Abstract
Autism spectrum disorder (ASD) is a prevalent neurodevelopmental disorder characterized by impairments in social communication and social interaction, and the presence of repetitive behaviors and/or restricted interests. In the past few years, large-scale whole-exome sequencing and genome-wide association studies have made enormous progress in our understanding of the genetic risk architecture of ASD. While showing a complex and heterogeneous landscape, these studies have led to the identification of genetic loci associated with ASD risk. The intersection of genetic and transcriptomic analyses have also begun to shed light on functional convergences between risk genes, with the mid-fetal development of the cerebral cortex emerging as a critical nexus for ASD. In this review, we provide a concise summary of the latest genetic discoveries on ASD. We then discuss the studies in postmortem tissues, stem cell models, and rodent models that implicate recently identified ASD risk genes in cortical development.
1. Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder estimated to affect up to ~1% of the population. The condition is clinically diagnosed by persistent deficits in social communication and interaction, and the presence of repetitive behaviors and/or restricted interests. ASD severity varies greatly and can evolve along different trajectories over time [1,2,3]. Along with ASD severity, cognitive development and language contribute to an individual’s adaptive behavior, as ~30% of affected individuals are minimally verbal [4]. Further, ASD can co-occur with other neurodevelopmental manifestations [e.g., intellectual disability (ID), attention deficit hyperactivity disorder (ADHD)], neurological disorders (e.g., hypotonia, epilepsy, movement disorders), and medical co-morbidities (e.g., congenital anomalies, gastrointestinal problems) [5]. Over the past few years, large-scale genomic studies have brought to light hundreds of genetic loci conferring risk. Through orthogonal transcriptomic datasets and follow-up analyses in cell and animal models, studies are uncovering the functional convergence of risk genes on biological processes. One such process is the development of the neocortex.
In this review, we link recent genetic findings in large-scale ASD genomic studies to functional and mechanistic observations in stem cell models and rodent models. We begin by reviewing the current knowledge of the risk architecture, with a specific focus on the contribution of sequencing studies. We then discuss the most recent findings in human neuroanatomical studies and transcriptomic analyses from human postmortem samples, supporting the importance of cortical dysregulation in ASD pathophysiology. We then focus on the most recent discoveries of the mechanisms and pathways surfacing from manipulations of highly penetrant ASD risk genes in rodent models. The specific genes discussed in each section do not create an exhaustive list of all the ASD risk genes found to be involved in cortical development. We specifically selected autosomal dominant risk genes emerging from the latest exome sequencing studies on ASD [6,7,8] and prioritized the discussion of findings published within the past 3 years. We also included a discussion of known and emerging X-linked ASD risk genes with new evidence implicating them in cortical development.
2. ASD Risk Architecture
Enormous progress has been made in our understanding of ASD risk architecture. It is now clear that at least half of the difference in susceptibility amongst individuals depends on genetic variants that are common in the population (common variation, in the form of single-nucleotide polymorphisms or SNPs) [9]. Each of these SNPs has a small effect size, but their additive action determines the difference in liability within the population [9]. While population-based studies [9] and twin studies [10,11,12,13] have provided compelling evidence for a role of common variation in ASD risk, the reproducibility of candidate loci has been hampered by the small sample size (and consequent limited statistical power) of early genome-wide associated studies (GWAS) [14,15,16]. Recently, a much larger GWAS meta-analysis on >18,000 ASD cases contrasted to >27,900 ancestry-matched controls has pinpointed 5 genome-wide significant loci [17].
In parallel, early studies using microarrays [14,18,19,20,21,22,23,24], whole-exome sequencing (WES) on increasingly larger cohorts [6,7,8,25,26,27,28,29,30,31,32,33,34,35,36,37], and more recently whole-genome sequencing (WGS) [38,39,40] have shown the significant contribution of rare genetic variation. Collectively, these efforts have led to the identification of hundreds of genes that when impacted by rare and disruptive genetic variants confer risk to ASD. These variants can be deletions or duplications of intragenic regions, entire genes, or multiple genes (copy number variation, CNV) [14,18,19,20,21,22,23,24,33,34,35]. Alternatively, they can impact as little as one nucleotide (single-nucleotide variation, SNV) or a few nucleotides (indels) [6,7,8,25,26,27,28,29,30,31,32,41]. CNV and SNV/indels conferring risk can be autosomal dominant [6,7,8,25,26,27,28,41], autosomal recessive [25,29], X-linked [25], or mosaic [30,31,32,42]. High-penetrance, autosomal dominant genes, in particular, have been successfully identified by sequencing ASD families, specifically trios (one affected individual and their unaffected parents) and/or quads (one affected individual, their unaffected parents, and their unaffected sibling) [6,7,8,26,28,37,41,43]. To associate discrete genes to ASD risk, early analyses [26,27,28,37] have harnessed the rarity of disruptive variants arising de novo, namely incurring in the parental gametes—in most case paternal [43,44]—and therefore detected only in the affected individual and not in their parents. These studies sequenced ~1000 ASD families and yielded 9 risk genes [26,27,28,37]. Beyond the obvious gain in power due to a larger sample size, newer analyses [6,8,41] have further boosted gene discovery by using refined association frameworks that go beyond de novo variation [45] and/or exploiting evolutionary constraint scores [8,46,47,48]. For example, a recent sequencing study has integrated de novo, inherited, and case-control variation from >35,000 exomes, >11,000 of which from individuals with an ASD diagnosis, and found 102 risk genes [8]. In addition to exome sequencing, genome sequencing is being harnessed to understand the role of rare non-coding variation in liability. A study on genomes from ~1900 quads showed that de novo mutations in promoter regions, especially in evolutionarily conserved binding sites for transcription factors located distally to the transcription start site, contribute to ASD risk [40]. Further, WGS on >2300 families with more than one child affected have been instrumental in identifying rare high-risk inherited variants, which have been elusive due to their modest effect size [38].
Analyses are now probing the interaction between rare and common variation in shaping risk architecture [49,50]. The overlap between loci identified by GWAS [17] and genes pinpointed by exome sequencing [8] is limited to one gene (KDM5B), likely due to power limitations [8].
Non-genetic risk factors, and in some cases their interactions with genetic factors, are also emerging [51,52]. One of the most replicated associations for ASD is advanced paternal age, with increased risk in the offspring of fathers in their mid-to-late 30s, further increasing with more advanced paternal ages [52,53,54,55]. The rate of de novo mutations in sperm increases with paternal age [43,44,56,57,58,59], and most de novo mutations found in ASD studies are of paternal origin [26,27]. However, studies based on simulated [60] and empirical [59] data indicate that de novo variation is likely not the major driver for the association between advanced paternal age and ASD risk. Rather, the common polygenic risk for adverse psychiatric outcomes might contribute to later age of childbearing [60,61].
3. ASD Risk Converging on Cortical Development—Evidence from Human Studies
Gene discovery has brought major advances in our understanding of the biological processes and pathways associated with risk. ASD risk genes implicated by de novo variation cluster to two main biological processes: synaptic function and gene expression, most prominently chromatin remodeling [6,8]. KDM5B, the gene with both GWAS [17] and sequencing signal [8], functions in chromatin remodeling. Some evidence for the role of the cytoskeleton has also begun to surface from de novo [8] and rare inherited [38] variation.
Transcriptomic analyses on postmortem brains sampled from unaffected individuals [62,63,64] or ASD cases contrasted with controls [65,66,67,68,69] have been instrumental to dissect the regions and developmental windows most vulnerable for ASD risk. These studies have shown that ASD genes are often co-expressed during brain development. Further, the spatiotemporal expression patterns of the risk genes implicate the mid-fetal development of the neocortex as a major nexus of risk. The master regulators of this concerted co-expression are also beginning to emerge, including the chromodomain helicase DNA binding protein 8 (CHD8) and T-Box brain transcription factor 1 (TBR1) [26,27,28,37,70,71,72,73,74,75,76,77]. For example, there is a significant enrichment of risk genes amongst the CHD8-bound genes identified in human neural stem cells (NSCs), mouse developing cortices, and human mid-fetal brain tissues [71,72] and the genes found differentially expressed in Chd8-depleted human and mouse progenitors [71,72,73,74,75]. Beyond transcriptional co-regulation, interesting findings are also emerging at the level of post-transcriptional regulation. First, postmortem brains from individuals with ASD show alterations in RNA editing, splicing, and isoform usage [65,66,68,69]. Second, ASD risk genes are enriched in targets of post-transcriptional regulators that are in turn encoded by ASD risk genes, including RNA-binding proteins FMRP (Fragile X Mental Retardation Protein) [6], CELF4 (CUGBP Elav-like family member 4) [8,78], and RBFOX1 (RNA Binding Fox-1 Homolog 1) [6,65,79]. Importantly, many of the mRNAs with altered splicing are targets of the splicing regulator RBFOX1, and 6 genes with isoform-level changes in ASD postmortem brains encode splicing factors [65]. These intriguing observations pose some risk genes as master regulators of downstream risk genes.
Within the cortex, single-cell RNA sequencing (scRNAseq) studies provide more granular resolution on cell types involved. Analyses on scRNAseq data from four fetal samples at mid-fetal development (post-conception weeks 17–18) show that ASD risk genes are predominantly expressed in developing glutamatergic neurons, both upper and lower layers, with interesting exceptions of genes highly expressed in oligodendrocyte precursors and pericytes [64]. Interestingly, ID risk genes are enriched not only in glutamatergic neurons but also in the radial glia (RG) [64]. By performing single-nucleus RNAseq on the prefrontal and anterior cingulate cortices of 15 ASD cases without co-morbid ID and 16 matched controls, Velmeshev and colleagues identified cell-type specific changes in the expression of >500 genes [67]. Interestingly, the degree of differential expression in upper-layer glutamatergic neurons or microglia was the most predictive of clinical severity [67]. Analyses of scRNAseq in human specimens have also the potential to identify species-specific changes. For example, outer radial glia (oRG) is a subtype of radial glia located in the outer subventricular zone (oSVZ) in gyrencephalic brains, such as those of primates and ferrets. The oRGC population is highly abundant in gyrencephalic brains, while it is minimal in lissencephalic species, such as rodents. This indicates that this specific type of progenitors is important for the generation of cortical folds (gyrus and sulci). Interestingly, two ASD risk genes (GFAP and TCF7L2) [8] are amongst the genes proposed to mark oRG specificity based on scRNAseq on the human cortex at gestational weeks 16–18 [80].
In parallel to sequencing studies, neuroimaging and neuroanatomical studies have provided support for structural changes in cortical development in individuals diagnosed with ASD. For example, a study on postmortem brains from 11 affected children and 11 unaffected controls in the same age range revealed focal patches of altered laminar cytoarchitecture in the prefrontal and temporal cortices of the ASD group [81]. Additional evidence for the role of cortical dynamics in ASD risk come from a prospective neuroimaging study that followed longitudinally 106 high-risk and 42 low-risk infants, whereby risk was defined based on the relationship to an older affected or unaffected sibling. Of the 106 high-risk infants, the 15 that went on to meet diagnostic criteria for ASD at 24 months displayed hyperexpansion of the cortical surface between 6 and 12 months of age, followed by a brain volume overgrowth between 12 and 24 months [82]. Further, a recent study has found that the expression of TBR1 overlaps with that of visuomotor integrators in the visual and motor cortices, showing the relevance of gene expression co-regulation at a circuit level [83].
4. ASD Risk Converging on Cortical Development—Evidence from Cellular and Rodent Models
During corticogenesis, a sequence of cellular events occurs to shape the developing cortex [84,85] (Figure 1). The first event to take place is cortical neurogenesis, at embryonic day (E) 10 in mice (33 in human). During this process, neuroepithelial progenitor cells (NPCs), which had previously undergone rapid expansion through symmetrical divisions lining the ventricular zone (VZ), transform into radial glial cells (RGCs). RGCs divide asymmetrically. In each division, RGCs give rise to another RGC, and a postmitotic neuron or an intermediate progenitor (IP). Apical IPs (aIPs) will stay at the VZ, while basal IPs (bIPs) will migrate to the SVZ. IPs divide symmetrically to form two identical neurons. Later in development, RGCs will also give rise to astrocytes and oligodendrocytes. Postmitotic neurons derived from RGCs are excitatory glutamatergic neurons called projection neurons, which project inside the cortex or to other brain regions. These neurons acquire a laminar distribution across the cortex, and morphogenesis and synaptogenesis begin to define their morphology and physiology. Inhibitory GABAergic interneurons do not develop from RGCs, but migrate from the ganglionic eminences, as discussed below.
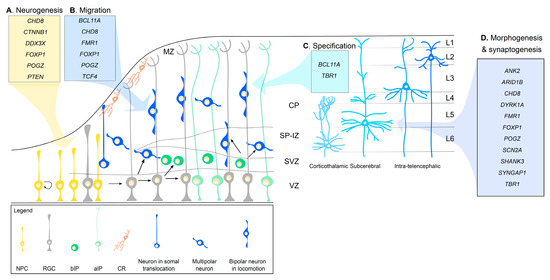
Figure 1.
Schematic representation of ASD risk genes involved in corticogenesis reviewed here. Migrating neurons and laminar specific neurons are indicated in different blue tones. NPC, neural progenitor cells; RGC, radial glial cells; bIP, basal intermediate progenitors; aIP, apical intermediate progenitors; CR, Cajal-Retzius cells; MZ, marginal zone; CP, cortical plate; SP-IZ, subplate-intermediate zone; SVZ, subventricular zone; VZ, ventricular zone; L1-6, layers 1-6. Interneurons, cortical afferents, and gliogenesis are not shown for simplicity.
In the next sections, we will discuss the evidence in rodent models implicating high-risk ASD genes [6,7,8,38] in cortical development. We note that cortical alterations have also emerged in mouse [86,87,88] and stem cell models (reviewed in [89]) of CNVs associated with ASD risk. We will not discuss the circuit changes and behavioral outcomes of the rodent models with mutations in ASD risk genes, which are discussed elsewhere [90,91,92]. We will primarily focus on the cellular and molecular functions of high-risk genes in (1) neurogenesis, including both progenitor proliferation and differentiation into neurons; (2) migration of the newborn projection neurons to the cortical plate; (3) cell specification of projection neurons, defining cortical lamination; (4) morphogenesis and synaptogenesis of projection neurons; (5) migration and development of cortical GABAergic interneurons (Figure 1, Table 1).
4.1. Neurogenesis
Alterations in neurogenesis can lead to both microcephaly and megalencephaly, which are frequently observed in ASD [93]. Proper neurogenesis requires different signaling pathways, including the Wnt, PI3K/AKT/mTOR, and Notch pathways. These pathways can lead to changes in gene expression programs, including transcription (e.g., CTNBB1, CHD8, FOXP1, POGZ) and mRNA translation (e.g., CELF4, DDX3X) (Figure 1A). While we will discuss these pathways and genes in relationship to cortical neurogenesis, they are also important in earlier neural patterning and later stages of brain development.
Beyond a well-established role in early brain patterning [94], the Wnt pathway modulates progenitor proliferation by inhibiting cell cycle exit and differentiation through the downstream component β-catenin [95]. Mutations in CTNNB1, the gene encoding for β-catenin, are associated with ASD [8], ID [96,97], and other syndromic manifestations [98] (a neurodevelopmental disorder with spastic diplegia and visual defects, OMIM 615075). Mutations or altered expression of Ctnnb1, even via manipulation of upstream regulators, affect cortical neurogenesis [95,99,100,101,102,103,104,105]. For example, transgenic mice expressing a constitutively active β-catenin have excessive RGCs, to the expenses of IPs population [99], and consequent enlargement of the ventricles [95]. Further, in utero silencing of the histone cell cycle regulator HIRA, an activator of β-catenin expression, induces terminal mitosis, thus reducing NPC proliferation in VZ/SVZ and IZ [100]. On the other hand, increased β-catenin levels also disrupt RGC proliferation, as observed in mice with a deletion in Apc, a gene necessary for β-catenin degradation [101]. In both studies, restoring β-catenin levels rescued the proliferation defects [100,101].
CHD8 [26,27,28,37] is also part of the Wnt signaling pathway [74]. Both human and mouse CHD8 are highly expressed in the cortex at the onset of neurogenesis (with a peak at E12 in mice) [74], and decrease with the progression of corticogenesis and in adulthood [74,106]. In line with these expression data, CHD8 is indispensable for cortical neurogenesis [74]. For example, in utero knockdown of Chd8 with short hairpin RNAs (shRNAs) at E13 results in a decrease in progenitors from VZ/SVZ and an increase in neurons at E16, indicating a premature depletion of the NPC pool [74]. Co-expression of an shRNA-resistant Chd8 rescued this phenotype [74]. However, several Chd8 mutant lines have consistently shown megalencephaly [75,107,108,109], which is in line with the macrocephaly observed in CHD8 patients ([110] but see also [111]). While the megalencephalic phenotype seems contradictory with the reduced progenitor proliferation, Suetterlin and colleagues have shown that the brain overgrowth starts postnatally [75]. One of the proposed mechanisms for the role of CHD8 in cortical development is the regulation of downstream genes: CHD8 is in fact an ATP-dependent chromatin remodeler enriched in the promoters of transcriptionally active genes [71,72,73]. RNA sequencing analyses on Chd8 mutant mice [74,109] and CHD8-depleted human progenitors [71,72] have shown that many downregulated genes are implicated in Wnt signaling [72,74,109] and/or cell cycle progression [71,74,109]. Further, Chd8 knockdown reduces Wnt-mediated transcriptional activity in mouse NPC in a luciferase assay, and co-expression of a resistant β-catenin construct normalized Wnt activity, the alterations in the number of NPC, and the gene expression dysregulation [74]. Further, as mentioned above, genes regulated by CHD8 are enriched in ASD risk genes [71,72,73,74,75].
DDX3X (DEAD-box helicase 3 X-linked) is another ASD risk gene [7,38,39,112] that has recently emerged as critical for cortical neurogenesis [113], possibly through interactions with the Wnt pathway. Mutations in DDX3X are associated with DDX3X syndrome (OMIM 300958), an X-linked condition that manifests with ID, brain anomalies, movement disorders, and behavioral problems, including ASD [113,114]. Ddx3x is highly expressed early during neurogenesis (E12.5–E14.5) in RGCs, and by E16.5 in both RGCs and neurons in the cortical plate (CP) [113]. In postnatal brains, Ddx3x is expressed in all cortical layers and in neurons, astrocytes, oligodendrocytes, microglia, and ependymal cells [113]. In vivo silencing of Ddx3x at E14.5 led to an increase of Pax6+ RGCs and Tbr2+ IPs, and a decrease in differentiated neurons. This imbalance is not caused by increased RGC apoptosis or reduced migration of the neurons to the CP, suggesting that Ddx3x is necessary for the differentiation of progenitors into neurons. While Ddx3x-depleted cortices have alterations in Wnt targets [113], a causal relationship has yet to be established. A more compelling mechanism relates to the role of DDX3X in modulating mRNA translation. First, DDX3X acts on specific mRNAs that are needed for neurogenesis, including RAC1 (Rac Family Small GTPase 1) [113]. Second, DDX3X regulates the formation of RNA-protein granules that are thought to affect mRNA translation, as shown by the formation of ectopic granules in the mouse neural progenitor line N2A or primary neurons over-expressing DDX3X clinical mutants [113].
Another signaling pathway crucial for cell growth, proliferation, and survival is the PI3K/AKT/mTOR pathway. PTEN (phosphatase and tensin homolog) is a tumor suppressor gene part of this cascade [115,116]. Mutations in PTEN are found in individuals with ASD [8] and are associated with an autosomal dominant macrocephaly/autism syndrome (OMIM 605309). Mouse models with conditional homozygous ablation of Pten (Pten−/−) have provided compelling evidence for a critical role of PTEN in progenitor proliferation and brain overgrowth [117]. Recent studies in Pten haploinsufficient mice (Pten+/−) are calling the attention on carefully considering gene dosage when modeling ASD mutations in rodents. Pten+/− mice display brain overgrowth that appears perinatally and is maintained throughout adulthood. This phenotype is accompanied, in early phases, by increased progenitor proliferation and excess of neurons at birth, followed by postnatal apoptosis of excess neurons and excess glia in adulthood [105]. Interestingly, there seems to be a gene dosage effect, with brain overgrowth primarily led by hyperplasia (increased cell number) in Pten+/− mice [105] and by both hyperplasia and hypertrophia (increased cell size) in Pten−/− mice [105,118,119]. These distinct phenotypes seem to track to distinct pathways: increased β-catenin signaling in Pten+/− mice, again supporting the role of β-catenin in proliferation, as opposed to both increase β-catenin and mTOR activity in Pten−/− mice [105]. Compatible with this hypothesis, brain overgrowth can be suppressed by Ctnnb1 haploinsufficiency, but not Mtor in Pten+/− mice [105], or by mTORC1 knockout [120] or inhibition [121] in Pten−/− mice.
Brain organoids derived from human and mouse embryonic stem cells engineered with homozygous deletions in PTEN also support a key role for this gene in progenitor proliferation. In the human organoids, PTEN loss resulted in increased protein kinase B (AKT) signaling, which delayed cell cycle exit resulting in prolonged proliferation of neural progenitor, including the oRG population [122]. This led to increased organoid size, outgrowth of neuroepithelial tissue, and folding [122]. Rescue of PTEN levels or AKT inhibition in mutant organoids normalized proliferation rate and neuronal differentiation. In addition to direct mutations in PTEN, also manipulating upstream regulators converge on defects in proliferation. For example, Lei and colleagues [123] described the increased proliferation of Pax6+ RGCs in VZ and reduced NPCs exiting the cell cycle in a conditional knockout mouse for Kdm6a (Lysine Demethylase 6A, also known as Utx), a lysine demethylase implicated in ID (Kabuki syndrome, OMIM 300867) affecting Pten transcription. These mutant mice have increased methylation of the Pten promoter and, consequently, decreased PTEN expression. Re-expression of PTEN rescued the abnormalities in NPC proliferation, reinforcing the importance of this phosphatase in the regulation of NPCs proliferation and differentiation [123].
Finally, ASD risk genes are also associated with the Notch signaling pathway, which modulates fate decisions in embryonic and adult NSCs [124,125]. FOXP1 (forkhead box P1) is part of a subfamily of transcription factors that temporally and spatially control the expression of a wide range of genes by binding to their promoter and enhancer sequences via a forkhead DNA binding motif [126]. FOXP1 is a high-risk gene for ASD [8] and mutations in the gene result in a neurodevelopmental condition called FOXP1 syndrome (OMIM 613670; [127] and references therein). Foxp1 expression peaks in the VZ of the mouse cortex between E10 and E12.5, and then decreases between E13.5 and E16.5 [128]. Foxp1 knockdown by in utero electroporation (IUE) results in reduced post-mitotic cortical neurons and increased IP in the intermediate zone (IZ) [129]. In line with these findings, experiments in murine cultured NSCs found that FOXP1 affects NSC differentiation into neurons and astrocytes by repressing Jagged1—a Notch ligand expressed in NSCs that is required for their maintenance in SVZ [129]. In vitro treatment with a Jagged1 inhibitor restores differentiation of Foxp1-depleted NSCs [129]. More recent findings indicate that Foxp1 is needed early during neurogenesis for the maintenance of the apical RG (aRG) pool [128]. Mice overexpressing Foxp1 had increased aRG self-renewal and their differentiation into early-born neurons at the detriment of late-born neurons and glia, while mice knocked out for Foxp1 disrupted early-born populations, with no effects on late-born neurons [128]. These observations indicate that sustained Foxp1 expression can extend the temporal window of early neurogenesis [128].
Another top ASD gene, POGZ (Pogo transposable element-derived protein with zinc finger domain; OMIM 616364; [26,27,28,37]), also favors neuronal differentiation by inhibiting the Notch signaling. IUE-mediated knockdown of Pogz leads to an increased proportion of NSCs, and reduced IPs and late-born neurons [130]. A murine Pogz rescued this phenotype, while mutants carrying ASD pathogenic mutations did not [130]. In line with these data, NSCs derived from induced pluripotent stem cells of a patient with ASD carrying a de novo missense mutation in POGZ also show increased proliferation and reduced degree of neuronal differentiation [130].
4.2. Migration
Newly generated glutamatergic neurons migrate radially from the SVZ to the preplate, situated at the pial surface. Initially, neurons acquire a unipolar morphology by extending a basal process towards the pial surface and then they translocate their nucleus and cytoplasm towards it (somal translocation). Later, neurons adopt a multipolar morphology while in the IZ that allows them to search for cell polarity cues in the environment. Then, they acquire a bipolar morphology and use the radial fiber of RGCs to migrate towards the pial surface (locomotion). At E50–75 in humans and E14 in mice, as neurogenesis progresses and more projection neurons reach the preplate, this region splits into the marginal zone and the cortical plate (CP). Postmitotic neurons continue to occupy the CP in an “inside-out” fashion, eventually forming six cortical layers (L1–L6). Early-born neurons will constitute deep layers (first L6 and later L5); late-born neurons will populate upper layers (first L4, then L3 and L2) [84,85] (Figure 1).
Defects in neuronal migration can cause cortical malformations (e.g., lissencephaly, heterotopia, polymicrogyria) that are frequent in individuals with ASD [81,113,131,132,133,134,135,136]. Not surprisingly, neuronal migration is regulated by several ASD genes [106,137,138,139] (Figure 1B). Some of these genes can impact proliferation as well, making it difficult to disentangle the impact of earlier proliferation defects on migration. For example, CHD8 regulates both proliferation (see above, [74]) and migration [106]. Interestingly, Chd8 downregulation during embryonic stages does not totally abolish neuronal migration, but it delays it until the early postnatal period [106]. Similarly, the ASD risk gene TCF4 (transcription factor 4) [8], associated with Pitt–Hopkins syndrome (OMIM 610954), regulates both proliferation and migration, as shown in a Tcf4 haploinsufficient mouse model [140]. Matsumura and colleagues [130] described that knockdown of Pogz delayed migration to the CP, but that defect tracked down to impaired neuronal differentiation (see above). Importantly, the study also showed that mice engineered to carry a de novo missense mutation found in a patient with ASD had mislocalization of upper-layer neurons into lower layers, a condition that was maintained until adulthood. Interestingly, this mutation did not affect GABA+ interneurons [130].
One of the emerging complications in ASD is that some risk genes might act with both a loss and a gain-of-function mechanism [8], as in the case of FOXP1 syndrome [127]. Additionally, there are other ASD risk genes where loss-of-function and missense mutations yield different clinical severity (e.g., DDX3X [113]) or where mutations in the same gene segregate into distinct biological signatures (e.g., ADNP [141]). In the case of Foxp1, loss-of-function and gain-of-function mutations appear to impact cortical migration [137,142]. Mice with forebrain-specific knockout of Foxp1 present reduced migration of late-born neurons from the SVZ/VZ to CP. The defect could be rescued by wild-type FOXP1, but not a mutant preventing sumoylation [137]. Interestingly, sumoylation modulates the interaction of FOXP1 with the chromatin remodeling complex NuRD, thus likely affecting gene expression [137]. Further, cortical migration was impacted by the expression of a Foxp1 mutant mouse homolog to the human recurrent nonsense mutation p.Arg525* ([127] and references therein) [142]. In both human cell lines [143,144] and mouse progenitors in vivo [142], the over-expressed mutant Foxp1 fails to reach the nucleus and form cytoplasmic aggregates. In utero expression of the mutant Foxp1 resulted in a higher proportion of cells with multipolar morphology in the IZ, and a consequent decrease of postmitotic neurons reaching the CP [142].
Other ASD risk genes, including the BAF chromatin remodeling complex subunit BCL11A (MIM 617101, [6,145]) and FMR1 (MIM 300624), are necessary for the switch of newborn neurons from multipolar to bipolar in the IZ. Failing in this process also underlies the delayed migration of late-born neurons to the upper layers in Bcl11a-deficient mice [139] and Fmr1 knockout mice [138]. Impaired multipolar-to-bipolar transition is driven by increased Sema3c expression resulting from Bcl11a loss [139] or decreased Chd2 levels in Fmr1 knockout mice [138]. In both of these models, the genetic manipulations did not affect progenitor proliferation or differentiation, indicating that the migration phenotype was not a secondary effect of earlier alterations.
4.3. Laminar Specification
Laminar specification is an essential process for shaping the cortex into six layers, completed at around 7 months of gestation in humans and at postnatal day 7 in mice. With cell fate specification, the developing glutamatergic projection neurons acquire subtype specificity. Two main subtypes of excitatory neurons are generated: corticofugal and intra-telencephalic projections neurons. Corticofugal projection neurons extend their axons away from the cortex into the thalamus (corticothalamic projection neurons, mostly in L6) or subcerebral nuclei (subcerebral projection neurons, mostly in L5). On the contrary, intra-telencephalic projection neurons (mostly located in upper L2 and L3) extend their axons within the cortex, to the ipsilateral (associative projection neurons) or contralateral (commissural projection neurons) hemisphere. Most of the latter projections cross through the corpus callosum (callosal projection neurons) (Figure 1).
Cell type specification is orchestrated by a set of transcription factors [146,147,148,149,150,151,152,153,154,155,156,157,158,159] (Figure 1C). Some of these transcription factors are associated with ASD, including BCL11A and TBR1 (OMIM 606053, [8,160,161]). In addition to regulating migration [139], Bcl11a contributes to specifying subcerebral projection neurons by repressing Tbr1 [159]. In fact, the IUE-mediated knockdown of Bcl11a at E13.5 results in an increase of TBR1+ neurons and a reduction of BCL11B+ neurons in the postnatal cortex [159]. Further, Bcl11a-deficient mice show a reduced pyramidal tract due to defective development of the corticospinal projections, and this defect can be rescued by increasing Bcl11b expression or silencing Tbr1 [159]. Tbr1 is a major driver of corticothalamic specification [70,158,162,163,164]. In fact, L6 neurons in the cortex of Tbr1 null mice misroute their axons to aberrantly innervate the corticospinal tract [162]. Further, L6 neurons in a mouse with conditional Tbr1 ablation show a shift towards L5 identity, with repercussions on, among others, dendritic patterning (see below) [70]. Recently, post-transcriptional mechanisms influencing specification have also begun to emerge [78,165,166], casting a scenario where proper corticogenesis requires a fine-tuning of gene expression.
4.4. Morphogenesis and Synaptogenesis
During early mid-fetal development, after neurogenesis has peaked and migration has begun, dendritogenesis [167,168], axonogenesis [169], and synaptogenesis [170] ensue. Although these three processes start prenatally, they remain active until adolescence and early adulthood to refine brain circuits. In the neocortex, dendritogenesis and axonogenesis begin when a neuron is in the IZ, after leaving the VZ, and obtains multipolar morphology. At this stage, neurite polarity is determined, where one process extends tangentially within the IZ to become the axon and another process extends towards the pial surface, serving as the leading process for radial migration, and will become the apical dendrite once the neuron has migrated to the CP. Basal dendrites sprout from the soma perpendicular to the pial surface and grow towards the VZ. As the apical and basal dendrites elongate, they will arborize creating many branched processes and synaptogenesis will then begin to form (Figure 1). Throughout childhood and adolescence, long thin and filopodia-like protrusions (immature spines) will be pruned and stabilized to stubby and mushroom-like protrusions (mature spines) to refine brain circuits [170,171].
There is a large body of evidence implicating altered neuronal morphogenesis and synaptogenesis in ASD risk [171,172] (Figure 1D). ASD risk genes that have been recently implicated in axonogenesis include ankyrin 2 (ANK2), CHD8, and the dual-specificity tyrosine phosphorylation regulated kinase 1A (DYRK1A; OMIM 614104) [6,8,26,27,28,37]. Cultured hippocampal neurons from a mouse expressing an Ank2 carrying an ASD-pathogenic mutation show increased axon branching, without a concomitant change in axon length or dendritic arborization [173]. The proposed mechanism suggests that Ank2 functions in controlling axon branching by suppressing microtubule invasion into newly formed filopodia [173]. However, this did not translate into gross differences of the major axon tract, including the corpus callosum [173], but resulted in a more subtle increase in cortical connectivity. Suppression of callosal projections, on the contrary, is observed in cortices electroporated with an anti-Chd8 shRNA [106]. One potential mechanism is the Chd8-dependent regulation of genes implicated in axonal development, as detected in human neural progenitor cells [72]. Furthermore, knocking down Dyrk1a or overexpressing it as wild-type or missense mutants homologous to those found in ASD individuals reduce the axonal length in mouse cortical neurons, revealing that the dosage of Dyrk1a is critical for proper neurite and axon development [174].
Dendritogenesis requires an intact expression of ASD risk genes. In addition to defective axonogenesis, Chd8 knockdown in L2/3 pyramidal neurons results in reduced dendritic density and complexity [74,106], which could be alleviated upon induction of the Wnt signaling [74]. ARID1B (AT-rich interaction domain 1B; OMIM 135900, [6,8]) is necessary for the proper growth of apical and basal dendrites, as Arid1b-deficient cortical mouse neurons display shorter dendrites and defective orientation of basal dendrites, but not apical dendrites, [175]. Arid1b-deficient L2–4 pyramidal neurons show smooth apical dendrites without branches and a reduced number of primary, secondary, and tertiary apical dendrites [175]. However, these results were obtained with shRNA-mediated knockdown and were not replicated in an Arid1b haploinsufficient mouse [176], emphasizing again the importance of taking dosage into account. One example where dendritogenesis defects are secondary to altered cell identity relates to TBR1, which, as discussed above, drives corticothalamic specification in L6 [70,158,162,163,164]. L6 pyramidal neurons with conditional haploinsufficiency of Tbr1 extend their apical dendrites to L1 and L2/3 instead of growing into L4. These defects in dendritic patterning indicate that L6 neurons have L5-like properties [70], further corroborating the importance of Tbr1 for identity specification (see above). Dendritogenesis of L5 neurons requires, among other factors, Foxp1, as the embryonic expression of an ASD-relevant Foxp1 mutant causes shorter apical dendrite in mature L5 pyramidal neurons [142]. FOXP1 sumoylation is critical for its role in dendritic morphogenesis [137,177]. For example, the ablation of Foxp1 in rat cortical neurons results in dendritic malformation and shorter dendrites, with no change in axonal length [177]. This phenotype can be rescued with wild-type FOXP1, but not with a non-sumoylatable FOXP1 mutant [177].
As mentioned, the synaptic function is a nexus of liability for ASD [6,8,170,171]. Mutations in many risk genes encoding synaptic proteins, including scaffolding protein SynGAP (encoded by SYNGAP1) [178,179,180] and SHANK3 [181,182], glutamate ionotropic receptor GluN2B (encoded by GRIN2B) [183], voltage-gated sodium channel Nav1.2 (encoded by SCN2A) [184], synaptic translational regulator FMRP (encoded by FMR1) [185,186] all result in defects in the formation and/or the maturation of the synapses. Mutations in ASD risk genes encoding transcription factors can also impact synaptogenesis. For example, heterozygous mice for an ASD-associated mutation in Pogz show an increased density of dendritic spines in L2/3 pyramidal neurons [130]. Mice haploinsufficient for Tbr1 in L5 or L6 have, on the contrary, reduced density of mature excitatory and inhibitory synapses accompanied by an exuberance of immature filopodia, [70,76], indicating suppressed maturation.
4.5. Migration and Development of Cortical Interneurons
Cortical inhibitory interneurons develop predominantly from areas in the subpallium: the medial and caudal ganglionic eminences (MGE and CGE, respectively) of the ventral telencephalon and the preoptic region [187]. After differentiation—a process based on both place of origin and time—the interneurons migrate tangentially to target locations in the neocortex, populating all cortical layers, where they act as regulators of glutamatergic projection neurons by providing local inhibition. The most common types of interneurons in the cortex are parvalbumin (PV), somatostatin (SST), and ionotropic serotonin receptor 5HT3a (5HT3aR) cells (reviewed in [187]). Each of these subtypes has different laminar distributions, in addition to their own subgroups of cells based on morphology and function, creating a diverse array of critical cortical cell populations.
The mouse homolog of the risk gene CHD2 (chromodomain helicase DNA binding protein 2; OMIM 615369, [8,188]) controls the genesis of cortical interneurons [189]. In the somatosensory cortex of Chd2 haploinsufficient mice, there is a significant reduction in the density of PV and SST neurons, due, at least partially, to a reduction in interneuron progenitors expressing Nkx2-1 in the MGE [189]. Nkx2-1 is fundamental for MGE neurogenesis [190,191] and evidence in human embryonic stem cells indicates that this gene is directly regulated by CHD2 [192]. The lamination of the interneurons is altered in Tbr1 mutants [70,77]. A mouse heterozygous for an ASD-associated missense mutation displays a disproportional distribution of PV interneurons in the deeper cortical layers [77], with no changes in other interneurons populations. However, Tbr1 haploinsufficiency in L6 results in a similarly abnormal patterning, but only at the expense of SST interneurons [70]. The mechanisms behind these changes in the tangential migration of interneurons from deep to superficial cortical layers—and thus the reasons behind the incongruences in cell type between the two studies [70,77]—are still unknown, but might be potentially due to alterations of signaling from the excitatory signal. An astonishing example of how interneurons can be regulated by an ASD gene in a non-cell-autonomous fashion relates to PTEN. Wong and colleagues found that interneurons in the primary somatosensory cortex undergo cell death between postnatal days 5 and 10 [193]. Interneurons that received robust excitatory inputs from pyramidal cells at the beginning of this interval escaped cell death, indicating that pyramidal cell activity orchestrates interneuron survival [193]. This regulation requires Pten: (1) PTEN levels were significantly decreased in interneurons following the activation of pyramidal cells at postnatal day 8; and, (2) there was a higher density of PV and SST interneurons after postnatal day 10 in a mouse with ablation of Pten in postmitotic MGE interneurons or with pharmacological inhibition of PTEN [193].
While all of these ASD risk genes affect both pyramidal and interneurons populations in the developing cortex, there is one gene, ARID1B, which seems to affect most predominantly the birth and migration of interneurons. Haploinsufficiency of Arid1b in mice has been shown to suppress the generation of inhibitory interneurons both in vivo and in vitro, due to the combination of decreased proliferation and increased apoptosis of ventral progenitor cells in the MGE [176]. One proposed mechanism relates to the direct regulation of the Pvalb gene (encoding parvalbumin) [176]. These alterations are not accompanied by changes in the density or lamination of cortical excitatory neurons (see above for a discussion about phenotypes in Arid1b-deficient pyramidal neurons; [175]). In support that Arid1b pathophysiology depends on the interneuron populations is a recent study using mice with Arid1b haploinsufficiency selectively in PV or SST interneurons [194]. While both models displayed behavioral phenotypes relevant to ASD, they did have distinct traits, with the PV-specific haploinsufficient mice showing sociability and social recognition deficits, anxiety, and a depression-like phenotype, and the SST-specific haploinsufficient mice exhibiting excessive repetitive behaviors [194].
5. Conclusions
In this review, we argue that findings in rodent models, along with human genetic, imaging, and neuroanatomical evidence, pinpoint neocortical development as a critical spatiotemporal window for ASD liability. Most ASD risk genes are involved in multiple steps of corticogenesis, emphasizing again the heterogeneity of the risk architecture (Table 1).

Table 1.
Summary of the findings discussed in the review. Acronyms in the glossary.
Many risk genes are functionally interrelated: perturbations in one risk gene can have repercussions on the expression of other downstream risk genes. Tackling the function of such master regulators of other risk genes is a promising future avenue to understand convergences and divergences between risk genes. Further, observations in human stem cell-derived models (e.g., organoids) will be key to identify species-specific alterations that define the unique features of social behavior in humans.
Author Contributions
Conceptualization: S.D.R., M.G.-F. and A.B.; writing—original draft: S.D.R., M.G.-F., A.B.; writing—review and editing: S.D.R., M.G.F., A.B. and Z.A. All authors have read and agreed to the published version of the manuscript.
Funding
M.G.-F. is supported by the Beatrice and Samuel A. Seaver Foundation. S.D.R. is supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number R21HD097561. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S.D.R. is also supported by the Beatrice and Samuel A. Seaver Foundation and is the recipient of the 2020 Fascitelli Research Scholar Award.
Conflicts of Interest
The authors declare no conflict of interest.
Glossary
| ADHD | attention deficit hyperactivity disorder |
| aIP | apical IP |
| ASD | autism spectrum disorder |
| bIP | basal IP |
| CGE | caudal ganglionic eminence |
| CNV | copy number variation |
| CP | cortical plate |
| GWAS | genome-wide association studies |
| ID | intellectual disability |
| IP | intermediate progenitor |
| IUE | in utero electroporation |
| IZ | intermediate zone |
| KD | knockdown |
| KI | knockin |
| KO | knockout |
| L1-6 | cortical layers 1-6 |
| MGE | medial ganglionic eminences |
| NPCs | neural progenitor cells |
| NSCs | neural stem cells |
| OMIM | Online Mendelian Inheritance in Man (https://www.omim.org) |
| oRG | outer radial glia |
| oSVZ | outer subventricular zone |
| PV | parvalbumin |
| RGC | radial glial cells |
| scRNAseq | single-cell RNA sequencing |
| shRNA | short hairpin RNA |
| SNP | single-nucleotide polymorphism |
| SNV | single-nucleotide variation |
| SP-IZ | subplate-intermediate zone |
| SST | somatostatin |
| SVZ | subventricular zone |
| VZ | ventricular zone |
| WES | whole-exome sequencing |
References
- Szatmari, P.; Georgiades, S.; Duku, E.; Bennett, T.A.; Bryson, S.; Fombonne, E.; Mirenda, P.; Roberts, W.; Smith, I.M.; Vaillancourt, T.; et al. Developmental trajectories of symptom severity and adaptive functioning in an inception cohort of preschool children with autism spectrum disorder. JAMA Psychiatry 2015, 72, 276–283. [Google Scholar] [CrossRef]
- Waizbard-Bartov, E.; Ferrer, E.; Young, G.S.; Heath, B.; Rogers, S.; Wu Nordahl, C.; Solomon, M.; Amaral, D.G. Trajectories of Autism Symptom Severity Change During Early Childhood. J. Autism. Dev. Disord. 2020. [Google Scholar] [CrossRef] [PubMed]
- McDonald, N.M.; Senturk, D.; Scheffler, A.; Brian, J.A.; Carver, L.J.; Charman, T.; Chawarska, K.; Curtin, S.; Hertz-Piccioto, I.; Jones, E.J.H.; et al. Developmental Trajectories of Infants With Multiplex Family Risk for Autism: A Baby Siblings Research Consortium Study. JAMA Neurol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tager-Flusberg, H.; Kasari, C. Minimally verbal school-aged children with autism spectrum disorder: The neglected end of the spectrum. Autism. Res. 2013, 6, 468–478. [Google Scholar] [CrossRef]
- Doshi-Velez, F.; Ge, Y.; Kohane, I. Comorbidity clusters in autism spectrum disorders: An electronic health record time-series analysis. Pediatrics 2014, 133, e54–e63. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef]
- Ritvo, E.R.; Freeman, B.J.; Mason-Brothers, A.; Mo, A.; Ritvo, A.M. Concordance for the syndrome of autism in 40 pairs of afflicted twins. Am. J. Psychiatry 1985, 142, 74–77. [Google Scholar] [PubMed]
- Steffenburg, S.; Gillberg, C.; Hellgren, L.; Andersson, L.; Gillberg, I.C.; Jakobsson, G.; Bohman, M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J. Child. Psychol. Psychiatry 1989, 30, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Folstein, S.; Rutter, M. Infantile autism: A genetic study of 21 twin pairs. J. Child. Psychol. Psychiatry 1977, 18, 297–321. [Google Scholar] [CrossRef] [PubMed]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.A.; Arking, D.E.; Daly, M.J.; Chakravarti, A. A genome-wide linkage and association scan reveals novel loci for autism. Nature 2009, 461, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Anney, R.; Klei, L.; Pinto, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; Sykes, N.; Pagnamenta, A.T.; et al. A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet. 2010, 19, 4072–4082. [Google Scholar] [CrossRef]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef]
- Devlin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Ronemus, M.; Yamrom, B.; Lee, Y.H.; Leotta, A.; Kendall, J.; Marks, S.; Lakshmi, B.; Pai, D.; Ye, K.; et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 2011, 70, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef]
- Lim, E.T.; Raychaudhuri, S.; Sanders, S.J.; Stevens, C.; Sabo, A.; MacArthur, D.G.; Neale, B.M.; Kirby, A.; Ruderfer, D.M.; Fromer, M.; et al. Rare complete knockouts in humans: Population distribution and significant role in autism spectrum disorders. Neuron 2013, 77, 235–242. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef]
- Doan, R.N.; Lim, E.T.; De Rubeis, S.; Betancur, C.; Cutler, D.J.; Chiocchetti, A.G.; Overman, L.M.; Soucy, A.; Goetze, S.; Autism Sequencing, C.; et al. Recessive gene disruptions in autism spectrum disorder. Nat. Genet. 2019, 51, 1092–1098. [Google Scholar] [CrossRef]
- Lim, E.T.; Uddin, M.; De Rubeis, S.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D’Gama, A.M.; Kim, S.N.; Hill, R.S.; Goldberg, A.P.; et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci. 2017, 20, 1217–1224. [Google Scholar] [CrossRef]
- Krupp, D.R.; Barnard, R.A.; Duffourd, Y.; Evans, S.A.; Mulqueen, R.M.; Bernier, R.; Riviere, J.B.; Fombonne, E.; O’Roak, B.J. Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am. J. Hum. Genet. 2017, 101, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Yang, X.; Li, Z.; Wang, S.; Zhang, Z.; Ye, A.Y.; Yan, L.; Yang, C.; Wu, Q.; Li, J.; et al. Postzygotic single-nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations. Hum. Mutat. 2017, 38, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Poultney, C.S.; Goldberg, A.P.; Drapeau, E.; Kou, Y.; Harony-Nicolas, H.; Kajiwara, Y.; De Rubeis, S.; Durand, S.; Stevens, C.; Rehnstrom, K.; et al. Identification of Small Exonic CNV from Whole-Exome Sequence Data and Application to Autism Spectrum Disorder. Am. J. Hum. Genet. 2013, 93, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Krumm, N.; O’Roak, B.J.; Karakoc, E.; Mohajeri, K.; Nelson, B.; Vives, L.; Jacquemont, S.; Munson, J.; Bernier, R.; Eichler, E.E. Transmission disequilibrium of small CNVs in simplex autism. Am. J. Hum. Genet. 2013, 93, 595–606. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fromer, M.; Moran, J.L.; Chambert, K.; Banks, E.; Bergen, S.E.; Ruderfer, D.M.; Handsaker, R.E.; McCarroll, S.A.; O’Donovan, M.C.; Owen, M.J.; et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 2012, 91, 597–607. [Google Scholar] [CrossRef]
- Feliciano, P.; Zhou, X.; Astrovskaya, I.; Turner, T.N.; Wang, T.; Brueggeman, L.; Barnard, R.; Hsieh, A.; Snyder, L.G.; Muzny, D.M.; et al. Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom. Med. 2019, 4, 19. [Google Scholar] [CrossRef]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef]
- Ruzzo, E.K.; Perez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178, 850–866.e26. [Google Scholar] [CrossRef]
- Yuen, R.K.; Merico, D.; Bookman, M.; Howe, J.L.; Thiruvahindrapuram, B.; Patel, R.V.; Whitney, J.; Deflaux, N.; Bingham, J.; Wang, Z.; et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 2017, 20, 602–611. [Google Scholar] [CrossRef]
- An, J.Y.; Lin, K.; Zhu, L.; Werling, D.M.; Dong, S.; Brand, H.; Wang, H.Z.; Zhao, X.; Schwartz, G.B.; Collins, R.L.; et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 2018, 362. [Google Scholar] [CrossRef]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [PubMed]
- Freed, D.; Pevsner, J. The Contribution of Mosaic Variants to Autism Spectrum Disorder. PLoS Genet. 2016, 12, e1006245. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Walker, M.F.; Carriero, N.J.; DiCola, M.; Willsey, A.J.; Ye, A.Y.; Waqar, Z.; Gonzalez, L.E.; Overton, J.D.; Frahm, S.; et al. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell. Rep. 2014, 9, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Sanders, S.J.; Liu, L.; De Rubeis, S.; Lim, E.T.; Sutcliffe, J.S.; Schellenberg, G.D.; Gibbs, R.A.; Daly, M.J.; Buxbaum, J.D.; et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013, 9, e1003671. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Kosmicki, J.A.; Samocha, K.E.; Howrigan, D.P.; Sanders, S.J.; Slowikowski, K.; Lek, M.; Karczewski, K.J.; Cutler, D.J.; Devlin, B.; Roeder, K.; et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 2017, 49, 504–510. [Google Scholar] [CrossRef]
- Robinson, E.B.; Samocha, K.E.; Kosmicki, J.A.; McGrath, L.; Neale, B.M.; Perlis, R.H.; Daly, M.J. Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc. Natl. Acad. Sci. USA 2014, 111, 15161–15165. [Google Scholar] [CrossRef]
- Weiner, D.J.; Wigdor, E.M.; Ripke, S.; Walters, R.K.; Kosmicki, J.A.; Grove, J.; Samocha, K.E.; Goldstein, J.I.; Okbay, A.; Bybjerg-Grauholm, J.; et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 2017, 49, 978–985. [Google Scholar] [CrossRef]
- Janecka, M.; Kodesh, A.; Levine, S.Z.; Lusskin, S.I.; Viktorin, A.; Rahman, R.; Buxbaum, J.D.; Schlessinger, A.; Sandin, S.; Reichenberg, A. Association of Autism Spectrum Disorder With Prenatal Exposure to Medication Affecting Neurotransmitter Systems. JAMA Psychiatry 2018, 75, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Janecka, M.; Mill, J.; Basson, M.A.; Goriely, A.; Spiers, H.; Reichenberg, A.; Schalkwyk, L.; Fernandes, C. Advanced paternal age effects in neurodevelopmental disorders-review of potential underlying mechanisms. Transl. Psychiatry 2017, 7, e1019. [Google Scholar] [CrossRef] [PubMed]
- Reichenberg, A.; Gross, R.; Weiser, M.; Bresnahan, M.; Silverman, J.; Harlap, S.; Rabinowitz, J.; Shulman, C.; Malaspina, D.; Lubin, G.; et al. Advancing paternal age and autism. Arch. Gen. Psychiatry 2006, 63, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Schendel, D.; Magnusson, P.; Hultman, C.; Suren, P.; Susser, E.; Gronborg, T.; Gissler, M.; Gunnes, N.; Gross, R.; et al. Autism risk associated with parental age and with increasing difference in age between the parents. Mol. Psychiatry 2016, 21, 693–700. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.J.; Petersen, L.; Agerbo, E.; Mors, O.; Mortensen, P.B.; Pedersen, C.B. A comprehensive assessment of parental age and psychiatric disorders. JAMA Psychiatry 2014, 71, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, J.M.; Wong, W.S.; Pinelli, M.; Farrah, T.; Bodian, D.; Stittrich, A.B.; Glusman, G.; Vissers, L.E.; Hoischen, A.; Roach, J.C.; et al. Parent-of-origin-specific signatures of de novo mutations. Nat. Genet. 2016, 48, 935–939. [Google Scholar] [CrossRef]
- Jonsson, H.; Sulem, P.; Kehr, B.; Kristmundsdottir, S.; Zink, F.; Hjartarson, E.; Hardarson, M.T.; Hjorleifsson, K.E.; Eggertsson, H.P.; Gudjonsson, S.A.; et al. Parental influence on human germline de novo mutations in 1548 trios from Iceland. Nature 2017, 549, 519–522. [Google Scholar] [CrossRef]
- Rahbari, R.; Wuster, A.; Lindsay, S.J.; Hardwick, R.J.; Alexandrov, L.B.; Turki, S.A.; Dominiczak, A.; Morris, A.; Porteous, D.; Smith, B.; et al. Timing, rates and spectra of human germline mutation. Nat. Genet. 2016, 48, 126–133. [Google Scholar] [CrossRef]
- Taylor, J.L.; Debost, J.P.G.; Morton, S.U.; Wigdor, E.M.; Heyne, H.O.; Lal, D.; Howrigan, D.P.; Bloemendal, A.; Larsen, J.T.; Kosmicki, J.A.; et al. Paternal-age-related de novo mutations and risk for five disorders. Nat. Commun. 2019, 10, 3043. [Google Scholar] [CrossRef]
- Gratten, J.; Wray, N.R.; Peyrot, W.J.; McGrath, J.J.; Visscher, P.M.; Goddard, M.E. Risk of psychiatric illness from advanced paternal age is not predominantly from de novo mutations. Nat. Genet. 2016, 48, 718–724. [Google Scholar] [CrossRef]
- Mehta, D.; Tropf, F.C.; Gratten, J.; Bakshi, A.; Zhu, Z.; Bacanu, S.A.; Hemani, G.; Magnusson, P.K.; Barban, N.; Esko, T.; et al. Evidence for Genetic Overlap Between Schizophrenia and Age at First Birth in Women. JAMA Psychiatry 2016, 73, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Werling, D.M.; Pochareddy, S.; Choi, J.; An, J.Y.; Sheppard, B.; Peng, M.; Li, Z.; Dastmalchi, C.; Santpere, G.; Sousa, A.M.M.; et al. Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex. Cell. Rep. 2020, 31, 107489. [Google Scholar] [CrossRef]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Polioudakis, D.; de la Torre-Ubieta, L.; Langerman, J.; Elkins, A.G.; Shi, X.; Stein, J.L.; Vuong, C.K.; Nichterwitz, S.; Gevorgian, M.; Opland, C.K.; et al. A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron 2019, 103, 785–801.e8. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef]
- Tran, S.S.; Jun, H.I.; Bahn, J.H.; Azghadi, A.; Ramaswami, G.; Van Nostrand, E.L.; Nguyen, T.B.; Hsiao, Y.E.; Lee, C.; Pratt, G.A.; et al. Widespread RNA editing dysregulation in brains from autistic individuals. Nat. Neurosci. 2019, 22, 25–36. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Swarup, V.; Belgard, T.G.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppa, V.; Ubieta, L.T.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef]
- Darbandi, S.F.; Schwartz, S.E.R.; Qi, Q.; Catta-Preta, R.; Pai, E.L.; Mandell, J.D.; Everitt, A.; Rubin, A.; Krasnoff, R.A.; Katzman, S.; et al. Neonatal Tbr1 Dosage Controls Cortical Layer 6 Connectivity. Neuron 2018, 100, 831–845.e7. [Google Scholar] [CrossRef]
- Cotney, J.; Muhle, R.A.; Sanders, S.J.; Liu, L.; Willsey, A.J.; Niu, W.; Liu, W.; Klei, L.; Lei, J.; Yin, J.; et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun. 2015, 6, 6404. [Google Scholar] [CrossRef] [PubMed]
- Sugathan, A.; Biagioli, M.; Golzio, C.; Erdin, S.; Blumenthal, I.; Manavalan, P.; Ragavendran, A.; Brand, H.; Lucente, D.; Miles, J.; et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4468–E4477. [Google Scholar] [CrossRef] [PubMed]
- Subtil-Rodriguez, A.; Vazquez-Chavez, E.; Ceballos-Chavez, M.; Rodriguez-Paredes, M.; Martin-Subero, J.I.; Esteller, M.; Reyes, J.C. The chromatin remodeller CHD8 is required for E2F-dependent transcription activation of S-phase genes. Nucleic Acids Res. 2014, 42, 2185–2196. [Google Scholar] [CrossRef]
- Durak, O.; Gao, F.; Kaeser-Woo, Y.J.; Rueda, R.; Martorell, A.J.; Nott, A.; Liu, C.Y.; Watson, L.A.; Tsai, L.H. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci. 2016, 19, 1477–1488. [Google Scholar] [CrossRef]
- Suetterlin, P.; Hurley, S.; Mohan, C.; Riegman, K.L.H.; Pagani, M.; Caruso, A.; Ellegood, J.; Galbusera, A.; Crespo-Enriquez, I.; Michetti, C.; et al. Altered Neocortical Gene Expression, Brain Overgrowth and Functional Over-Connectivity in Chd8 Haploinsufficient Mice. Cereb. Cortex. 2018, 28, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Darbandi, S.F.; Schwartz, S.E.R.; Pai, E.L.; Everitt, A.; Turner, M.L.; Cheyette, B.N.R.; Willsey, A.J.; State, M.W.; Sohal, V.S.; Rubenstein, J.L.R. Enhancing WNT Signaling Restores Cortical Neuronal Spine Maturation and Synaptogenesis in Tbr1 Mutants. Cell. Rep. 2020, 31, 107495. [Google Scholar] [CrossRef]
- Yook, C.; Kim, K.; Kim, D.; Kang, H.; Kim, S.G.; Kim, E.; Kim, S.Y. A TBR1-K228E Mutation Induces Tbr1 Upregulation, Altered Cortical Distribution of Interneurons, Increased Inhibitory Synaptic Transmission, and Autistic-Like Behavioral Deficits in Mice. Front. Mol. Neurosci. 2019, 12, 241. [Google Scholar] [CrossRef]
- Popovitchenko, T.; Park, Y.; Page, N.F.; Luo, X.; Krsnik, Z.; Liu, Y.; Salamon, I.; Stephenson, J.D.; Kraushar, M.L.; Volk, N.L.; et al. Translational derepression of Elavl4 isoforms at their alternative 5′ UTRs determines neuronal development. Nat. Commun. 2020, 11, 1674. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Damianov, A.; Lin, C.H.; Fontes, M.; Parikshak, N.N.; Anderson, E.S.; Geschwind, D.H.; Black, D.L.; Martin, K.C. Cytoplasmic Rbfox1 Regulates the Expression of Synaptic and Autism-Related Genes. Neuron 2016, 89, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Pollen, A.A.; Nowakowski, T.J.; Chen, J.; Retallack, H.; Sandoval-Espinosa, C.; Nicholas, C.R.; Shuga, J.; Liu, S.J.; Oldham, M.C.; Diaz, A.; et al. Molecular identity of human outer radial glia during cortical development. Cell 2015, 163, 55–67. [Google Scholar] [CrossRef]
- Stoner, R.; Chow, M.L.; Boyle, M.P.; Sunkin, S.M.; Mouton, P.R.; Roy, S.; Wynshaw-Boris, A.; Colamarino, S.A.; Lein, E.S.; Courchesne, E. Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med. 2014, 370, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Hazlett, H.C.; Gu, H.; Munsell, B.C.; Kim, S.H.; Styner, M.; Wolff, J.J.; Elison, J.T.; Swanson, M.R.; Zhu, H.; Botteron, K.N.; et al. Early brain development in infants at high risk for autism spectrum disorder. Nature 2017, 542, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Bueicheku, E.; Aznarez-Sanado, M.; Diez, I.; Uquillas, F.D.; Ortiz-Teran, L.; Qureshi, A.Y.; Sunol, M.; Basaia, S.; Ortiz-Teran, E.; Pastor, M.A.; et al. Central neurogenetic signatures of the visuomotor integration system. Proc. Natl. Acad. Sci. USA 2020, 117, 6836–6843. [Google Scholar] [CrossRef]
- De Rubeis, S.; Roeder, K.; Devlin, B. Charney & Nestler’s Neurobiology of Mental Illness. In Neurodevelopmental Mechanisms of Pediatric Psychiatric Disorders Animal and Human Studies; Charney, D.S., Nestler, E.J., Sklar, P., Buxbaum, J.D., Eds.; Oxford University Press: Oxford, UK, 2017. [Google Scholar] [CrossRef]
- Bystron, I.; Blakemore, C.; Rakic, P. Development of the human cerebral cortex: Boulder Committee revisited. Nat. Rev. Neurosci. 2008, 9, 110–122. [Google Scholar] [CrossRef]
- Pucilowska, J.; Vithayathil, J.; Tavares, E.J.; Kelly, C.; Karlo, J.C.; Landreth, G.E. The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J. Neurosci. 2015, 35, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Nakai, N.; Nagano, M.; Saitow, F.; Watanabe, Y.; Kawamura, Y.; Kawamoto, A.; Tamada, K.; Mizuma, H.; Onoe, H.; Watanabe, Y.; et al. Serotonin rebalances cortical tuning and behavior linked to autism symptoms in 15q11-13 CNV mice. Sci. Adv. 2017, 3, e1603001. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Yokoyama, K.; Seiriki, K.; Naka, Y.; Matsumura, K.; Kondo, M.; Yamamoto, K.; Hayashida, M.; Kasai, A.; Ago, Y.; et al. Psychiatric-disorder-related behavioral phenotypes and cortical hyperactivity in a mouse model of 3q29 deletion syndrome. Neuropsychopharmacology 2019, 44, 2125–2135. [Google Scholar] [CrossRef]
- Gordon, A.; Geschwind, D.H. Human in vitro models for understanding mechanisms of autism spectrum disorder. Mol. Autism. 2020, 11, 26. [Google Scholar] [CrossRef]
- Golden, C.E.; Buxbaum, J.D.; De Rubeis, S. Disrupted circuits in mouse models of autism spectrum disorder and intellectual disability. Curr. Opin. Neurobiol. 2018, 48, 106–112. [Google Scholar] [CrossRef]
- Sullivan, J.M.; De Rubeis, S.; Schaefer, A. Convergence of spectrums: Neuronal gene network states in autism spectrum disorder. Curr. Opin. Neurobiol. 2019, 59, 102–111. [Google Scholar] [CrossRef]
- Drapeau, E.; Harony-Nicolas, H.; Crawley, J.N. Charney & Nestler’s Neurobiology of Mental Illness. In Animal and Cellular Models of Pediatric Psychiatric Disorders; Charney, D.S., Nestler, E.J., Sklar, P., Buxbaum, J.D., Eds.; Oxford University Press: Oxford, UK, 2017. [Google Scholar] [CrossRef]
- Pirozzi, F.; Nelson, B.; Mirzaa, G. From microcephaly to megalencephaly: Determinants of brain size. Dialogues Clin. Neurosci. 2018, 20, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Brafman, D.; Willert, K. Wnt/beta-catenin signaling during early vertebrate neural development. Dev. Neurobiol. 2017, 77, 1239–1259. [Google Scholar] [CrossRef] [PubMed]
- Chenn, A.; Walsh, C.A. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 2002, 297, 365–369. [Google Scholar] [CrossRef] [PubMed]
- de Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef]
- Kuechler, A.; Willemsen, M.H.; Albrecht, B.; Bacino, C.A.; Bartholomew, D.W.; van Bokhoven, H.; van den Boogaard, M.J.; Bramswig, N.; Buttner, C.; Cremer, K.; et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: Expanding the mutational and clinical spectrum. Hum. Genet. 2015, 134, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, C.N.; Mutch, C.A.; Swaminathan, S.; Taketo, M.M.; Chenn, A. Persistent expression of stabilized beta-catenin delays maturation of radial glial cells into intermediate progenitors. Dev. Biol. 2007, 309, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiao, J. Histone chaperone HIRA regulates neural progenitor cell proliferation and neurogenesis via beta-catenin. J. Cell. Biol. 2017, 216, 1975–1992. [Google Scholar] [CrossRef]
- Nakagawa, N.; Li, J.; Yabuno-Nakagawa, K.; Eom, T.Y.; Cowles, M.; Mapp, T.; Taylor, R.; Anton, E.S. APC sets the Wnt tone necessary for cerebral cortical progenitor development. Genes Dev. 2017, 31, 1679–1692. [Google Scholar] [CrossRef]
- Mao, Y.; Ge, X.; Frank, C.L.; Madison, J.M.; Koehler, A.N.; Doud, M.K.; Tassa, C.; Berry, E.M.; Soda, T.; Singh, K.K.; et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 2009, 136, 1017–1031. [Google Scholar] [CrossRef]
- Woodhead, G.J.; Mutch, C.A.; Olson, E.C.; Chenn, A. Cell-autonomous beta-catenin signaling regulates cortical precursor proliferation. J. Neurosci. 2006, 26, 12620–12630. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Itoh, Y.; Tabata, H.; Nakajima, K.; Akiyama, T.; Masuyama, N.; Gotoh, Y. The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 2004, 131, 2791–2801. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, W.C.; Sejourne, J.; Clipperton-Allen, A.E.; Page, D.T. Pten Mutations Alter Brain Growth Trajectory and Allocation of Cell Types through Elevated beta-Catenin Signaling. J. Neurosci. 2015, 35, 10252–10267. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, Y.Y.; Wang, X.; Tan, G.H.; Li, H.P.; Hulbert, S.W.; Li, C.Y.; Hu, C.C.; Xiong, Z.Q.; Xu, X.; et al. Autism-associated CHD8 deficiency impairs axon development and migration of cortical neurons. Mol. Autism. 2018, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Nishiyama, M.; Shoji, H.; Ohkawa, Y.; Kawamura, A.; Sato, T.; Suyama, M.; Takumi, T.; Miyakawa, T.; Nakayama, K.I. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 2016, 537, 675–679. [Google Scholar] [CrossRef]
- Gompers, A.L.; Su-Feher, L.; Ellegood, J.; Copping, N.A.; Riyadh, M.A.; Stradleigh, T.W.; Pride, M.C.; Schaffler, M.D.; Wade, A.A.; Catta-Preta, R.; et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci. 2017, 20, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Zhou, Y.; Slaymaker, I.M.; Shetty, A.S.; Weisbach, N.R.; Kim, J.A.; Sharma, J.; Desai, M.; Sood, S.; Kempton, H.R.; et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell. Rep. 2017, 19, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-van Silfhout, A.T.; et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, 158, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Chaste, P.; Klei, L.; Sanders, S.J.; Murtha, M.T.; Hus, V.; Lowe, J.K.; Willsey, A.J.; Moreno-De-Luca, D.; Yu, T.W.; Fombonne, E.; et al. Adjusting head circumference for covariates in autism: Clinical correlates of a highly heritable continuous trait. Biol. Psychiatry 2013, 74, 576–584. [Google Scholar] [CrossRef]
- Takata, A.; Miyake, N.; Tsurusaki, Y.; Fukai, R.; Miyatake, S.; Koshimizu, E.; Kushima, I.; Okada, T.; Morikawa, M.; Uno, Y.; et al. Integrative Analyses of De Novo Mutations Provide Deeper Biological Insights into Autism Spectrum Disorder. Cell. Rep. 2018, 22, 734–747. [Google Scholar] [CrossRef]
- Lennox, A.L.; Hoye, M.L.; Jiang, R.; Johnson-Kerner, B.L.; Suit, L.A.; Venkataramanan, S.; Sheehan, C.J.; Alsina, F.C.; Fregeau, B.; Aldinger, K.A.; et al. Pathogenic DDX3X Mutations Impair RNA Metabolism and Neurogenesis during Fetal Cortical Development. Neuron 2020, 106, 404–420.e8. [Google Scholar] [CrossRef] [PubMed]
- Snijders Blok, L.; Madsen, E.; Juusola, J.; Gilissen, C.; Baralle, D.; Reijnders, M.R.; Venselaar, H.; Helsmoortel, C.; Cho, M.T.; Hoischen, A.; et al. Mutations in DDX3X Are a Common Cause of Unexplained Intellectual Disability with Gender-Specific Effects on Wnt Signaling. Am. J. Hum. Genet. 2015, 97, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Carracedo, A.; Salmena, L.; Song, S.J.; Egia, A.; Malumbres, M.; Pandolfi, P.P. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 2011, 144, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Ostrowski, M.C.; Romigh, T.; Minaguchi, T.; Waite, K.A.; Eng, C. The ERK1/2 pathway modulates nuclear PTEN-mediated cell cycle arrest by cyclin D1 transcriptional regulation. Hum. Mol. Genet. 2006, 15, 2553–2559. [Google Scholar] [CrossRef] [PubMed]
- Groszer, M.; Erickson, R.; Scripture-Adams, D.D.; Lesche, R.; Trumpp, A.; Zack, J.A.; Kornblum, H.I.; Liu, X.; Wu, H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 2001, 294, 2186–2189. [Google Scholar] [CrossRef]
- Backman, S.A.; Stambolic, V.; Suzuki, A.; Haight, J.; Elia, A.; Pretorius, J.; Tsao, M.S.; Shannon, P.; Bolon, B.; Ivy, G.O.; et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat. Genet. 2001, 29, 396–403. [Google Scholar] [CrossRef]
- Kwon, C.H.; Zhu, X.; Zhang, J.; Knoop, L.L.; Tharp, R.; Smeyne, R.J.; Eberhart, C.G.; Burger, P.C.; Baker, S.J. Pten regulates neuronal soma size: A mouse model of Lhermitte-Duclos disease. Nat. Genet. 2001, 29, 404–411. [Google Scholar] [CrossRef]
- Chen, C.J.; Sgritta, M.; Mays, J.; Zhou, H.; Lucero, R.; Park, J.; Wang, I.C.; Park, J.H.; Kaipparettu, B.A.; Stoica, L.; et al. Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat. Med. 2019, 25, 1684–1690. [Google Scholar] [CrossRef]
- Zhou, J.; Blundell, J.; Ogawa, S.; Kwon, C.H.; Zhang, W.; Sinton, C.; Powell, C.M.; Parada, L.F. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J. Neurosci. 2009, 29, 1773–1783. [Google Scholar] [CrossRef]
- Li, Y.; Muffat, J.; Omer, A.; Bosch, I.; Lancaster, M.A.; Sur, M.; Gehrke, L.; Knoblich, J.A.; Jaenisch, R. Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell 2017, 20, 385–396.e3. [Google Scholar] [CrossRef]
- Lei, X.; Jiao, J. UTX Affects Neural Stem Cell Proliferation and Differentiation through PTEN Signaling. Stem Cell. Rep. 2018, 10, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Louvi, A.; Artavanis-Tsakonas, S. Notch signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ables, J.L.; Breunig, J.J.; Eisch, A.J.; Rakic, P. Not(ch) just development: Notch signalling in the adult brain. Nat. Rev. Neurosci. 2011, 12, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.; Brosens, J.J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef]
- Siper, P.M.; De Rubeis, S.; Trelles, M.D.P.; Durkin, A.; Di Marino, D.; Muratet, F.; Frank, Y.; Lozano, R.; Eichler, E.E.; Kelly, M.; et al. Prospective investigation of FOXP1 syndrome. Mol. Autism. 2017, 8, 57. [Google Scholar] [CrossRef]
- Pearson, C.A.; Moore, D.M.; Tucker, H.O.; Dekker, J.D.; Hu, H.; Miquelajauregui, A.; Novitch, B.G. Foxp1 Regulates Neural Stem Cell Self-Renewal and Bias Toward Deep Layer Cortical Fates. Cell. Rep. 2020, 30, 1964–1981.e3. [Google Scholar] [CrossRef]
- Braccioli, L.; Vervoort, S.J.; Adolfs, Y.; Heijnen, C.J.; Basak, O.; Pasterkamp, R.J.; Nijboer, C.H.; Coffer, P.J. FOXP1 Promotes Embryonic Neural Stem Cell Differentiation by Repressing Jagged1 Expression. Stem Cell. Rep. 2017, 9, 1530–1545. [Google Scholar] [CrossRef]
- Matsumura, K.; Seiriki, K.; Okada, S.; Nagase, M.; Ayabe, S.; Yamada, I.; Furuse, T.; Shibuya, H.; Yasuda, Y.; Yamamori, H.; et al. Pathogenic POGZ mutation causes impaired cortical development and reversible autism-like phenotypes. Nat. Commun. 2020, 11, 859. [Google Scholar] [CrossRef]
- Guerrini, R.; Marini, C. Genetic malformations of cortical development. Exp. Brain Res. 2006, 173, 322–333. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Leventer, R.J. Listen carefully: LIS1 and DCX MLPA in lissencephaly and subcortical band heterotopia. Eur. J. Hum. Genet. 2009, 17, 701–702. [Google Scholar] [CrossRef]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.W.; et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.E.; Kerr, M.P.; Gibbon, F.; Turnpenny, P.D.; Hamandi, K.; Stoodley, N.; Robertson, S.P.; Pilz, D.T. Neuropsychiatric disease in patients with periventricular heterotopia. J. Neuropsychiatry Clin. Neurosci. 2013, 25, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Mavili, E.; Coskun, A.; Per, H.; Donmez, H.; Kumandas, S.; Yikilmaz, A. Polymicrogyria: Correlation of magnetic resonance imaging and clinical findings. Childs Nerv. Syst. 2012, 28, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Scala, M.; Torella, A.; Severino, M.; Morana, G.; Castello, R.; Accogli, A.; Verrico, A.; Vari, M.S.; Cappuccio, G.; Pinelli, M.; et al. Three de novo DDX3X variants associated with distinctive brain developmental abnormalities and brain tumor in intellectually disabled females. Eur. J. Hum. Genet. 2019, 27, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Usui, N.; Araujo, D.J.; Kulkarni, A.; Co, M.; Ellegood, J.; Harper, M.; Toriumi, K.; Lerch, J.P.; Konopka, G. Foxp1 regulation of neonatal vocalizations via cortical development. Genes Dev. 2017, 31, 2039–2055. [Google Scholar] [CrossRef] [PubMed]
- La Fata, G.; Gartner, A.; Dominguez-Iturza, N.; Dresselaers, T.; Dawitz, J.; Poorthuis, R.B.; Averna, M.; Himmelreich, U.; Meredith, R.M.; Achsel, T.; et al. FMRP regulates multipolar to bipolar transition affecting neuronal migration and cortical circuitry. Nat. Neurosci. 2014, 17, 1693–1700. [Google Scholar] [CrossRef]
- Wiegreffe, C.; Simon, R.; Peschkes, K.; Kling, C.; Strehle, M.; Cheng, J.; Srivatsa, S.; Liu, P.; Jenkins, N.A.; Copeland, N.G.; et al. Bcl11a (Ctip1) Controls Migration of Cortical Projection Neurons through Regulation of Sema3c. Neuron 2015, 87, 311–325. [Google Scholar] [CrossRef]
- Li, H.; Zhu, Y.; Morozov, Y.M.; Chen, X.; Page, S.C.; Rannals, M.D.; Maher, B.J.; Rakic, P. Disruption of TCF4 regulatory networks leads to abnormal cortical development and mental disabilities. Mol. Psychiatry 2019, 24, 1235–1246. [Google Scholar] [CrossRef]
- Breen, M.S.; Garg, P.; Tang, L.; Mendonca, D.; Levy, T.; Barbosa, M.; Arnett, A.B.; Kurtz-Nelson, E.; Agolini, E.; Battaglia, A.; et al. Episignatures Stratifying Helsmoortel-Van Der Aa Syndrome Show Modest Correlation with Phenotype. Am. J. Hum. Genet. 2020, 107, 555–563. [Google Scholar] [CrossRef]
- Li, X.; Han, X.; Tu, X.; Zhu, D.; Feng, Y.; Jiang, T.; Yang, Y.; Qu, J.; Chen, J.G. An Autism-Related, Nonsense Foxp1 Mutant Induces Autophagy and Delays Radial Migration of the Cortical Neurons. Cereb. Cortex. 2019, 29, 3193–3208. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Daoud, H.; Rochefort, D.; Piton, A.; Gauthier, J.; Langlois, M.; Foomani, G.; Dobrzeniecka, S.; Krebs, M.O.; Joober, R.; et al. De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am. J. Hum. Genet. 2010, 87, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Sollis, E.; Graham, S.A.; Vino, A.; Froehlich, H.; Vreeburg, M.; Dimitropoulou, D.; Gilissen, C.; Pfundt, R.; Rappold, G.A.; Brunner, H.G.; et al. Identification and functional characterization of de novo FOXP1 variants provides novel insights into the etiology of neurodevelopmental disorder. Hum. Mol. Genet. 2016, 25, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.; Estruch, S.B.; Graham, S.A.; McRae, J.; Sawiak, S.J.; Hurst, J.A.; Joss, S.K.; Holder, S.E.; Morton, J.E.; Turner, C.; et al. BCL11A Haploinsufficiency Causes an Intellectual Disability Syndrome and Dysregulates Transcription. Am. J. Hum. Genet. 2016, 99, 253–274. [Google Scholar] [CrossRef] [PubMed]
- Lodato, S.; Arlotta, P. Generating neuronal diversity in the mammalian cerebral cortex. Ann. Rev. Cell. Dev. Biol. 2015, 31, 699–720. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, M.H.; Ayoub, A.E.; Rakic, P. POU-III transcription factors (Brn1, Brn2, and Oct6) influence neurogenesis, molecular identity, and migratory destination of upper-layer cells of the cerebral cortex. Cereb. Cortex. 2013, 23, 2632–2643. [Google Scholar] [CrossRef]
- Sugitani, Y.; Nakai, S.; Minowa, O.; Nishi, M.; Jishage, K.; Kawano, H.; Mori, K.; Ogawa, M.; Noda, T. Brn-1 and Brn-2 share crucial roles in the production and positioning of mouse neocortical neurons. Genes Dev. 2002, 16, 1760–1765. [Google Scholar] [CrossRef]
- Oishi, K.; Aramaki, M.; Nakajima, K. Mutually repressive interaction between Brn1/2 and Rorb contributes to the establishment of neocortical layer 2/3 and layer 4. Proc. Natl. Acad. Sci. USA 2016, 113, 3371–3376. [Google Scholar] [CrossRef]
- Harb, K.; Magrinelli, E.; Nicolas, C.S.; Lukianets, N.; Frangeul, L.; Pietri, M.; Sun, T.; Sandoz, G.; Grammont, F.; Jabaudon, D.; et al. Area-specific development of distinct projection neuron subclasses is regulated by postnatal epigenetic modifications. Elife 2016, 5, e09531. [Google Scholar] [CrossRef]
- Leone, D.P.; Heavner, W.E.; Ferenczi, E.A.; Dobreva, G.; Huguenard, J.R.; Grosschedl, R.; McConnell, S.K. Satb2 Regulates the Differentiation of Both Callosal and Subcerebral Projection Neurons in the Developing Cerebral Cortex. Cereb. Cortex. 2015, 25, 3406–3419. [Google Scholar] [CrossRef]
- Alcamo, E.A.; Chirivella, L.; Dautzenberg, M.; Dobreva, G.; Farinas, I.; Grosschedl, R.; McConnell, S.K. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron 2008, 57, 364–377. [Google Scholar] [CrossRef]
- Arlotta, P.; Molyneaux, B.J.; Chen, J.; Inoue, J.; Kominami, R.; Macklis, J.D. Neuronal Subtype-Specific Genes that Control Corticospinal Motor Neuron Development In Vivo. Neuron 2005, 45, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Baranek, C.; Dittrich, M.; Parthasarathy, S.; Bonnon, C.G.; Britanova, O.; Lanshakov, D.; Boukhtouche, F.; Sommer, J.E.; Colmenares, C.; Tarabykin, V.; et al. Protooncogene Ski cooperates with the chromatin-remodeling factor Satb2 in specifying callosal neurons. Proc. Natl. Acad. Sci. USA 2012, 109, 3546–3551. [Google Scholar] [CrossRef] [PubMed]
- Britanova, O.; de Juan Romero, C.; Cheung, A.; Kwan, K.Y.; Schwark, M.; Gyorgy, A.; Vogel, T.; Akopov, S.; Mitkovski, M.; Agoston, D.; et al. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron 2008, 57, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Srivatsa, S.; Parthasarathy, S.; Britanova, O.; Bormuth, I.; Donahoo, A.L.; Ackerman, S.L.; Richards, L.J.; Tarabykin, V. Unc5C and DCC act downstream of Ctip2 and Satb2 and contribute to corpus callosum formation. Nat. Commun. 2014, 5, 3708. [Google Scholar] [CrossRef] [PubMed]
- Woodworth, M.B.; Greig, L.C.; Liu, K.X.; Ippolito, G.C.; Tucker, H.O.; Macklis, J.D. Ctip1 Regulates the Balance between Specification of Distinct Projection Neuron Subtypes in Deep Cortical Layers. Cell. Rep. 2016, 15, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Hevner, R.F.; Shi, L.; Justice, N.; Hsueh, Y.; Sheng, M.; Smiga, S.; Bulfone, A.; Goffinet, A.M.; Campagnoni, A.T.; Rubenstein, J.L. Tbr1 regulates differentiation of the preplate and layer 6. Neuron 2001, 29, 353–366. [Google Scholar] [CrossRef]
- Canovas, J.; Berndt, F.A.; Sepulveda, H.; Aguilar, R.; Veloso, F.A.; Montecino, M.; Oliva, C.; Maass, J.C.; Sierralta, J.; Kukuljan, M. The Specification of Cortical Subcerebral Projection Neurons Depends on the Direct Repression of TBR1 by CTIP1/BCL11a. J. Neurosci. 2015, 35, 7552–7564. [Google Scholar] [CrossRef]
- Deriziotis, P.; O’Roak, B.J.; Graham, S.A.; Estruch, S.B.; Dimitropoulou, D.; Bernier, R.A.; Gerdts, J.; Shendure, J.; Eichler, E.E.; Fisher, S.E. De novo TBR1 mutations in sporadic autism disrupt protein functions. Nat. Commun. 2014, 5, 4954. [Google Scholar] [CrossRef]
- den Hoed, J.; Sollis, E.; Venselaar, H.; Estruch, S.B.; Deriziotis, P.; Fisher, S.E. Functional characterization of TBR1 variants in neurodevelopmental disorder. Sci. Rep. 2018, 8, 14279. [Google Scholar] [CrossRef]
- Greig, L.C.; Woodworth, M.B.; Galazo, M.J.; Padmanabhan, H.; Macklis, J.D. Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 2013, 14, 755–769. [Google Scholar] [CrossRef]
- Bedogni, F.; Hodge, R.D.; Elsen, G.E.; Nelson, B.R.; Daza, R.A.; Beyer, R.P.; Bammler, T.K.; Rubenstein, J.L.; Hevner, R.F. Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc. Natl. Acad. Sci. USA 2010, 107, 13129–13134. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.L.; Betancourt, J.; Larkin, K.A.; Abrams, B.; Guo, C.; Rubenstein, J.L.; Chen, B. Tbr1 and Fezf2 regulate alternate corticofugal neuronal identities during neocortical development. J. Neurosci. 2011, 31, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Zahr, S.K.; Yang, G.; Kazan, H.; Borrett, M.J.; Yuzwa, S.A.; Voronova, A.; Kaplan, D.R.; Miller, F.D. A Translational Repression Complex in Developing Mammalian Neural Stem Cells that Regulates Neuronal Specification. Neuron 2018, 97, 520–537.e6. [Google Scholar] [CrossRef] [PubMed]
- Kraushar, M.L.; Viljetic, B.; Wijeratne, H.R.; Thompson, K.; Jiao, X.; Pike, J.W.; Medvedeva, V.; Groszer, M.; Kiledjian, M.; Hart, R.P.; et al. Thalamic WNT3 Secretion Spatiotemporally Regulates the Neocortical Ribosome Signature and mRNA Translation to Specify Neocortical Cell Subtypes. J. Neurosci. 2015, 35, 10911–10926. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Bonni, A. Cell-intrinsic drivers of dendrite morphogenesis. Development 2013, 140, 4657–4671. [Google Scholar] [CrossRef] [PubMed]
- Valnegri, P.; Puram, S.V.; Bonni, A. Regulation of dendrite morphogenesis by extrinsic cues. Trends Neurosci. 2015, 38, 439–447. [Google Scholar] [CrossRef]
- Cheng, P.L.; Poo, M.M. Early events in axon/dendrite polarization. Ann. Rev. Neurosci. 2012, 35, 181–201. [Google Scholar] [CrossRef]
- Sudhof, T.C. Towards an Understanding of Synapse Formation. Neuron 2018, 100, 276–293. [Google Scholar] [CrossRef]
- Neniskyte, U.; Gross, C.T. Errant gardeners: Glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat. Rev. Neurosci. 2017, 18, 658–670. [Google Scholar] [CrossRef]
- Tavazoie, S.F.; Alvarez, V.A.; Ridenour, D.A.; Kwiatkowski, D.J.; Sabatini, B.L. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 2005, 8, 1727–1734. [Google Scholar] [CrossRef]
- Yang, R.; Walder-Christensen, K.K.; Kim, N.; Wu, D.; Lorenzo, D.N.; Badea, A.; Jiang, Y.H.; Yin, H.H.; Wetsel, W.C.; Bennett, V. ANK2 autism mutation targeting giant ankyrin-B promotes axon branching and ectopic connectivity. Proc. Natl. Acad. Sci. USA 2019, 116, 15262–15271. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.; Duan, W.Y.; Yu, B.; Tong, D.L.; Cheng, C.; Zhang, Y.F.; Wu, W.; Ye, K.; Zhang, W.X.; Wu, M.; et al. Autism-associated Dyrk1a truncation mutants impair neuronal dendritic and spine growth and interfere with postnatal cortical development. Mol. Psychiatry 2018, 23, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Ka, M.; Chopra, D.A.; Dravid, S.M.; Kim, W.Y. Essential Roles for ARID1B in Dendritic Arborization and Spine Morphology of Developing Pyramidal Neurons. J. Neurosci. 2016, 36, 2723–2742. [Google Scholar] [CrossRef]
- Jung, E.M.; Moffat, J.J.; Liu, J.; Dravid, S.M.; Gurumurthy, C.B.; Kim, W.Y. Arid1b haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat. Neurosci. 2017, 20, 1694–1707. [Google Scholar] [CrossRef]
- Rocca, D.L.; Wilkinson, K.A.; Henley, J.M. SUMOylation of FOXP1 regulates transcriptional repression via CtBP1 to drive dendritic morphogenesis. Sci. Rep. 2017, 7, 877. [Google Scholar] [CrossRef] [PubMed]
- Clement, J.P.; Aceti, M.; Creson, T.K.; Ozkan, E.D.; Shi, Y.; Reish, N.J.; Almonte, A.G.; Miller, B.H.; Wiltgen, B.J.; Miller, C.A.; et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 2012, 151, 709–723. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Hong, I.; Gamache, T.R.; Ju, S.; Collado-Torres, L.; Shin, J.H.; Huganir, R.L. SynGAP isoforms differentially regulate synaptic plasticity and dendritic development. Elife 2020, 9. [Google Scholar] [CrossRef]
- Michaelson, S.D.; Ozkan, E.D.; Aceti, M.; Maity, S.; Llamosas, N.; Weldon, M.; Mizrachi, E.; Vaissiere, T.; Gaffield, M.A.; Christie, J.M.; et al. SYNGAP1 heterozygosity disrupts sensory processing by reducing touch-related activity within somatosensory cortex circuits. Nat. Neurosci. 2018, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Chen, J.; Chen, Q.; Ren, K.; Feng, D.; Mao, H.; Yao, H.; Yang, J.; Liu, H.; Liu, Y.; et al. Anterior cingulate cortex dysfunction underlies social deficits in Shank3 mutant mice. Nat. Neurosci. 2019, 22, 1223–1234. [Google Scholar] [CrossRef]
- Peca, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef]
- Brigman, J.L.; Wright, T.; Talani, G.; Prasad-Mulcare, S.; Jinde, S.; Seabold, G.K.; Mathur, P.; Davis, M.I.; Bock, R.; Gustin, R.M.; et al. Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J. Neurosci. 2010, 30, 4590–4600. [Google Scholar] [CrossRef] [PubMed]
- Spratt, P.W.E.; Ben-Shalom, R.; Keeshen, C.M.; Burke, K.J., Jr.; Clarkson, R.L.; Sanders, S.J.; Bender, K.J. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron 2019, 103, 673–685.e5. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Martin, A.; Crespo, M.; Portera-Cailliau, C. Delayed stabilization of dendritic spines in fragile X mice. J. Neurosci. 2010, 30, 7793–7803. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, E.D.; Fiole, D.; Mantri, S.S.; Huang, C.; Portera-Cailliau, C. Dendritic Spines in Early Postnatal Fragile X Mice Are Insensitive to Novel Sensory Experience. J. Neurosci. 2019, 39, 412–419. [Google Scholar] [CrossRef]
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 2016, 91, 260–292. [Google Scholar] [CrossRef]
- Carvill, G.L.; Heavin, S.B.; Yendle, S.C.; McMahon, J.M.; O’Roak, B.J.; Cook, J.; Khan, A.; Dorschner, M.O.; Weaver, M.; Calvert, S.; et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 2013, 45, 825–830. [Google Scholar] [CrossRef]
- Kim, Y.J.; Khoshkhoo, S.; Frankowski, J.C.; Zhu, B.; Abbasi, S.; Lee, S.; Wu, Y.E.; Hunt, R.F. Chd2 Is Necessary for Neural Circuit Development and Long-Term Memory. Neuron 2018, 100, 1180–1193.e6. [Google Scholar] [CrossRef]
- Butt, S.J.; Sousa, V.H.; Fuccillo, M.V.; Hjerling-Leffler, J.; Miyoshi, G.; Kimura, S.; Fishell, G. The requirement of Nkx2-1 in the temporal specification of cortical interneuron subtypes. Neuron 2008, 59, 722–732. [Google Scholar] [CrossRef]
- Nobrega-Pereira, S.; Kessaris, N.; Du, T.; Kimura, S.; Anderson, S.A.; Marin, O. Postmitotic Nkx2-1 controls the migration of telencephalic interneurons by direct repression of guidance receptors. Neuron 2008, 59, 733–745. [Google Scholar] [CrossRef]
- Meganathan, K.; Lewis, E.M.A.; Gontarz, P.; Liu, S.; Stanley, E.G.; Elefanty, A.G.; Huettner, J.E.; Zhang, B.; Kroll, K.L. Regulatory networks specifying cortical interneurons from human embryonic stem cells reveal roles for CHD2 in interneuron development. Proc. Natl. Acad. Sci. USA 2017, 114, E11180–E11189. [Google Scholar] [CrossRef]
- Wong, F.K.; Bercsenyi, K.; Sreenivasan, V.; Portales, A.; Fernandez-Otero, M.; Marin, O. Pyramidal cell regulation of interneuron survival sculpts cortical networks. Nature 2018, 557, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Jung, E.M.; Jeon, B.T.; Kim, W.Y. Arid1b haploinsufficiency in parvalbumin- or somatostatin-expressing interneurons leads to distinct ASD-like and ID-like behavior. Sci. Rep. 2020, 10, 7834. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).