; Gerlach, M.; Youdim, M.B.; Riederer, P. Parkinson’s disease: A major hypokinetic basal ganglia disorder. J. Neural Transm. 1999, 106, 443–476. [Google Scholar] [CrossRef]
- Riederer, P.; Wuketich, S. Time course of nigrostriatal degeneration in parkinson’s disease. A detailed study of influential factors in human brain amine analysis. J. Neural Transm. 1976, 38, 277–301. [Google Scholar] [CrossRef]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef]
- Jellinger, K.A. Pathology of Parkinson’s disease. Changes other than the nigrostriatal pathway. Mol. Chem. Neuropathol. 1991, 14, 153–197. [Google Scholar] [CrossRef]
- Gerlach, M.; Gsell, W.; Kornhuber, J.; Jellinger, K.; Krieger, V.; Pantucek, F.; Vock, R.; Riederer, P. A post mortem study on neurochemical markers of dopaminergic, GABA-ergic and glutamatergic neurons in basal ganglia-thalamocortical circuits in Parkinson syndrome. Brain Res. 1996, 741, 142–152. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Whittaker, V.P.; Michaelson, I.A.; Kirkland, R.J. The separation of synaptic vesicles from nerve-ending particles (‘synaptosomes’). Biochem. J. 1964, 90, 293–303. [Google Scholar] [CrossRef]
- Whittaker, V.P. The application of subcellular fractionation techniques to the study of brain function. Prog. Biophys. Mol. Biol. 1965, 15, 39–96. [Google Scholar] [CrossRef]
- Jhou, J.-F.; Tai, H.-C. The Study of Postmortem Human Synaptosomes for Understanding Alzheimer’s Disease and Other Neurological Disorders: A Review. Neurol. Ther. 2017, 6, 57–68. [Google Scholar] [CrossRef]
- Whittaker, V.P. Thirty years of synaptosome research. J. Neurocytol. 1993, 22, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Cowburn, R.; Barton, A.; Reynolds, G.; Lofdahl, E.; O’Carroll, A.-M.; Wester, P.; Winblad, B. Region-specific loss of glutamate innervation in Alzheimer’s disease. Neurosci. Lett. 1987, 73, 77–80. [Google Scholar] [CrossRef]
- Hardy, J.; Adolfsson, R.; Alafuzoff, I.; Bucht, G.; Marcusson, J.; Nyberg, P.; Perdahl, E.; Wester, P.; Winblad, B. Transmitter deficits in Alzheimer’s disease. Neurochem. Int. 1985, 7, 545–563. [Google Scholar] [CrossRef]
- Nordberg, A.; Winblad, B. Reduced number of [3H]nicotine and [3H]acetylcholine binding sites in the frontal cortex of Alzheimer brains. Neurosci. Lett. 1986, 72, 115–120. [Google Scholar] [CrossRef]
- Wang, D.-S.; Lipton, R.B.; Katz, M.J.; Davies, P.; Buschke, H.; Kuslansky, G.; Verghese, J.; Younkin, S.G.; Eckman, C.; Dickson, D.W. Decreased neprilysin immunoreactivity in Alzheimer disease, but not in pathological aging. J. Neuropathol. Exp. Neurol. 2005, 64, 378–385. [Google Scholar] [CrossRef]
- Wood, J.D.; MacMillan, J.C.; Harper, P.S.; Lowenstein, P.R.; Jones, A.L. Partial characterisation of murine huntingtin and apparent variations in the subcellular localisation of huntingtin in human, mouse and rat brain. Hum. Mol. Genet. 1996, 5, 481–487. [Google Scholar] [CrossRef][Green Version]
- Postupna, N.O.; Latimer, C.S.; Dirk Keene, C.; Montine, K.S.; Montine, T.J.; Darvas, M. Synaptosomes, 1st ed.; Springer: New York, NY, USA, 2018. [Google Scholar]
- Pifl, C.; Rajput, A.; Reither, H.; Blesa, J.; Cavada, C.; Obeso, J.A.; Rajput, A.H.; Hornykiewicz, O. Is Parkinson’s disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J. Neurosci. 2014, 34, 8210–8218. [Google Scholar] [CrossRef]
- Haberland, N.; Hetey, L. Studies in postmortem dopamine uptake. II. Alterations of the synaptosomal catecholamine uptake in postmortem brain regions in schizophrenia. J. Neural Transm. 1987, 68, 303–313. [Google Scholar] [CrossRef]
- Scarr, E.; Gray, L.; Keriakous, D.; Robinson, P.J.; Dean, B. Increased levels of SNAP-25 and synaptophysin in the dorsolateral prefrontal cortex in bipolar I disorder. Bipolar Disord. 2006, 8, 133–143. [Google Scholar] [CrossRef]
- Goldstein, I.; Levy, T.; Galili, D.; Ovadia, H.; Yirmiya, R.; Rosen, H.; Lichtstein, D. Involvement of Na(+), K(+)-ATPase and endogenous digitalis-like compounds in depressive disorders. Biol. Psychiatry 2006, 60, 491–499. [Google Scholar] [CrossRef]
- Grant, S.G.N. The synapse proteome and phosphoproteome: A new paradigm for synapse biology. Biochem. Soc. Trans. 2006, 34, 59–63. [Google Scholar] [CrossRef]
- Bai, F.; Witzmann, F.A. Synaptosome proteomics. Subcell. Biochem. 2007, 43, 77–98. [Google Scholar] [CrossRef]
- Roy, M.; Sorokina, O.; Skene, N.; Simonnet, C.; Mazzo, F.; Zwart, R.; Sher, E.; Smith, C.; Armstrong, J.D.; Grant, S.G.N. Proteomic analysis of postsynaptic proteins in regions of the human neocortex. Nat. Neurosci. 2018, 21, 130–138. [Google Scholar] [CrossRef]
- DeGiorgis, J.A.; Jaffe, H.; Moreira, J.E.; Carlotti, C.G.; Leite, J.P.; Pant, H.C.; Dosemeci, A. Phosphoproteomic analysis of synaptosomes from human cerebral cortex. J. Proteome Res. 2005, 4, 306–315. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Etheridge, N.; Nouwens, A.S.; Dodd, P.R. SWATH analysis of the synaptic proteome in Alzheimer’s disease. Neurochem. Int. 2015, 87, 1–12. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Nouwens, A.S.; Dodd, P.R.; Etheridge, N. The synaptic proteome in Alzheimer’s disease. Alzheimers. Dement. 2013, 9, 499–511. [Google Scholar] [CrossRef]
- Velásquez, E.; Martins-de-Souza, D.; Velásquez, I.; Carneiro, G.R.A.; Schmitt, A.; Falkai, P.; Domont, G.B.; Nogueira, F.C.S. Quantitative Subcellular Proteomics of the Orbitofrontal Cortex of Schizophrenia Patients. J. Proteome Res. 2019, 18, 4240–4253. [Google Scholar] [CrossRef]
- Velásquez, E.; Nogueira, F.C.S.; Velásquez, I.; Schmitt, A.; Falkai, P.; Domont, G.B.; Martins-de-Souza, D. Synaptosomal Proteome of the Orbitofrontal Cortex from Schizophrenia Patients Using Quantitative Label-Free and iTRAQ-Based Shotgun Proteomics. J. Proteome Res. 2017, 16, 4481–4494. [Google Scholar] [CrossRef]
- Gajera, C.R.; Fernandez, R.; Postupna, N.; Montine, K.S.; Fox, E.J.; Tebaykin, D.; Angelo, M.; Bendall, S.C.; Keene, C.D.; Montine, T.J. Mass synaptometry: High-dimensional multi parametric assay for single synapses. J. Neurosci. Methods 2019, 312, 73–83. [Google Scholar] [CrossRef]
- Evans, G.J.O. Subcellular fractionation of the brain: Preparation of synaptosomes and synaptic vesicles. Cold Spring Harb. Protoc. 2015, 2015, 462–466. [Google Scholar] [CrossRef]
- Plum, S.; Steinbach, S.; Abel, L.; Marcus, K.; Helling, S.; May, C. Proteomics in neurodegenerative diseases: Methods for obtaining a closer look at the neuronal proteome. Proteom. Clin. Appl. 2015, 9, 848–871. [Google Scholar] [CrossRef]
- Dunkley, P.R.; Jarvie, P.E.; Robinson, P.J. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat. Protoc. 2008, 3, 1718–1728. [Google Scholar] [CrossRef]
- Plum, S.; Helling, S.; Theiss, C.; Leite, R.E.P.; May, C.; Jacob-Filho, W.; Eisenacher, M.; Kuhlmann, K.; Meyer, H.E.; Riederer, P.; et al. Combined enrichment of neuromelanin granules and synaptosomes from human substantia nigra pars compacta tissue for proteomic analysis. J. Proteom. 2013, 94, 202–206. [Google Scholar] [CrossRef]
- Braak, H.; Tredici, K.D.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Grinberg, L.T.; de Lucena Ferretti, R.E.; Farfel, J.M.; Leite, R.; Pasqualucci, C.A.; Rosemberg, S.; Nitrini, R.; Saldiva, P.H.N.; Filho, W.J. Brain bank of the Brazilian aging brain study group-A milestone reached and more than 1600 collected brains. Cell Tissue Bank. 2007, 8, 151–162. [Google Scholar] [CrossRef]
- Suemoto, C.K.; Ferretti-Rebustini, R.E.L.; Rodriguez, R.D.; Leite, R.E.P.; Soterio, L.; Brucki, S.M.D.; Spera, R.R.; Cippiciani, T.M.; Farfel, J.M.; Chiavegatto Filho, A.; et al. Neuropathological diagnoses and clinical correlates in older adults in Brazil: A cross-sectional study. PLoS Med. 2017, 14, e1002267. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 2010, 119, 1–4. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol. Appl. Neurobiol. 1989, 15, 13–26. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Uszkoreit, J.; Perez-Riverol, Y.; Eggers, B.; Marcus, K.; Eisenacher, M. Protein Inference Using PIA Workflows and PSI Standard File Formats. J. Proteome Res. 2019, 18, 741–747. [Google Scholar] [CrossRef]
- Uszkoreit, J.; Maerkens, A.; Perez-Riverol, Y.; Meyer, H.E.; Marcus, K.; Stephan, C.; Kohlbacher, O.; Eisenacher, M. PIA: An Intuitive Protein Inference Engine with a Web-Based User Interface. J. Proteome Res. 2015, 14, 2988–2997. [Google Scholar] [CrossRef]
- Plum, S.; Steinbach, S.; Attems, J.; Keers, S.; Riederer, P.; Gerlach, M.; May, C.; Marcus, K. Proteomic characterization of neuromelanin granules isolated from human substantia nigra by laser-microdissection. Sci. Rep. 2016, 6, 37139. [Google Scholar] [CrossRef]
- Schuld, J.; Orfanos, Z.; Chevessier, F.; Eggers, B.; Heil, L.; Uszkoreit, J.; Unger, A.; Kirfel, G.; van der Ven, P.F.M.; Marcus, K.; et al. Homozygous expression of the myofibrillar myopathy-associated p.W2710X filamin C variant reveals major pathomechanisms of sarcomeric lesion formation. Acta Neuropathol. Commun. 2020, 8, 154. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual account of protein investment in cellular functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef]
- Otto, A.; Bernhardt, J.; Meyer, H.; Schaffer, M.; Herbst, F.-A.; Siebourg, J.; Mäder, U.; Lalk, M.; Hecker, M.; Becher, D. Systems-wide temporal proteomic profiling in glucose-starved Bacillus subtilis. Nat. Commun. 2010, 1, 137. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- DiGiovanni, J.; Sun, T.; Sheng, Z.-H. Characterizing synaptic vesicle proteins using synaptosomal fractions and cultured hippocampal neurons. Curr. Protoc. Neurosci. 2012, 59, 2–7. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef]
- Kersey, J.H.; LeBien, T.W.; Abramson, C.S.; Newman, R.; Sutherland, R.; Greaves, M. P-24: A human leukemia-associated and lymphohemopoietic progenitor cell surface structure identified with monoclonal antibody. J. Exp. Med. 1981, 153, 726–731. [Google Scholar] [CrossRef]
- Nakamura, Y.; Iwamoto, R.; Mekada, E. Expression and distribution of CD9 in myelin of the central and peripheral nervous systems. Am. J. Pathol. 1996, 149, 575–583. [Google Scholar]
- Schenk, G.J.; Dijkstra, S.; van het Hof, A.J.; van der Pol, S.M.A.; Drexhage, J.A.R.; van der Valk, P.; Reijerkerk, A.; van Horssen, J.; de Vries, H.E. Roles for HB-EGF and CD9 in multiple sclerosis. Glia 2013, 61, 1890–1905. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Matsumine, H.; Saito, M.; Shimoda-Matsubayashi, S.; Tanaka, H.; Ishikawa, A.; Nakagawa-Hattori, Y.; Yokochi, M.; Kobayashi, T.; Igarashi, S.; Takano, H.; et al. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am. J. Hum. Genet. 1997, 60, 588–596. [Google Scholar]
- Leroy, E.; Anastasopoulos, D.; Konitsiotis, S.; Lavedan, C.; Polymeropoulos, M.H. Deletions in the Parkin gene and genetic heterogeneity in a Greek family with early onset Parkinson’s disease. Hum. Genet. 1998, 103, 424–427. [Google Scholar] [CrossRef]
- Lücking, C.B.; Abbas, N.; Dürr, A.; Bonifati, V.; Bonnet, A.-M.; de Broucker, T.; de Michele, G.; Wood, N.W.; Agid, Y.; Brice, A. Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. Lancet 1998, 352, 1355–1356. [Google Scholar] [CrossRef]
- Valente, E.M.; Bentivoglio, A.R.; Dixon, P.H.; Ferraris, A.; Ialongo, T.; Frontali, M.; Albanese, A.; Wood, N.W. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am. J. Hum. Genet. 2001, 68, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Theocharopoulou, G. The ubiquitous role of mitochondria in Parkinson and other neurodegenerative diseases. Aims Neurosci. 2020, 7, 43–65. [Google Scholar] [CrossRef] [PubMed]
- Basso, M.; Giraudo, S.; Lopiano, L.; Bergamasco, B.; Bosticco, E.; Cinquepalmi, A.; Fasano, M. Proteome analysis of mesencephalic tissues: Evidence for Parkinson’s disease. Neurol. Sci. 2003, 24, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Pienaar, I.S.; Daniels, W.M.U.; Götz, J. Neuroproteomics as a promising tool in Parkinson’s disease research. J. Neural Transm. 2008, 115, 1413–1430. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Licker, V.; Turck, N.; Kövari, E.; Burkhardt, K.; Côte, M.; Surini-Demiri, M.; Lobrinus, J.A.; Sanchez, J.-C.; Burkhard, P.R. Proteomic analysis of human substantia nigra identifies novel candidates involved in Parkinson’s disease pathogenesis. Proteomics 2014, 14, 784–794. [Google Scholar] [CrossRef]
- Licker, V.; Côte, M.; Lobrinus, J.A.; Rodrigo, N.; Kövari, E.; Hochstrasser, D.F.; Turck, N.; Sanchez, J.-C.; Burkhard, P.R. Proteomic profiling of the substantia nigra demonstrates CNDP2 overexpression in Parkinson’s disease. J. Proteom. 2012, 75, 4656–4667. [Google Scholar] [CrossRef]
- Werner, C.J.; Heyny-von Haussen, R.; Mall, G.; Wolf, S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci. 2008, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Basso, M.; Giraudo, S.; Corpillo, D.; Bergamasco, B.; Lopiano, L.; Fasano, M. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteomics 2004, 4, 3943–3952. [Google Scholar] [CrossRef] [PubMed]
- Monti, C.; Lane, L.; Fasano, M.; Alberio, T. Update of the Functional Mitochondrial Human Proteome Network. J. Proteome Res. 2018, 17, 4297–4306. [Google Scholar] [CrossRef] [PubMed]
- Alberio, T.; Bondi, H.; Colombo, F.; Alloggio, I.; Pieroni, L.; Urbani, A.; Fasano, M. Mitochondrial proteomics investigation of a cellular model of impaired dopamine homeostasis, an early step in Parkinson’s disease pathogenesis. Mol. Biosyst. 2014, 10, 1332–1344. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. Lancet 1989, 333, 1269. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Sherer, T.B.; Betarbet, R.; Panov, A.V. Complex I and Parkinson’s disease. IUBMB Life 2001, 52, 135–141. [Google Scholar] [CrossRef]
- Grünblatt, E.; Ruder, J.; Monoranu, C.M.; Riederer, P.; Youdim, M.B.; Mandel, S.A. Differential Alterations in Metabolism and Proteolysis-Related Proteins in Human Parkinson’s Disease Substantia Nigra. Neurotox. Res. 2018, 33, 560–568. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, I.; Lin, M.T.; Zheng, K.; Keller-McGandy, C.E.; Betensky, R.A.; Johns, D.R.; Beal, M.F.; Standaert, D.G.; Simon, D.K. Somatic mitochondrial DNA mutations in single neurons and glia. Neurobiol. Aging 2005, 26, 1343–1355. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; La Morgia, C.; Rizzo, G.; Carelli, V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Mov. Disord. 2017, 32, 346–363. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 2017, 32, 1264–1310. [Google Scholar] [CrossRef]
- Lesko, N.; Naess, K.; Wibom, R.; Solaroli, N.; Nennesmo, I.; von Döbeln, U.; Karlsson, A.; Larsson, N.-G. Two novel mutations in thymidine kinase-2 cause early onset fatal encephalomyopathy and severe mtDNA depletion. Neuromuscul. Disord. 2010, 20, 198–203. [Google Scholar] [CrossRef]
- Knierim, E.; Seelow, D.; Gill, E.; von Moers, A.; Schuelke, M. Clinical application of whole exome sequencing reveals a novel compound heterozygous TK2-mutation in two brothers with rapidly progressive combined muscle-brain atrophy, axonal neuropathy, and status epilepticus. Mitochondrion 2015, 20, 1–6. [Google Scholar] [CrossRef]
- Bartesaghi, S.; Betts-Henderson, J.; Cain, K.; Dinsdale, D.; Zhou, X.; Karlsson, A.; Salomoni, P.; Nicotera, P. Loss of thymidine kinase 2 alters neuronal bioenergetics and leads to neurodegeneration. Hum. Mol. Genet. 2010, 19, 1669–1677. [Google Scholar] [CrossRef]
- Brown, A.; Amunts, A.; Bai, X.-C.; Sugimoto, Y.; Edwards, P.C.; Murshudov, G.; Scheres, S.H.W.; Ramakrishnan, V. Structure of the large ribosomal subunit from human mitochondria. Science 2014, 346, 718–722. [Google Scholar] [CrossRef]
- Amunts, A.; Brown, A.; Toots, J.; Scheres, S.H.W.; Ramakrishnan, V. The structure of the human mitochondrial ribosome. Science 2015, 348, 95–98. [Google Scholar] [CrossRef]
- Sundquist, K.; Ahmad, A.; Svensson, P.J.; Zöller, B.; Sundquist, J.; Memon, A.A. Polymorphisms in PARK2 and MRPL37 are associated with higher risk of recurrent venous thromboembolism in a sex-specific manner. J. Thromb. Thrombolysis 2018, 46, 154–165. [Google Scholar] [CrossRef]
- Bayat, V.; Thiffault, I.; Jaiswal, M.; Tétreault, M.; Donti, T.; Sasarman, F.; Bernard, G.; Demers-Lamarche, J.; Dicaire, M.-J.; Mathieu, J.; et al. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012, 10, e1001288. [Google Scholar] [CrossRef]
- Teixeira, P.F.; Masuyer, G.; Pinho, C.M.; Branca, R.M.M.; Kmiec, B.; Wallin, C.; Wärmländer, S.K.T.S.; Berntsson, R.P.-A.; Ankarcrona, M.; Gräslund, A.; et al. Mechanism of Peptide Binding and Cleavage by the Human Mitochondrial Peptidase Neurolysin. J. Mol. Biol. 2018, 430, 348–362. [Google Scholar] [CrossRef]
- Castro, L.M.; Cavalcanti, D.M.L.P.; Araujo, C.B.; Rioli, V.; Icimoto, M.Y.; Gozzo, F.C.; Juliano, M.; Juliano, L.; Oliveira, V.; Ferro, E.S. Peptidomic analysis of the neurolysin-knockout mouse brain. J. Proteom. 2014, 111, 238–248. [Google Scholar] [CrossRef]
- Checler, F.; Vincent, J.P.; Kitabgi, P. Purification and characterization of a novel neurotensin-degrading peptidase from rat brain synaptic membranes. J. Biol. Chem. 1986, 261, 11274–11281. [Google Scholar]
- Fontenele-Neto, J.D.; Massarelli, E.E.; Gurgel Garrido, P.A.; Beaudet, A.; Ferro, E.S. Comparative fine structural distribution of endopeptidase 24.15 (EC3.4.24.15) and 24.16 (EC3.4.24.16) in rat brain. J. Comp. Neurol. 2001, 438, 399–410. [Google Scholar] [CrossRef]
- Boczonadi, V.; Ricci, G.; Horvath, R. Mitochondrial DNA transcription and translation: Clinical syndromes. Essays Biochem. 2018, 62, 321–340. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Gender | Age (in years) | Ø Age ± SD | DS | PMI (h) | Ø PMI (h) ± SD | Tissue Weight (g) | PD Braak/ AD Braak | CDR | CERAD |
|---|---|---|---|---|---|---|---|---|---|---|
| Control | Female | 88 | 78.8 ± 6.9 | NA | ~13:00 | 14:04 ± 0.13 | 0.2306 | NA | NA | NA |
| Male | 80 | NA | 19:39 | 0.4616 | NA | NA | NA | |||
| Female | 73 | NA | 13:30 | 0.4076 | NA | NA | NA | |||
| Male | 82 | NA | 12:11 | 0.2530 | NA | NA | NA | |||
| Female | 71 | NA | 12:00 | 0.3925 | NA | NA | NA | |||
| PD | Female | 87 | 80.8 ± 6.8 | 3 | 13:08 | 14:19 ± 0.12 | 0.2461 | 3/3 | 0 | none |
| Male | 88 | 5 | 11:20 | 0.3607 | 5/3 | 0.5 | moderate | |||
| Female | 75 | 3 | 14:40 | 0.6556 | 3/2 | 0 | moderate | |||
| Male | 81 | 6 | 19:10 | 1.1524 | 6/2 | 3 | moderate | |||
| Female | 73 | 3 | 13:20 | 0.4838 | 3/2 | 0 | none |
| Protein Family | CTRL 1134511 | CTRL 12449/11 | CTRL 3887/11 | CTRL 6614/11 | CTRL 6878/11 | PD 10158/11 | PD 10488/11 | PD 11321/09 | PD 11749/09 | PD 4406/09 |
|---|---|---|---|---|---|---|---|---|---|---|
| ATP Synthase | 16.59 | 16.53 | 13.24 | 15.49 | 13.76 | 12.30 | 12.03 | 14.17 | 14.82 | 16.51 |
| TIM/TOM complex | 0.23 | 0.31 | 0.30 | 0.29 | 0.26 | 0.29 | 0.11 | 0.27 | 0.27 | 0.38 |
| Entry | Protein Names | Gene Names | Subcellular Location [CC] |
|---|---|---|---|
| P21926 | CD9 antigen (5H9 antigen) * | CD9 MIC3 TSPAN29 GIG2 | Cell membrane |
| O00142 | Thymidine kinase 2, mitochondrial | TK2 | Mitochondrion. |
| Q9BZE1 | 39S ribosomal protein L37, mitochondrial | MRPL37 MRPL2 RPML2 HSPC235 | Mitochondrion |
| Q9BYT8 | Neurolysin, mitochondrial | NLN AGTBP KIAA1226 | Mitochondrion intermembrane space |
| P33316 | Deoxyuridine 5′-triphosphate nucleotidohydrolase, mitochondrial | DUT | Nucleus |
| Q99798 | Aconitate hydratase, mitochondrial | ACO2 | Mitochondrion |
| P80404 | 4-aminobutyrate aminotransferase, mitochondrial | ABAT GABAT | Mitochondrion matrix. |
| Q9H6V9 | Lipid droplet-associated hydrolase | LDAH C2orf43 | Lipid droplet |
| P40926 | Malate dehydrogenase, mitochondrial | MDH2 | Mitochondrion matrix |
| P42126 | Enoyl-CoA delta isomerase 1, mitochondrial | ECI1 DCI | Mitochondrion matrix |
| Q92665 | 28S ribosomal protein S31, mitochondrial | MRPS31 IMOGN38 | Mitochondrion |
| O00330 | Pyruvate dehydrogenase protein X component, mitochondrial | PDHX PDX1 | Mitochondrion matrix. |
| P31930 | Cytochrome b-c1 complex subunit 1, mitochondrial | UQCRC1 | Mitochondrion inner membrane |
| Q96GW9 | Methionine--tRNA ligase, mitochondrial | MARS2 | Mitochondrion matrix |
| O14744 | Protein arginine N-methyltransferase 5 | PRMT5 HRMT1L5 IBP72 JBP1 SKB1 | Cytoplasm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plum, S.; Eggers, B.; Helling, S.; Stepath, M.; Theiss, C.; Leite, R.E.P.; Molina, M.; Grinberg, L.T.; Riederer, P.; Gerlach, M.; et al. Proteomic Characterization of Synaptosomes from Human Substantia Nigra Indicates Altered Mitochondrial Translation in Parkinson’s Disease. Cells 2020, 9, 2580. https://doi.org/10.3390/cells9122580
Plum S, Eggers B, Helling S, Stepath M, Theiss C, Leite REP, Molina M, Grinberg LT, Riederer P, Gerlach M, et al. Proteomic Characterization of Synaptosomes from Human Substantia Nigra Indicates Altered Mitochondrial Translation in Parkinson’s Disease. Cells. 2020; 9(12):2580. https://doi.org/10.3390/cells9122580
Chicago/Turabian StylePlum, Sarah, Britta Eggers, Stefan Helling, Markus Stepath, Carsten Theiss, Renata E. P. Leite, Mariana Molina, Lea T. Grinberg, Peter Riederer, Manfred Gerlach, and et al. 2020. "Proteomic Characterization of Synaptosomes from Human Substantia Nigra Indicates Altered Mitochondrial Translation in Parkinson’s Disease" Cells 9, no. 12: 2580. https://doi.org/10.3390/cells9122580
APA StylePlum, S., Eggers, B., Helling, S., Stepath, M., Theiss, C., Leite, R. E. P., Molina, M., Grinberg, L. T., Riederer, P., Gerlach, M., May, C., & Marcus, K. (2020). Proteomic Characterization of Synaptosomes from Human Substantia Nigra Indicates Altered Mitochondrial Translation in Parkinson’s Disease. Cells, 9(12), 2580. https://doi.org/10.3390/cells9122580