Optimization of Enzyme Essays to Enhance Reliability of Activity Measurements in Leukocyte Lysates for the Diagnosis of Metachromatic Leukodystrophy and Gangliosidoses

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Separation of Leukocyte Subpopulations and Degranulation Experiments
2.1.1. Isolation of Granulocytes and Mononuclear Blood Cells
2.1.2. MiniMACS Separation of Lymphocytes and Monocytes from Mononuclear Cells
2.1.3. Degranulation Experiments Using Isolated Granulocytes
2.1.4. Measurement of Myeloperoxidase in Supernatants of Granulocytes after Degranulation
2.2. Leukocyte Preparations and Blood Cell Analysis
2.3. Enzyme Assays of Sphingolipidoses
2.3.1. Arylsulfatase A
2.3.2. β-Galactosidase
2.3.3. β-Hexosaminidase A
2.3.4. β-Hexosaminidase A+B
2.4. Statistical Analyses
3. Results
3.1. Leukocytes as Enzyme Source—Potential Pitfalls
3.1.1. Different Leukocytes Contain Different Lysosomal Enzymes Activities
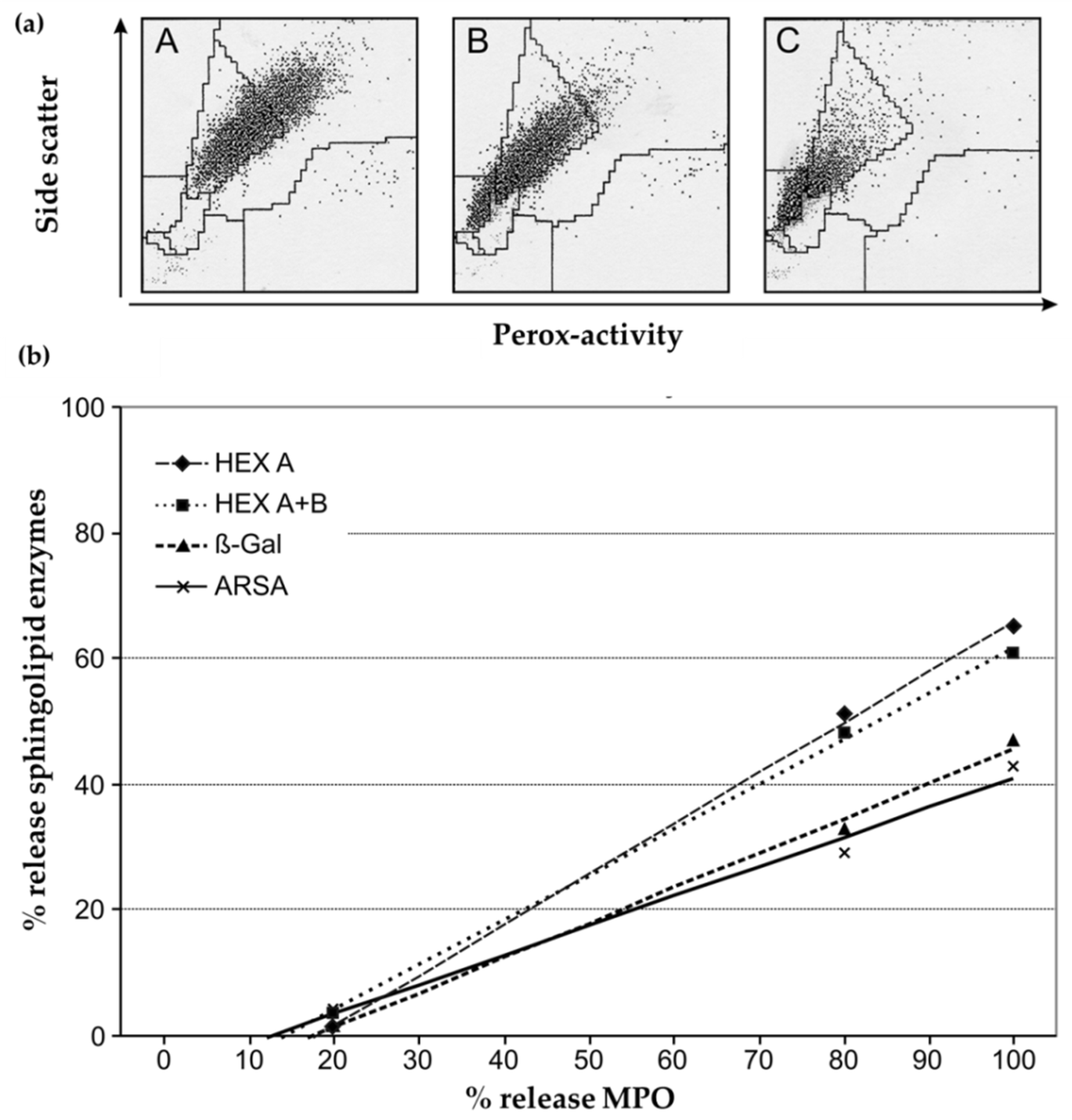
3.1.2. Loss of MPO and Other Lysosomal Enzymes in Granulocytes during Leukocyte Preparation
3.2. Choice of a Suitable Reference System
3.3. Statistical Evaluation during a Seven-Year Observation Period
3.3.1. Inter-assay Variability (Internal Control Person)
3.3.2. Demographic Characterization of the Cohort
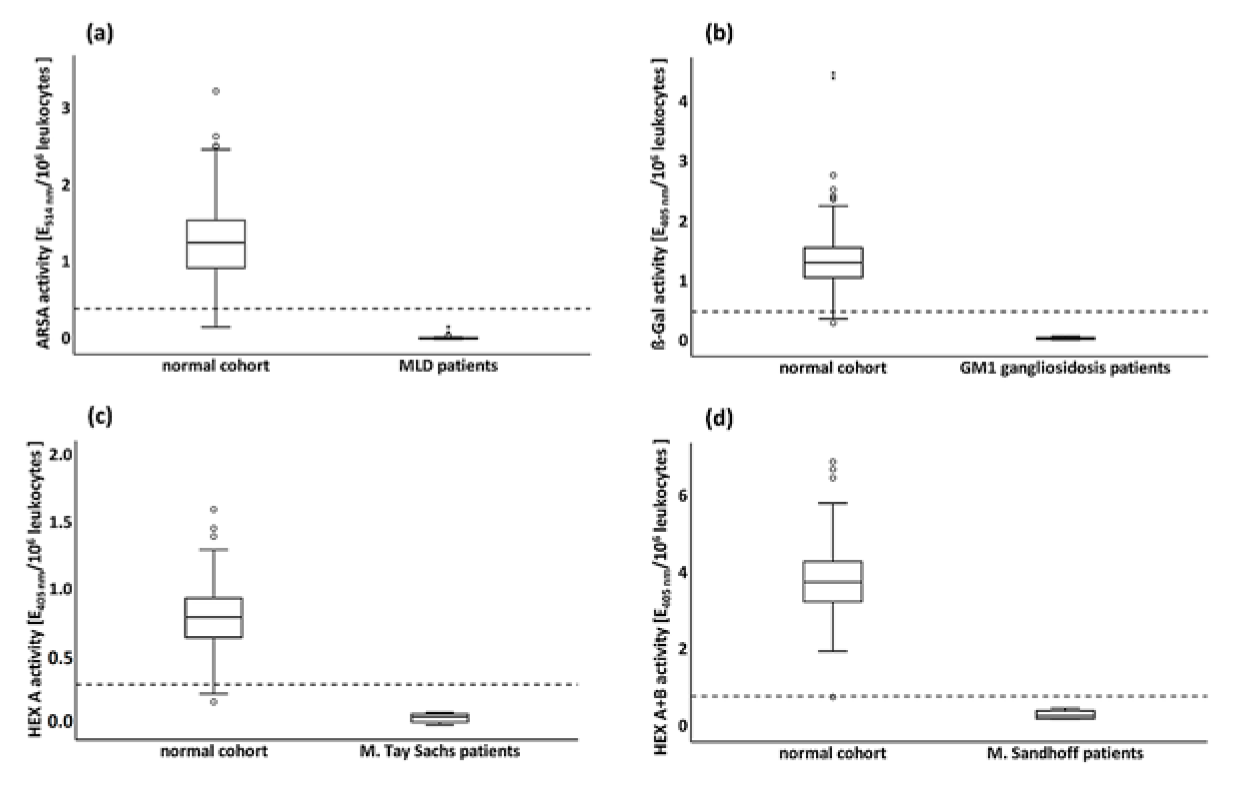
3.3.3. Enzyme Activity in the Normal Cohort and Minimal Permissible Value
Effects of Age and Transport Time on Enzyme Activity in the Normal Cohort
3.3.4. Enzyme Activities in Pathological Cohorts
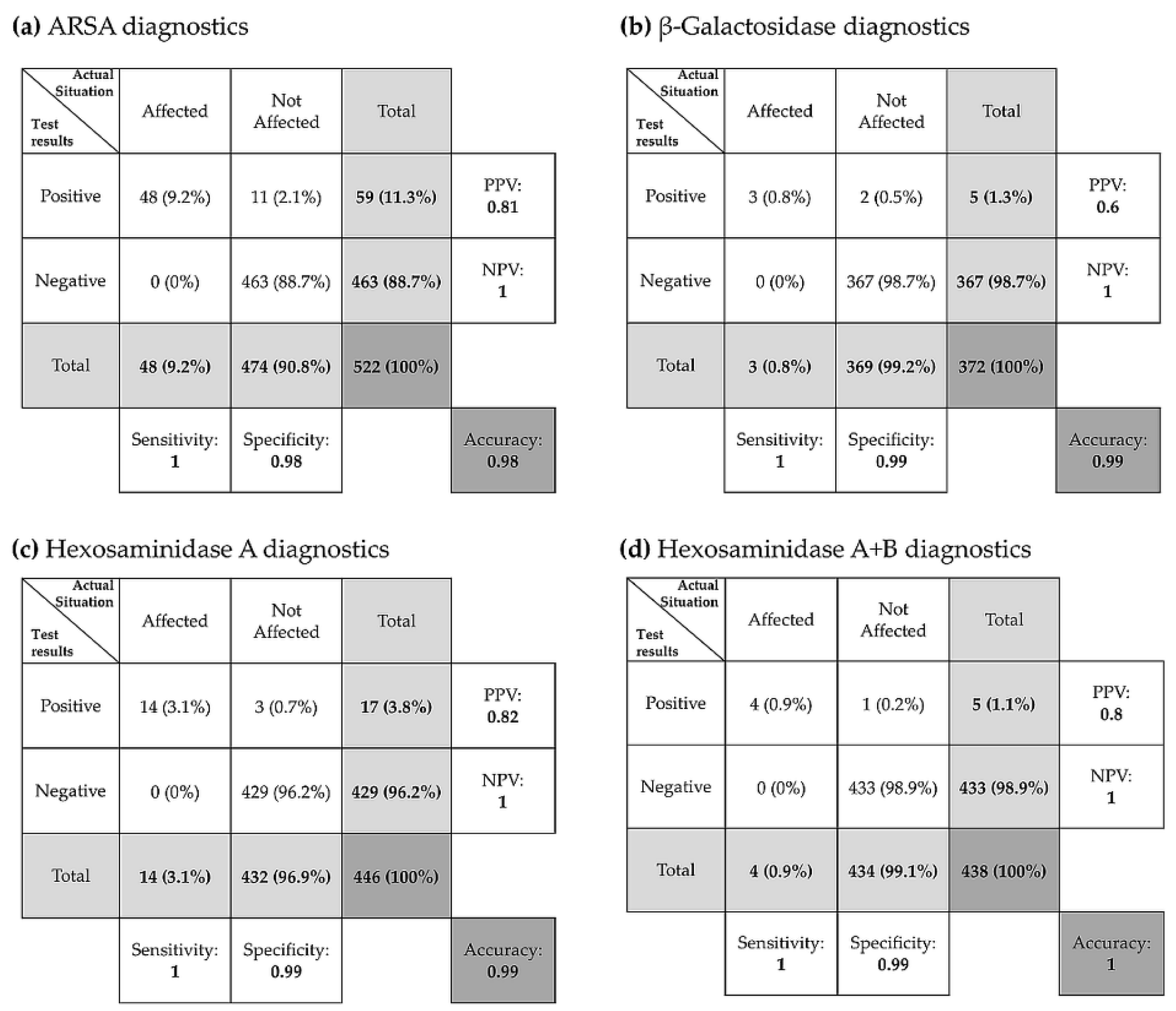
3.3.5. Evaluation of Diagnostic Tests
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mehta, A.B.; Winchester, B. Lysosomal Storage Disorders: A Practical Guide, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Bouwman, M.G.; Teunissen, Q.G.A.; Wijburg, F.A.; Linthorst, G.E. ‘Doctor Google’ ending the diagnostic odyssey in lysosomal storage disorders: Parents using internet search engines as an efficient diagnostic strategy in rare diseases. Arch. Dis. Child. 2010, 95, 642–644. [Google Scholar] [CrossRef] [PubMed]
- Groeschel, S.; Kühl, J.-S.; Bley, A.E.; Kehrer, C.; Weschke, B.; Döring, M.; Böhringer, J.; Schrum, J.; Santer, R.; Kohlschütter, A.; et al. Long-term Outcome of Allogeneic Hematopoietic Stem Cell Transplantation in Patients with Juvenile Metachromatic Leukodystrophy Compared With Nontransplanted Control Patients. JAMA Neurol. 2016, 73, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Dali, C.Í.; Sevin, C.; Krägeloh-Mann, I.; Giugliani, R.; Sakai, N.; Wu, J.; Wasilewski, M. Safety of intrathecal delivery of recombinant human arylsulfatase A in children with metachromatic leukodystrophy: Results from a phase 1/2 clinical trial. Mol. Genet. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Van Rappard, D.F.; Boelens, J.J.; Wolf, N.I. Metachromatic leukodystrophy: Disease spectrum and approaches for treatment. Best Pr. Res. Clin. Endocrinol. Metab. 2015, 29, 261–273. [Google Scholar] [CrossRef]
- Kehrer, C.; Elgün, S.; Raabe, C.; Böhringer, J.; Beck-Wödl, S.; Bevot, A.; Kaiser, N.; Schöls, L.; Krägeloh-Mann, I.; Groeschel, S. Association of age at onset and first symptoms with disease progression in patients with metachromatic leukodystrophy. Neurology 2020. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, G.J.; Böer, M.; Weykamp, C.W.; De Vries, R.; van den Berg, I.; Janssens-Puister, J.; Niezen-Koning, K.E.; Wevers, R.A.; Poorthuis, B.J.H.M.; Van Diggelen, O.P. External quality assurance programme for enzymatic analysis of lysosomal storage diseases: A pilot study. J. Inherit. Metab. Dis. 2005, 28, 979–990. [Google Scholar] [CrossRef]
- Gieselmann, V.; Wenger, D.A.; Krägeloh-Mann, I. Metachromatic Leukodystrophy and Globoid Cell Leukodystrophy. In Lysosomal Storage Disorders; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 70–79. [Google Scholar]
- Gravel, R.C.J.; Kaback, M.; Mahuran, D.; Sandhoff, K.; Suzuki, K. The GM2 gangliosidoses. In The Metabolic and Molecular Bases of Inherited Disease; Scriever, C.B.A., Valle, D., Sly, W., Childs, B., Kinzler, K., Beaudet, A., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 1995; pp. 2839–2879. [Google Scholar]
- Kolodny, E.F.A. Metachromatic leukodystrophy and multiple sulfatase deficiency: Sulfatide lipidosis. In The Metabolic and Molecular Bases of Inherited Disease; Scriever, C.B.A., Valle, D., Sly, W., Childs, B., Kinzler, K., Eds.; McGraw-Hill: New York, NY, USA, 1995; pp. 2693–2739. [Google Scholar]
- Suzuki, K. Enzymic diagnosis of sphingolipidoses. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1978; Volume 50, pp. 456–488. [Google Scholar]
- Suzuki, Y.; Nanba, E.; Matsuda, J.; Higaki, K.; Oshima, A. β-Galactosidase deficiency (β-galactosidosis): GM1 gangliosidosis and Morquio B disease. In The Metabolic and Molecular Bases of Inherited Disease; Scriever, C.B.A., Valle, D., Sly, W., Childs, B., Kinzler, K., Beaudet, A., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 1995; pp. 2785–2823. [Google Scholar]
- Beutler, E.; Kuhl, W.; Matsumoto, F.; Pangalis, G. Acid hydrolases in leukocytes and platelets of normal subjects and in patients with Gaucher’s and Fabry’s disease. J. Exp. Med. 1976, 143, 975–980. [Google Scholar] [CrossRef]
- Nakagawa, S.; Kumin, S.; Fox, D.; Nitowsky, H.M. Human hexosaminidase isozymes. III. Distribution and activity of isozymes in peripheral blood leukocytes and platelets. J. Lab. Clin. Med. 1978, 91, 922–928. [Google Scholar]
- Schmidt, R.; Von Figura, K.; Paschke, E.; Kresse, H. Sanfilippo’s disease type a: Sulfamidase activity in peripheral leukocytes of normal, heterozygous and homozygous individuals. Clin. Chim. Acta 1977, 80, 7–15. [Google Scholar] [CrossRef]
- Gieselmann, V.; Krägeloh-Mann, I. Metachromatic Leukodystrophy—An Update. Neuropediatrics 2010, 41, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.T.R. The Gangliosidoses. In Lysosomal Storage Disorders: A Practical Guide, 1st ed.; Mehta, A.B., Winchester, B., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 63–69. [Google Scholar]
- Sandhoff, K.; Harzer, K. Gangliosides and Gangliosidoses: Principles of Molecular and Metabolic Pathogenesis. J. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-Y.; Wu, T.-L.; Hung, C.-C.; Tsao, K.-C.; Sun, C.-F.; Wu, L.L.; Wu, J.T. Development of an ELISA for myeloperoxidase on microplate: Normal reference values and effect of temperature on specimen preparation. Clin. Chim. Acta 2006, 373, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.; Datwyler, S.A.; Hsu, S.C.; Matias, M.S.; Pacenti, D.P.; Lueders, C.; Mueller, C.; Danne, O.; Möckel, M. Effect of Collection Tube Type and Preanalytical Handling on Myeloperoxidase Concentrations. Clin. Chem. 2008, 54, 1076–1079. [Google Scholar] [CrossRef]
- Zipfel, M.; Carmine, T.C.; Gerber, C.; Niethammer, D.; Bruchelt, G. Evidence for the Activation of Myeloperoxidase by f-Meth-Leu-Phe Prior to Its Release from Neutrophil Granulocytes. Biochem. Biophys. Res. Commun. 1997, 232, 209–212. [Google Scholar] [CrossRef]
- Lee-Vaupel, M.; Conzelmann, E. A simple chromogenic assay for arylsulfatase A. Clin. Chim. Acta 1987, 164, 171–180. [Google Scholar] [CrossRef]
- Böhringer, J.; Santer, R.; Schumacher, N.; Gieseke, F.; Cornils, K.; Pechan, M.; Kustermann-Kuhn, B.; Handgretinger, R.; Schöls, L.; Harzer, K.; et al. Enzymatic characterization of novel arylsulfatase A variants using human arylsulfatase A-deficient immortalized mesenchymal stromal cells. Hum. Mutat. 2017, 38, 1511–1520. [Google Scholar] [CrossRef]
- Leckie, M.; Gomma, A.; Purcell, I.; Nyawo, B.; Dewar, A.; Okrongly, D.; Burman, J.; Hooper, J.; Barnes, P.; Clague, J.; et al. Automated quantitation of peripheral blood neutrophil activation in patients with myocardial ischaemia. Int. J. Cardiol. 2004, 95, 307–313. [Google Scholar] [CrossRef]
- Dörner, K. Klinische Chemie und Hämatologie; Georg Thieme Verlag: New York, NY, USA, 2006. [Google Scholar]
- Simon, M.R.; Roi, L.D.; Desai, S.; Salberg, D.J.; Rosec, N.R. Comparison of Cul Tures of Human Lymphocytes Obtained Following Nh4Cl Induced Red Blood Cell Lysis and Ficoll-Hypaque Density Gradient Centrifugation. Immunol. Commun. 1983, 12, 301–314. [Google Scholar] [CrossRef]
- Leinekugel, P.; Michel, S.; Conzelmann, E.; Sandhoff, K. Quantitative correlation between the residual activity of β-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Qual. Life Res. 1992, 88, 513–523. [Google Scholar] [CrossRef]
- Burin, M.G.; Dutra-Filho, C.; Brum, J.; Mauricio, T.; Amorim, M.; Giugliani, R. Effect of collection, transport, processing and storage of blood specimens on the activity of lysosomal enzymes in plasma and leukocytes. Braz. J. Med Biol. Res. 2000, 33, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Elbin, C.S.; Olivova, P.; Marashio, C.A.; Cooper, S.K.; Cullen, E.; Keutzer, J.M.; Zhang, X.K. The effect of preparation, storage and shipping of dried blood spots on the activity of five lysosomal enzymes. Clin. Chim. Acta 2011, 412, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Covarrubias, V.; Dane, A.; Hankemeier, T.; Vreeken, R.J. The influence of citrate, EDTA, and heparin anticoagulants to human plasma LC–MS lipidomic profiling. Metabolomics 2013, 9, 337–348. [Google Scholar] [CrossRef]
- Yatziv, S.; Kahane, I.; Abeliuk, P.; Cividalli, G.; Rachmilewitz, E.A. “Lysosomal” enzyme activities in red blood cells of normal individuals and patients with homozygous β-thalassaemia. Clin. Chim. Acta 1979, 96, 67–72. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Fraction | ||||
|---|---|---|---|---|
| n1 | Lymphocytes | Monocytes | Granulocytes | |
| Hexosaminidase A+B | 9 | 1.0 | 4.36 ± 1.05 | 1.81 ± 0.56 |
| Hexosaminidase A | 8 | 1.0 | 4.57 ± 1.59 | 2.81 ± 0.73 |
| β-Galactosidase | 8 | 1.0 | 4.47 ± 1.00 | 1.64 ± 0.41 |
| Arylsulfatase A | 9 | 1.0 | 2.31 ± 0.68 | 1.64 ± 0.38 |
| MLD | GM1 | Tay–Sachs Disease | Sandhoff Disease | |
|---|---|---|---|---|
| Number | 48 | 3 | 14 | 4 |
| ♂ | 24 | 2 | 8 | 2 |
| ♀ | 24 | 1 | 6 | 2 |
| Age at onset (years) | 11.7 ± 12 | 7 ± 10 | 16 ± 17 | 0.9 ± 0.1 |
| Median | 6.2 | 1.5 | 10 | 0.9 |
| Min. | 0.08 | 1.1 | 0.08 | 0.8 |
| Max. | 41.6 | 18.1 | 41 | 1 |
| Minimal permissible value(E/106 leukocyte) | 0.4 | 0.5 | 0.3 | 0.8 |
| Average of measured enzyme activity 1 (E/106 leukocytes) | 0.02 ± 0.03 | 0.05 ± 0.03 | 0.05 ± 0.03 | 0.32 ± 0.13 |
| ARSA | β-Gal | HEX A | HEX A+B | |
|---|---|---|---|---|
| Number | 474 | 369 | 432 | 434 |
| Measured enzyme activity 1 (E/106 leukocytes) | 1.27 ± 0.46 | 1.36 ± 0.44 | 0.81 ± 0.27 | 3.78 ± 0.80 |
| Minimal permissible value 2 (E/106 leukocytes) | 0.4 | 0.5 | 0.3 | 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strobel, S.; Hesse, N.; Santhanakumaran, V.; Groeschel, S.; Bruchelt, G.; Krägeloh-Mann, I.; Böhringer, J. Optimization of Enzyme Essays to Enhance Reliability of Activity Measurements in Leukocyte Lysates for the Diagnosis of Metachromatic Leukodystrophy and Gangliosidoses. Cells 2020, 9, 2553. https://doi.org/10.3390/cells9122553
Strobel S, Hesse N, Santhanakumaran V, Groeschel S, Bruchelt G, Krägeloh-Mann I, Böhringer J. Optimization of Enzyme Essays to Enhance Reliability of Activity Measurements in Leukocyte Lysates for the Diagnosis of Metachromatic Leukodystrophy and Gangliosidoses. Cells. 2020; 9(12):2553. https://doi.org/10.3390/cells9122553
Chicago/Turabian StyleStrobel, Sebastian, Naomi Hesse, Vidiyaah Santhanakumaran, Samuel Groeschel, Gernot Bruchelt, Ingeborg Krägeloh-Mann, and Judith Böhringer. 2020. "Optimization of Enzyme Essays to Enhance Reliability of Activity Measurements in Leukocyte Lysates for the Diagnosis of Metachromatic Leukodystrophy and Gangliosidoses" Cells 9, no. 12: 2553. https://doi.org/10.3390/cells9122553
APA StyleStrobel, S., Hesse, N., Santhanakumaran, V., Groeschel, S., Bruchelt, G., Krägeloh-Mann, I., & Böhringer, J. (2020). Optimization of Enzyme Essays to Enhance Reliability of Activity Measurements in Leukocyte Lysates for the Diagnosis of Metachromatic Leukodystrophy and Gangliosidoses. Cells, 9(12), 2553. https://doi.org/10.3390/cells9122553