CRISPR FokI Dead Cas9 System: Principles and Applications in Genome Engineering


Abstract
1. Introduction
2. Gene-Editing Tools
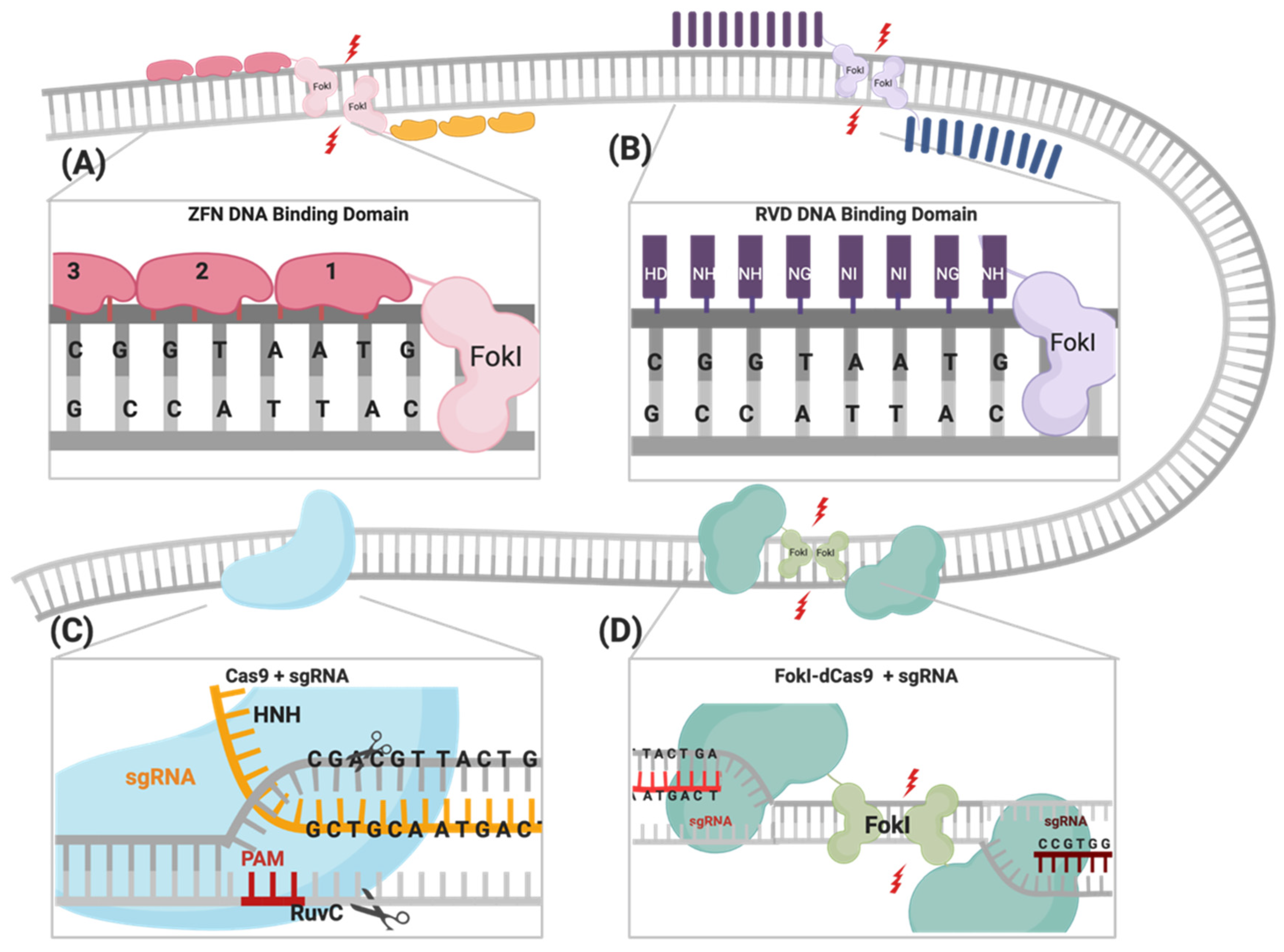
2.1. Zinc Finger Nucleases (ZFNs)
2.2. Transcription Activator-Like Effector Nucleases (TALENs)
2.3. CRISPR/Cas Systems
2.3.1. Cas9 Variants
Cas9 Nickases
Inactive Cas9 (Dead Cas9)
Base Editors
FokI Endonuclease Fused to dCas9
3. Engineering FokI–dCas9
3.1. FokI Fusion to dCas9
3.2. Nuclear Localization Sequence (NLS)
3.3. Linker
4. Principles of Gene Editing Using FokI–dCas9
4.1. sgRNA Design
4.2. PAM Sequences and fdCas9 Variants
4.3. Obligate FokI Dimerization
4.4. fdCas9 System Delivery
5. Applications of FokI–dCas9
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, H.-X.; Zhang, Y.; Yin, H. Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol. Ther. 2019, 27, 735–746. [Google Scholar] [CrossRef]
- Carroll, D. Genome Editing: Past, Present, and Future. Yale J. Boil. Med. 2017, 90, 653–659. [Google Scholar]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Chandrasegaran, S. Recent advances in the use of ZFN-mediated gene editing for human gene therapy. Cell Gene Ther. Insights 2017, 3, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; Chen, T.; De Haas, M.; Holland, H.A.; Akhtar, W.; Van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813.e6. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef]
- Song, J.; Yang, D.; Xu, J.; Zhu, T.; Chen, Y.E.; Zhang, J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016, 7, 10548. [Google Scholar] [CrossRef]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.M.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Fine, E.J.; Antico, C.J.; Bao, G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013, 41, 9584–9592. [Google Scholar] [CrossRef]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef]
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014, 32, 677–683. [Google Scholar] [CrossRef]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Wyvekens, N.; Khayter, C.; Foden, J.A.; Thapar, V.; Reyon, D.; Goodwin, M.J.; Aryee, M.J.; Joung, J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014, 32, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Aouida, M.; Eid, A.; Ali, Z.; Cradick, T.J.; Lee, C.; Deshmukh, H.; Atef, A.; Abusamra, D.; Gadhoum, S.Z.; Merzaban, J.S.; et al. Efficient fdCas9 Synthetic Endonuclease with Improved Specificity for Precise Genome Engineering. PLoS ONE 2015, 10, e0133373. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Sakuma, T.; Sakamoto, T.; Ohmuraya, M.; Nakagata, N.; Yamamoto, T. Production of knockout mice by DNA microinjection of various CRISPR/Cas9 vectors into freeze-thawed fertilized oocytes. BMC Biotechnol. 2015, 15, 33. [Google Scholar] [CrossRef]
- Havlicek, S.; Shen, Y.; Alpagu, Y.; Bruntraeger, M.B.; Zufir, N.B.; Phuah, Z.Y.; Fu, Z.; Dunn, N.R.; Stanton, L.W. Re-engineered RNA-Guided FokI-Nucleases for Improved Genome Editing in Human Cells. Mol. Ther. 2017, 25, 342–355. [Google Scholar] [CrossRef]
- Cathomen, T.; Joung, J.K. Zinc-finger Nucleases: The Next Generation Emerges. Mol. Ther. 2008, 16, 1200–1207. [Google Scholar] [CrossRef]
- Klug, A. The discovery of zinc fingers and their development for practical applications in gene regulation and genome manipulation. Q. Rev. Biophys. 2010, 43, 1–21. [Google Scholar] [CrossRef]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.-L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007, 25, 778–785. [Google Scholar] [CrossRef]
- Chou, S.-T.; Leng, Q.; Mixson, J. Zinc finger nucleases: Tailor-made for gene therapy. Drugs Futur. 2012, 37, 183. [Google Scholar] [CrossRef][Green Version]
- Xiong, K.; Li, S.; Zhang, H.; Cui, Y.; Yu, D.; Li, Y.; Sun, W.; Fu, Y.; Teng, Y.; Liu, Z.; et al. Targeted editing of goat genome with modular-assembly zinc finger nucleases based on activity prediction by computational molecular modeling. Mol. Biol. Rep. 2013, 40, 4251–4256. [Google Scholar] [CrossRef] [PubMed]
- Wah, D.A.; Bitinaite, J.; Schildkraut, I.; Aggarwal, A.K. Structure of FokI has implications for DNA cleavage. Proc. Natl. Acad. Sci. USA 1998, 95, 10564–10569. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.M.; Musunuru, K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zhao, H. Transcription activator-like effector nucleases (TALENs): A highly efficient and versatile tool for genome editing. Biotechnol. Bioeng. 2013, 110, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Cuculis, L.; Abil, Z.; Zhao, H.; Schroeder, C.M. TALE proteins search DNA using a rotationally decoupled mechanism. Nat. Chem. Biol. 2016, 12, 831–837. [Google Scholar] [CrossRef]
- Bogdanove, A.J.; Voytas, D.F. TAL Effectors: Customizable Proteins for DNA Targeting. Science 2011, 333, 1843–1846. [Google Scholar] [CrossRef]
- Zhu, H.; Suh, Y.; Goh, S.-L.; Liang, Q.; Chen, C.; Du, S.; Phang, R.-Z.; Tay, F.C.; Tan, W.-K.; Li, Z.; et al. Baculoviral transduction facilitates TALEN-mediated targeted transgene integration and Cre/LoxP cassette exchange in human-induced pluripotent stem cells. Nucleic Acids Res. 2013, 41, e180. [Google Scholar] [CrossRef]
- Mussolino, C.; Alzubi, J.; Fine, E.J.; Morbitzer, R.; Cradick, T.J.; Lahaye, T.; Bao, G.; Cathomen, T. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014, 42, 6762–6773. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Bikard, D.; Cox, D.D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, H.; Chen, L.; Xie, K. Recent Advances in Genome Editing Using CRISPR/Cas9. Front. Plant Sci. 2016, 7, 703. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2, e00471. [Google Scholar] [CrossRef]
- O’Geen, H.; Yu, A.S.; Segal, D.J. How specific is CRISPR/Cas9 really? Curr. Opin. Chem. Biol. 2015, 29, 72–78. [Google Scholar] [CrossRef]
- Karvelis, T.; Gasiunas, G.; Miksys, A.; Barrangou, R.; Horvath, P.; Siksnys, V. crRNA and tracrRNA guide Cas9-mediated DNA interference inStreptococcus thermophilus. RNA Biol. 2013, 10, 841–851. [Google Scholar] [CrossRef]
- Oakes, B.L.; Nadler, D.C.; Savage, D.F. Protein engineering of Cas9 for enhanced function. Methods Enzym. 2014, 546, 491–511. [Google Scholar]
- Ho, B.X.; Loh, S.J.H.; Chan, W.K.; Soh, B.S. In Vivo Genome Editing as a Therapeutic Approach. Int. J. Mol. Sci. 2018, 19, 2721. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.C.; Weisbach, N.R.; Santinha, A.J.; Incarnato, D.; Platt, R.J. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 2019, 16, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Vo, T.D.; Mendel, M.C.; Greenberg, S.G.; Wang, J.; Xia, D.F.; Miller, J.C.; Urnov, F.D.; Gregory, P.D.; Holmes, M.C. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat. Chem. Biol. 2010, 8, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef]
- Gautron, A.-S.; Juillerat, A.; Guyot, V.; Filhol, J.-M.; Dessez, E.; Duclert, A.; Duchateau, P.; Poirot, L. Fine and Predictable Tuning of TALEN Gene Editing Targeting for Improved T Cell Adoptive Immunotherapy. Mol. Ther. Nucleic Acids 2017, 9, 312–321. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Bothmer, A.; Phadke, T.; Barrera, L.A.; Margulies, C.M.; Lee, C.S.; Buquicchio, F.; Moss, S.; Abdulkerim, H.S.; Selleck, W.; Jayaram, H.; et al. Characterization of the interplay between DNA repair and CRISPR/Cas9-induced DNA lesions at an endogenous locus. Nat. Commun. 2017, 8, 13905. [Google Scholar] [CrossRef]
- Wang, B.; Yan, S. When and How to Use Nickases for Efficient Genome Editing. Available online: https://www.idtdna.com/pages/education/decoded/article/when-and-how-to-use-nickases-for-efficient-genome-editing (accessed on 9 April 2020).
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; Dicarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Maximiliaan, H.; Tu, L.-C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat. Biotechnol. 2016, 34, 528–530. [Google Scholar] [CrossRef]
- Anton, T.; Karg, E.; Bultmann, S. Applications of the CRISPR/Cas system beyond gene editing. Biol. Methods Protoc. 2018, 3, bpy002. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Miller, B.R.; Langner, L.M.; Grünewald, J.; Joung, J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2020, 1–6. [Google Scholar] [CrossRef]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef]
- Li, C.; Zong, Y.; Wang, Y.; Jin, S.; Zhang, D.; Song, Q.; Zhang, R.; Gao, C. Expanded base editing in rice and wheat using a Cas9-adenosine deaminase fusion. Genome Biol. 2018, 19, 59. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Jin, S.; Zong, Y.; Gao, Q.; Zhu, Z.; Wang, Y.; Qin, P.; Liang, C.; Wang, D.; Qiu, J.-L.; Zhang, F.; et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 2019, 364, 292–295. [Google Scholar] [CrossRef]
- Mishra, R.; Joshi, R.K.; Zhao, K. Base editing in crops: Current advances, limitations and future implications. Plant Biotechnol. J. 2020, 18, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, N.; Salvaris, E.J.; Philip, G.K.; Wakefield, M.J.; Nottle, M.B.; Hawthorne, W.J.; Cowan, P.J. Fok I-dCas9 mediates high-fidelity genome editing in pigs. Xenotransplantation 2019, 27, e12551. [Google Scholar] [CrossRef] [PubMed]
- Andreas, S.; Schwenk, F.; Küter-Luks, B.; Faust, N.; Kühn, R. Enhanced efficiency through nuclear localization signal fusion on phage phiC31-integrase: Activity comparison with Cre and FLPe recombinase in mammalian cells. Nucleic Acids Res. 2002, 30, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Staahl, B.T.; Benekareddy, M.; Coulon-Bainier, C.; Banfal, A.A.; Floor, S.N.; Sabo, J.K.; Urnes, C.; Munares, G.A.; Ghosh, A.; Doudna, J.A. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol. 2017, 35, 431–434. [Google Scholar] [CrossRef]
- Han, H.A.; Pang, J.K.S.; Soh, B.-S. Mitigating off-target effects in CRISPR/Cas9-mediated in vivo gene editing. J. Mol. Med. 2020, 98, 615–632. [Google Scholar] [CrossRef]
- Wyvekens, N.; Topkar, V.V.; Khayter, C.; Joung, J.K.; Tsai, S.Q. Dimeric CRISPR RNA-Guided FokI-dCas9 Nucleases Directed by Truncated gRNAs for Highly Specific Genome Editing. Hum. Gene Ther. 2015, 26, 425–431. [Google Scholar] [CrossRef]
- Ma, D.; Xu, Z.; Zhang, Z.; Chen, X.; Zeng, X.; Zhang, Y.; Deng, T.; Ren, M.; Sun, Z.; Jiang, R.; et al. Engineer chimeric Cas9 to expand PAM recognition based on evolutionary information. Nat. Commun. 2019, 10, 560. [Google Scholar] [CrossRef]
- Catto, L.E.; Ganguly, S.; Milsom, S.E.; Welsh, A.J.; Halford, S.E. Protein assembly and DNA looping by the FokI restriction endonuclease. Nucleic Acids Res. 2006, 34, 1711–1720. [Google Scholar] [CrossRef]
- Jubair, L.; Fallaha, S.; McMillan, N.A. Systemic Delivery of CRISPR/Cas9 Targeting HPV Oncogenes Is Effective at Eliminating Established Tumors. Mol. Ther. 2019, 27, 2091–2099. [Google Scholar] [CrossRef]
- Cameron, P.; Coons, M.M.; Klompe, S.E.; Lied, A.M.; Smith, S.C.; Vidal, B.; Donohoue, P.D.; Rotstein, T.; Kohrs, B.W.; Nyer, D.B.; et al. Harnessing type I CRISPR–Cas systems for genome engineering in human cells. Nat. Biotechnol. 2019, 37, 1471–1477. [Google Scholar] [CrossRef]
- Wilbie, D.; Walther, J.; Mastrobattista, E. Delivery Aspects of CRISPR/Cas for in Vivo Genome Editing. Acc. Chem. Res. 2019, 52, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, K.; Aouida, M.; El-Agnaf, O.M. Tissue-Specific Delivery of CRISPR Therapeutics: Strategies and Mechanisms of Non-Viral Vectors. Int. J. Mol. Sci. 2020, 21, 7353. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Vigouroux, A.; Rousset, F.; Varet, H.; Khanna, V.; Bikard, D. A CRISPRi screen in E. coli reveals sequence-specific toxicity of dCas9. Nat. Commun. 2018, 9, 1912. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Troilo, P.J.; Wang, X.; Griffiths, T.G.; Pacchione, S.J.; Barnum, A.B.; Harper, L.B.; Pauley, C.J.; Niu, Z.; Denisova, L.; et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004, 11, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Hung, S.S.; Khalid, M.K.N.M.; Wang, J.-H.; Chrysostomou, V.; Wong, V.H.; Singh, V.; Wing, K.; Tu, L.; Bender, J.A.; et al. Utility of Self-Destructing CRISPR/Cas Constructs for Targeted Gene Editing in the Retina. Hum. Gene Ther. 2019, 30, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Semenova, N.; Bosnjak, M.; Markelc, B.; Znidar, K.; Cemazar, M.; Heller, L. Multiple cytosolic DNA sensors bind plasmid DNA after transfection. Nucleic Acids Res. 2019, 47, 10235–10246. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Nottle, M.B.; Salvaris, E.J.; Fisicaro, N.; McIlfatrick, S.; Vassiliev, I.; Hawthorne, W.J.; O’Connell, P.J.; Brady, J.L.; Lew, A.M.; Cowan, P.J. Targeted insertion of an anti-CD2 monoclonal antibody transgene into the GGTA1 locus in pigs using FokI-dCas9. Sci. Rep. 2017, 7, 8383. [Google Scholar] [CrossRef]
- Hara, S.; Tamano, M.; Yamashita, S.; Kato, T.; Saito, T.; Sakuma, T.; Yamamoto, T.; Inui, M.; Takada, S. Generation of mutant mice via the CRISPR/Cas9 system using FokI-dCas9. Sci. Rep. 2015, 5, 11221. [Google Scholar] [CrossRef]
- Terao, M.; Tamano, M.; Hara, S.; Kato, T.; Kinoshita, M.; Takada, S. Utilization of the CRISPR/Cas9 system for the efficient production of mutant mice using crRNA/tracrRNA with Cas9 nickase and FokI-dCas9. Exp. Anim. 2016, 65, 275–283. [Google Scholar] [CrossRef]
- Jung, C.J.; Zhang, J.; Trenchard, E.; Lloyd, K.C.; West, D.B.; Rosen, B.; DeJong, P.J. Efficient gene targeting in mouse zygotes mediated by CRISPR/Cas9-protein. Transgenic Res. 2016, 26, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Peng, S.; Xie, Z. Integration and exchange of split dCas9 domains for transcriptional controls in mammalian cells. Nat. Commun. 2016, 7, 13056. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Peng, S.; Huang, W.; Cai, Z.; Xie, Z. Rational Design of Mini-Cas9 for Transcriptional Activation. ACS Synth. Biol. 2018, 7, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Shen, N.; Jung-Klawitter, S.; Betzen, C.; Hoffmann, G.F.; Hoheisel, J.D.; Blau, N. CRISPR RNA-guided FokI nucleases repair a PAH variant in a phenylketonuria model. Sci. Rep. 2016, 6, 35794. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Zinc Finger Nucleases (ZFNs) | Transcription Activator-Like Effector Nucleases (TALENs) | CRISPR/Cas Systems | ||
|---|---|---|---|---|
| CRISPR Associated Endonuclease (Cas9) | FokI Dead Cas9 Endonuclease (FokI–dCas9) | |||
| DNA catalytic domain | FokI | FokI | RuvC and HNH | FokI |
| DNA recognition | DNA: Protein | DNA: RNA | ||
| unit of target recognition | Pairs of ZFNs (via the ZF motifs) | Pairs of TALENs (via RVD tandem repeat). | One 17–20 bp sgRNA | Pairs of 19–20 bp sgRNAs |
| Recognized target size | Recognizes 18-24 bp | Recognizes 30–40 bp. | Recognizes NGG PAM sequence + 17–20 bp | Recognizes two NGG PAM sequences + 38–40 bp |
| Specificity | Tolerates few positional mismatches | Tolerates both positional and multiple consecutive mismatches | Enhanced specificity due to the dual sgRNAs requirement of | |
| Spacer size | 5–7 bp | 14–16 bp | No spacer required | 13–18 bp and/or 26 bp |
| Ease of delivery | Limited delivery due to the difficulty of linking ZF modules | Difficult delivery due to cDNA size and extensive TALEs repeats | Easily delivered using standard delivery and cloning techniques | Harder to deliver due to increased size of construct and added components |
| Limitation | Off-target effects Limited delivery due to size constraints | Off-target effects Expensive | Off-target effects due to mismatch tolerance PAM sequence availability | Difficult to deliver A strict system with many obligatory requirements |
| Multiplexing | Difficult | Easy, can form multiplexes directed to multiple genes | ||
| Nickases (Pairs of D10 or H840) | FokI-dCas9 | |||
|---|---|---|---|---|
| Structure |  |  |  |  |
| Existing forms | Monomers | Dimers | Monomers | Dimers |
| Cleavage domain | HNH or RuvC | Pairs of HNH or pairs of RuvC | FokI (But inactive) | Pairs of FokI |
| Obligate dimerization | - | No | - | Yes |
| Spacer length | - | Up to 100bp | - | 16-18bp or 26bp (depending on variant used) |
| Target size | 17-20bp | 34-40bp + spacer length | 19-20bp | 38-40bp + spacer length |
| Linker | - | - | - | Required to link the FokI domain |
| Type of DNA damage | Single strand nicks | staggered double strand break | No damage induced | Double strands break |
| Type of mutations | Can induce point mutations | Additions or deletions of >2 bps | Non-mutagenic | Additions or deletions of >2 bps |
| Off-target effect (Compared to WT Cas9) | Low-moderate | Low | Nearly non-existing | Rare |
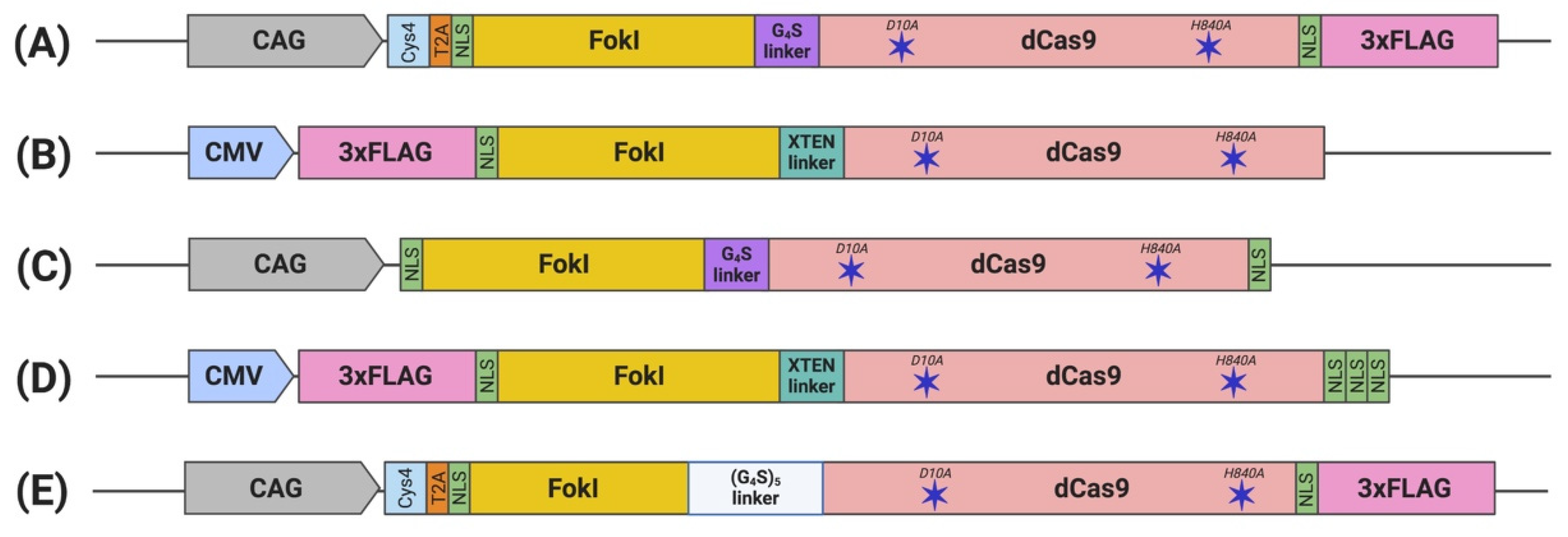
| sgRNA Delivery Method | Linker | Gene Editing Activity of fdCas9 | Optimal Spacer Distance | Off-Targets | ||||
|---|---|---|---|---|---|---|---|---|
| Compared to Negative Control | Compared to Single WT Cas9 | Compared to Paired Casas9 nickases | Genes Tested | Activity | ||||
| Tsai (2014) [22] (Figure 2A) | Csy4-based dual sgRNA expression system | GGGGS linker | 3–40% | Differences varied depending on gene tested | Similar to fdCas9 | 13–18 bp | VEGFA | Indistinguishable off-target mutation |
| Guilinger (2014) [23] (Figure 2B) | dual sgRNA expression plasmid | 17 linkers tested; best activity using XTEN linker | GFP disruption: 10% reported by flow cytometry and 20% by T7EI | eGFP disruption: 25% reported by flow cytometry and 2/3 of fdCas9 activity by T7EI | eGFP disruption: 15% reported by flow cytometry and similar activity to fdCas9 by T7EI | 15 and 25 bp | AAVS1, CLTA, EMX1, HBB, VEGFA | Fold increase of on/off-target editing: 140 compared to WT Cas9 1.3–8.8 compared to nickases |
| Average of 14.9% on human genes | Average of 28.2% on human genes | Average of 20.6% on human genes | ||||||
| Nakagawa (2015) [25] (Figure 2C) | All-in-one construct, included in the fdCas9 plasmid | GGGGS linker | Tested on mice fertilized oocyte | 13–18 bp | Top three candidates of used sgRNA | No off-target editing was reported | ||
| Average of 49%, and moderate birth rate | Average of 90%, and a low birth rate | Average of 2.9%, and a high birth rate | ||||||
| Aouida (2015) [24] (Figure 2D) | sgRNA expressing DNA fragments | XTEN linker | eGFP disruption: 5% reported by flow cytometry | eGFP disruption: 12.3% reported by flow cytometry | eGFP disruption: 1% reported by flow cytometry | 15–39 bp | CCR5, AAVS1, EMX1, HBB | Only WT Cas9 showed 25–30% off-target editing |
| Havlicek (2017) [26] (Figure 2E) | Csy4-based multiple sgRNA expression systems | (GGGGS)5 | Average of about 30% human gene editing | Cas9 orthologs including SpCas9-HF1 and eSpCas9 showed higher activity | Not tested | 13–18 and 26 bp | CLTA, EMX1, VEGFA | fdCas9 outperform limited off-target editing compared to all Cas9 orthologs tested |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saifaldeen, M.; Al-Ansari, D.E.; Ramotar, D.; Aouida, M. CRISPR FokI Dead Cas9 System: Principles and Applications in Genome Engineering. Cells 2020, 9, 2518. https://doi.org/10.3390/cells9112518
Saifaldeen M, Al-Ansari DE, Ramotar D, Aouida M. CRISPR FokI Dead Cas9 System: Principles and Applications in Genome Engineering. Cells. 2020; 9(11):2518. https://doi.org/10.3390/cells9112518
Chicago/Turabian StyleSaifaldeen, Maryam, Dana E. Al-Ansari, Dindial Ramotar, and Mustapha Aouida. 2020. "CRISPR FokI Dead Cas9 System: Principles and Applications in Genome Engineering" Cells 9, no. 11: 2518. https://doi.org/10.3390/cells9112518
APA StyleSaifaldeen, M., Al-Ansari, D. E., Ramotar, D., & Aouida, M. (2020). CRISPR FokI Dead Cas9 System: Principles and Applications in Genome Engineering. Cells, 9(11), 2518. https://doi.org/10.3390/cells9112518