Insulin/Glucose-Responsive Cells Derived from Induced Pluripotent Stem Cells: Disease Modeling and Treatment of Diabetes



Abstract
1. Introduction
2. Reprogramming of Somatic Cells into iPSCs
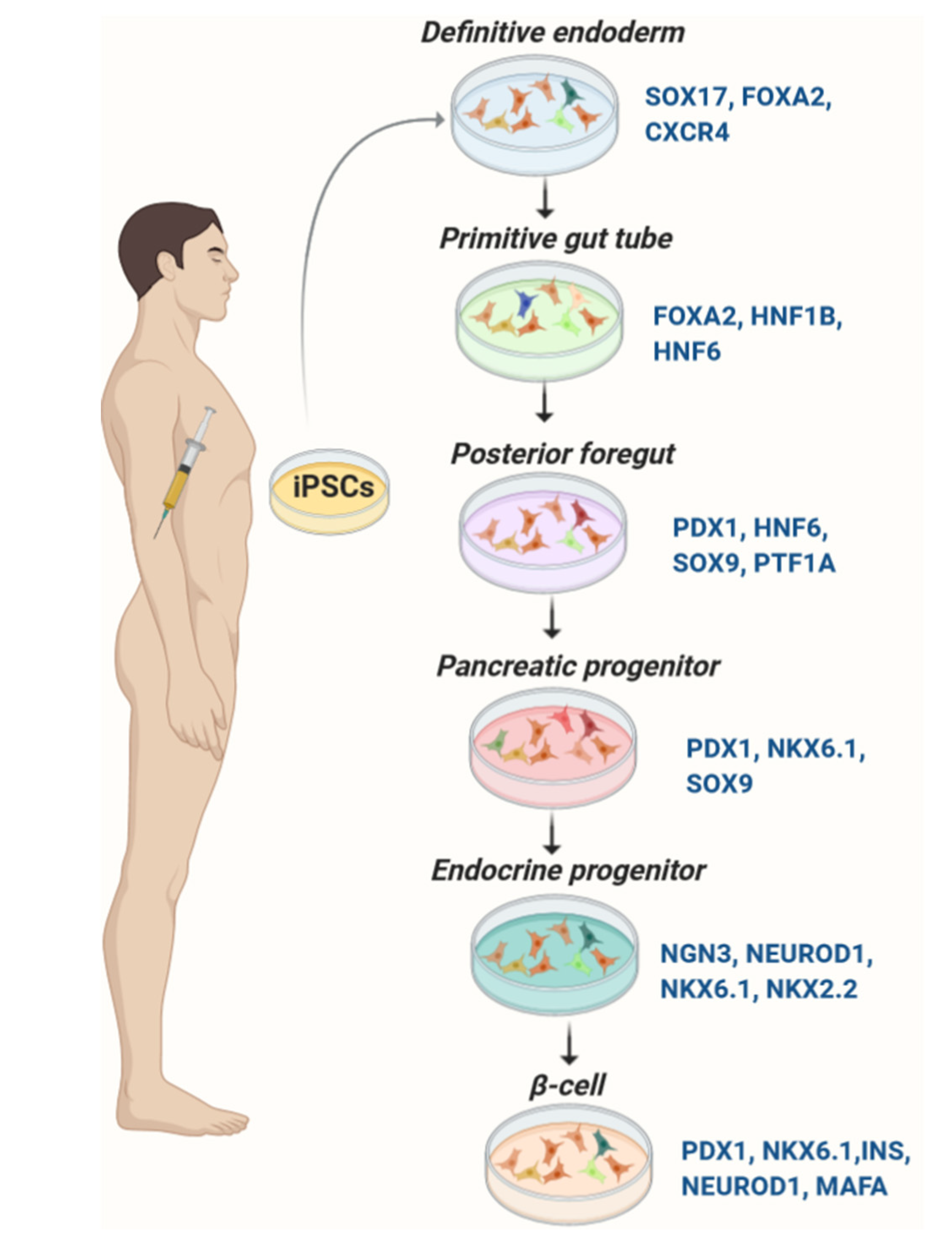
3. iPSC-Derived Pancreatic β-Like Cells
3.1. Development of the Pancreas
3.2. Differentiation of Pancreatic β-Like Cells from iPSC
3.2.1. Induction of Definitive Endoderm
3.2.2. Formation of Primitive Gut Tube and Development of Posterior Foregut
3.2.3. Development of Progenitor Cells

3.2.4. Production of Pancreatic β-Like Cells from iPSCs

3.2.5. Impact of Cellular Microenvironment in Differentiation of iPSC-Derived β-Cells
3.2.6. iPSC-Derived β-Like Cells: Treatment of Diabetes and Disease-in-a-Dish Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| iPSC Source | Protocol | In Vivo/Vitro Efficacy | Stage/Day | Ref. |
|---|---|---|---|---|
| Human iPSCs | Days 1–3: GDF8 and GSK3β inh. Days 4–5: FGF7 and VitC. Days 6–7: FGF7, VitC, 1 µM RA, SANT, TPB, LDN. Days 8–10: FGF7, VitC, 100 nM RA, SANT, TPB, LDN. Days 11–13: 50 nM RA, T3, SANT, ALK5 inh, LDN. Days 14–21/29: T3, ALK5 inh, LDN, γ-secretase inh. Days 21/29–28/36: T3, ALK5 inh, AXL inh, N-Acetylcysteine | ↑ GSIS and plasma human c-peptide levels after transplantation in mice | 7/28–36 | [78] |
| Human iPSCs | Days 1–2: Activin A, CHIR99021. Day 3: No feed. Day 4: KGF. Day5: No feed. Day 6: KGF. Days 7–8: LDN, KGF, SANT, Y-27632, RA, PdBU. Days 9–14: KGF, SANT, Y-27632, RA, Activin A. Days 15–22: Betacellulin, RA, T3, ALK5 inh, SANT, Heparin, γ-secretase inh. Days 22–29/36: T3, ALK5 inh | ↑ GSIS in vivo and in vitro | 6/29–36 | [132] |
| Human iPSCs | Days 1–2: Activin A, CHIR99021. Days 2–4: Activin A. Days 4–7: KGF. Days 7–9: KGF, SANT1, RA, LDN (only Day 7), PdBU. Days 9–14: KGF, SANT, RA. Days 14–18: RA, SANT1, ITS-X, VitC, Heparin, T3, ALK5 inh, Betacellulin, γ-secretase inh. Days 18–21: RA, ITS-X, VitC, Heparin, T3, ALK5 inh, Betacellulin, γ-secretase inh. Days 21–35: ALK5 inh, T3 | ↑ GSIS in vitro | 6/35 | [92] |
| Human iPSCs | Days 1–2: Activin A, Wnt3a, VitC. Days 3–4: Activin A. Days 5–12: dorsomorphin, RA, SB431542 (TGFβ inh). Days 13–23: Forskolin dex, ALK5 inh, Nicotinamide, T3. Days 24–27/29: overexpression of ERRγ | ↑ KCl stimulated insulin secretion without ERRγ overexpression. ↔ GSIS without ERRγ overexpression. ↑ KCl and glucose stimulated insulin secretion after ERRγ overexpression | 3/23 | [94] |
| Human iPSCs | Days 1–3: Activin A, CHIR99021. Days 4–7: KGF. Days 8–9: KGF, SANT1, RA, LDN, PdBU. Days 10–15: KGF, SANT, RA. Days 16–23: RA, SANT1, T3, ALK5 inh, Betacellulin, γ-secretase inh. Days 18–21: RA, ITS-X, VitC, Heparin, T3, ALK5 inh, Betacellulin, γ-secretase inh. Days 22–60: ALK5 inh, T3 | ↑ GSIS in vitro | 6/22–60 | [102] |
| Human iPSCs | Days 1–2: Activin A, CHIR99021. Days 2–3: Activin A. Days 4–5: VitC, KGF. Days 6–10: Insulin, ITS-X, KGF, SANT, RA, LDN, TPB | NR | 4/10 | [7] |
| Human iPSCs | Days 1–3: CHIR99021, GDF8, Activin A, Wnt3A. Days 4–5: FGF7, VitC. Days 6–10: FGF7, VitC, RA, TPB, LDN, Noggin, SANT. Days 11–13: RA, LDN, SANT, ALK5 inh, T3. Days 14–25: LDN, ALK5 inh, T3, γ-secretase inh, Heparin. Days 30–45: ALK5 inh, T3, Cyclopamine, AXL inh. | NR | 7/45 | [9] |
| Human iPSCs | Day 0: Wnt3A, Activin A, Y-27632, ITS-X. Day 1: Activin A, Y-27632, ITS-X. Day 2: ITS-X, KGF, ALK4 inh, Y-27632. Day 3: ITS-X, KGF, ALK4 inh. Day 4: KGF. Days 5–7: TTNBP, Noggin, Cyclopamine, Heregulin. Days 8–12: Noggin, Heregulin, EGF, KGF, Y-27632 | ↑ Fasting and glucose-stimulated human c-peptide levels in vivo | 4/12 | [11] |
| Human iPSCs | Days 1–3: Activin A, CHIR99021. Days 4–5: FGF7, VitC. Days 6–7: FGF7, VitC, RA, TPB, LDN, SANT. Days 8–10: FGF7, VitC, RA, LDN, SANT, EGE, Nicotinamide. Days 11–14: LDN, ALK5 inh, Betacellulin, Heparin, RA, ITS-X, GC1, ZnSO4. Days 14–28: LDN, ALK5 inh, Betacellulin, Heparin, γ-secretase inh, ITS-X, GC1, ZnSO4. Days 29–35: ALK5 inh, Heparin, ITS-X, GC1, ZnSO4, Trolox, JNK inh, Resveratrol, N-Acetylcysteine, AXL inh | ↑ GSIS and plasma human c-peptide levels after transplantation in mice | 7/35 | [133] |
| Human iPSCs | Days 1–3: Activin A, CHIR99021, Y-27632, DMSO. Days 3–7: KGF. Days 7–10: KGF, Noggin, VitC, TTNPB, Cyclopamine. Days 10–14: KGF, EGF, Nicotinamide, Y-27632, VitC. Days 14–17: SANT-1, RA, ALK5 inh, LDN, T3, γ-secretase inh, bFGF, XAV939 (Wnt inh), Y-27632 | ↓ Fasting blood glucose and ↑ plasma human c-peptide after transplantation into diabetic mice | 5/17 | [54] |
| Human iPSCs | Days 1–3: CHIR99021, GDF8. Days 4–5: FGF7, VitC. Days 6–7: FGF7, VitC, RA, TPB, LDN, Noggin, SANT, ITS-X. Days 8–10: ITS-X, Heparin, RA, LDN, SANT, ALK5 inh, T3. Days 11–13: ITS-X, SANT, LDN, ALK5 inh, T3, RA, Heparin. Days 14–20: ALK5 inh, T3, LDN, γ-secretase inh. Days 20–27: ALK5 inh, T3, N-Acetylcysteine, AXL inh. Day 28: Wnt 3/4/5 or Wnt inhibitor (G007-LK) for 4h. Days 28–30/37: ALK5 inh, T3, N-Acetylcysteine, AXL inh | ↔ GSIS ↔ KCl stimulated insulin secretion | 7/37 | [53] |
| Human iPSCs | Days 0–1: Activin A, B27, CHIR99021. Days 1–3: B27, Activin A. Days 3–10: B27, RA, Dorsomorphin, SB431542. Days 10–20: B27, Forskolin, Repsox, Nicotinamide, Dex, Testosterone | ↔ GSIS ↑KCl stimulated insulin secretion | 3/20 | [95] |
| Human iPSCs | Days 1–2: Activin A, 2-ME, CHIR99021. Days 3–5: Activin A, 2-ME. Days 6–7: 2-ME, Cyclopamine, FGF10. Days 8–13: Noggin, RA, Cyclopamine, ALK5 inh, 2-ME. Days 14–15: 2-ME, Noggin, AlLK5 inh, Indolactam V. Days 16–23: 2-ME, Exendin4, Nicotinamide, IBMX, Forskolin | ↑ c-peptide secretion in the presence of KCl, KATP channel blocker, LVDCC and muscarinic agonists | 5/23 | [67] |
| Human iPSCs | Days 1–2: Activin A, CHIR99021, FGF2, BMP4, 2-ME. Days 3–4: KSR, Activin A, 2-ME. Days 5–7: FGF7, ITS-X. Days 8–11: FGF7, ITS-X, SANT-1, LDN, EC23, Indolactam V. Days 12–14: FGF10, ITS-X, SANT-1, LDN, EC23, Indolactam V. Days 15–21: EGF, ITS-X, SANT-1, LDN, EC23, ZnSO4, Indolactam V, RepSox, Heparin, Nicotinamide, Exendin4, Y-27632, γ-secretase inh. Days 22–31: BMP4, HGF, IGF, ITS-X, ZnSO4, Indolactam V, RepSox, Heparin, Nicotinamide, Exendin4, Forskolin | ↓ Nonfasting blood glucose, improved glucose tolerance, ↑ plasma human c-peptide after transplantation into diabetic mice | 6/31 | [14] |
| Human iPSCs | Days 1–2: Activin A, CHIR99021. Days 2–4: Activin A. Days 4–7: KGF. Days 7–9: KGF, SANT1, RA, LDN (only Day 7), PdBU. Days 9–14: KGF, SANT, RA. Days 14–18: RA, SANT1, ITS-X, VitC, Heparin, T3, ALK5 inh, Betacellulin, γ-secretase inh. Days 18–21: RA, ITS-X, VitC, Heparin, T3, ALK5 inh, Betacellulin, γ-secretase inh. Days 21–35: ALK5 inh, T3 | ↑ GSIS both in vitro and in vivo | 6/35 | [90] |
| Human iPSCs | Days 1–5: Activin A, Wnt3a. Days 5–7: KGF, VitC, Y27632. Days 7–8: KGF, VitC. Days 8–12: SANT-1, RA, Noggin, TPB, VitC, KGF. Days 12–16: ALK5 inh, Noggin, GLP-1, SANT-1, RA, γ-secretase inh, Heparin, T3. Days 16–17: ALK5 inh, Noggin, GLP-1, γ-secretase inh, Heparin, T3. Days 17–27: Nicotinamide, IGF-1, GLP-1, ALK5 inh, T3, Heparin | ↑ GSIS and KCl stimulated insulin secretion both in vivo and in vitro | 5/27 | [91] |
4. iPSC-Derived Insulin-Responsive Cells and Insulin Resistance
4.1. Insulin Resistance
4.2. iPSC-Derived Hepatocytes
4.2.1. Development of the Liver
4.2.2. Differentiation of iPSCs into Hepatocytes
Differentiation of Definitive Endoderm and Immature Hepatocytes
| Molecule | Function | Ref. |
|---|---|---|
| Activin A | Member of the TGF-β family; induces DE lineage from stem cells | [144] |
| ALK5iII/RepSox | TGFβR-1/ALK5 inhibitor, upregulates expression of UCN3, MAFA, NKX6.1, and PDX1; induces hepatic and myocyte differentiation | [8,79] |
| B27 | Supports cell growth, viability and induction of endoderm lineage. Promotes β-cell differentiation, maturation, and increases the number of insulin+ cells. Also induces hepatocyte differentiation | [98,145,146] |
| Betacellulin | EGF receptor ligand. Maintains expression of NKX6.1 and PDX1 in endocrine progenitors. Aids in inducing MAFA expression in β-like cells | [147] |
| bFGF/FGF2 | Suppresses SHH signaling and initiates pancreatic differentiation by inducing PDX1 expression. Also induces hepatoblasts and myogenic differentiation by deriving mesoderm lineage | [63,72,148] |
| CHIR99021 | Inhibits GSK3α/β and promotes Wnt signaling for efficient induction of DE lineage | [79,149] |
| Cyclopamine | Blocks SHH signaling. Induces PE lineage and promotes PDX1 expression | [150,151] |
| Db-cAMP | Nerve growth factor. Induces expression of MAFA and insulin | [152] |
| Dexamethasone | Enhances β-like cell differentiation and proliferation. Increases the number of insulin+ cells at the end of differentiation. Aids maturation of hepatocytes and adipocytes | [72,79,153] |
| DMSO | Can be used in combination with activin A to stimulate the process of DE induction | [154] |
| Dorsomorphin | BMP inhibitor, enhances PDX1+ expression in the PE stage. Aids in maturing β-like cells | [79,152] |
| EC23 | Synthetic retinoic acid receptor agonist, used for the formation of PE | [14] |
| EGF | Stimulates cell growth, differentiation and maturation of several cell types including hepatocytes, myocytes and β-cells. Expands the PDX1+ PPs and promotes endocrine cell fate | [155] |
| Exendin-4 | Analog of GLP1. Promotes β-like cell maturation by enhancing the expression of GCK, GLUT2, and NEUROD1 | [156] |
| Fasudil | RhoA/Rho kinase (Rock) inhibitor; promotes DE | [157] |
| FGF10 | Aids in the induction of DE and enhances the characteristic markers of PE | [158] |
| FGF4 | At high concentration promotes endodermal cell fate and expansion | [65] |
| FGF7 | Induces expression of PDX1, PTF1A, and HLXB9. Aids in producing 3D cellular clusters | [159] |
| Forskolin | Increases the levels of cAMP. Derives differentiation and maturation of hepatocytes, myocytes and β-cells. Is required for priming β-cell differentiation and insulin expression | [8,79,160] |
| GDF8 | Belongs to the TGFβ family and induces DE | [78] |
| Glutamine | Induces myocytes characteristics while differentiating. Induces hepatic specification from DE lineage | [140,161] |
| Heparin | Co-factor for FGF2. Enhances generation of endocrine cells and mature β-like cells from PDX1+ PPs | [133] |
| Heregulin | Member of the EGF family used in deriving PE cells | [11] |
| HGF | Matures β-like cells, hepatocytes, and myocytes | [151,162] |
| Hydrocortisone | Matures hepatocytes while differentiating from DE lineage | [163] |
| IBMX | Phosphodiesterene inhibitor and an adenosine receptor antagonist. Induces adipocyte differentiation and maturation. Enhances insulin expression and proportion of differentiating β-like cells | [164] |
| IDE1/2 | Activator of the SMAD2/3 pathway and induces DE lineage | [165] |
| IGF-1 | Induces myogenic as well as β-like cell differentiation and maturation | [166] |
| Indolactam V | Activator of the PKC pathway; induces PDX+ PPs | [167] |
| ITS-X | Supports differentiation and maturation of hepatocytes and adipocytes. Also aids in formation of PPs to insulin-producing β-like cells | [72,168,169] |
| KGF/FGF7 | Generates PDX1+ PPs and PDX1+/NKX6.1+ endocrine progenitor cells. Drives hepatoblasts from foregut endoderm cells | [8,170] |
| LDN | BMP type 1 receptor inhibitor. Promotes PDX1+ PPs and maturation of β-like cells | [171] |
| LY294002 | Inhibits GSK3-β and PI3K activity for efficient induction of DE lineage | [144] |
| Lysophosphatidic acid | Acts through G protein-coupled receptors. Induces hepatoblast differentiation and expansion | [8] |
| N-acetyl cysteine | Functions as an antioxidant. Enhances expression of MAFA | [145,172] |
| NECA | Activates adenosine signaling and promotes β-like cell proliferation | [164] |
| Nicotinamide | A poly (ADP-ribose) synthetase inhibitor; promotes expression of PDX1 up to the later stages in β cell differentiation process. Crucial for hepatocyte differentiation, proliferation and maturation | [147,173] |
| Noggin | BMP inhibitor, induces PDX1+ PPs and NGN3+ endocrine progenitors by suppressing hepatic lineage differentiation | [67] |
| Oncostatin M | Member of IL-6 cytokine family and is crucial for liver development in the final stage of hepatocyte differentiation | [174] |
| PdBU | A phorbol ester, acts as an activator of PKC and is used in promoting pancreatic differentiation | [90] |
| Pioglitazone | An antidiabetic drug, induces lipid-accumulating adipocyte differentiation | [175] |
| Plasmanate | A plasma protein fraction used for inducing adipocyte differentiation | [176] |
| Resveratrol | A stilbenoid polyphenol, enhances the expression of key β-cell maturation genes | [177] |
| Retinoic acid | Crucial for generating NGN3+ endocrine progenitors and for the β-cell specification. Differentiates hepatoblasts into cholangiocyte progenitors. Depending on its concentration and stage administration, it can have a variable but crucial effect on adipocyte differentiation | [72,178,179,180] |
| RG108 | Inhibits DNA methyltransferase, Stimulate reprogramming from somatic cells to iPSCs | [39] |
| RKI-1447 | Rho-kinase inhibitor, induces DE lineage and aids differentiation into PDX1+ PPs | [157] |
| Rosiglitazone | An antidiabetic drug, derives adipogenesis by enhancing the expression of PPARγ and C/EBP-α as well as activation of MAPK and PI3K pathways | [181] |
| SANT-1 | SHH signaling inhibitor, enhances formation of PE and PDX1+ NKX6.1+ PPs | [74] |
| SB431542 | TGF-β receptor inhibitor, enhances number of NKX6.1+ NGN3+ endocrine progenitors | [145] |
| Sodium Butyrate | Inhibits histone deacetylation and aids in DE lineage induction | [182] |
| Sodium cromoglicate | Enhances NGN3+ endocrine precursors and insulin+ cells | [183] |
| Sphingosine-1-phosphate | A signaling sphingolipid metabolite, aids hepatoblast expansion during differentiation | [8] |
| Stauprimide | Belongs to the family of indolocarbazoles, derives DE lineage by downregulating c-Myc expression | [184] |
| Triiodothyronine (T3) | Induces MAFA expression and generates mono-hormonal insulin+ cells. Induces and maintains brown/beige adipogenesis | [104,145,164] |
| Taurine | Induces PE lineage, promotes insulin expression in β-like cells | [185] |
| Thiazovivin | Rho-kinase inhibitor, induces DE lineage | [157] |
| TPB | A PKC activator, enhances generation of NKX6.1+ PPs and endocrine progenitors | [78] |
| TTNPB | Analog of retinoic acid, aids in pancreas specification | [54] |
| Vitamin C | Induces PDX1+ NKX6.1+ PPs and prevents the formation of polyhormonal cells during β-cells differentiation. Also induces expression of hepatocyte-specific genes and aid in its maturation process. Induces mesoderm lineage in adipocyte differentiation | [78,186,187] |
| Wortmannin | Inhibits GSK3-β and PI3K activity and induces DE lineage | [149] |
| XAV939 | Tankyrase inhibitor which targets Wnt/β signaling and promotes β-like cell maturation | [53] |
| Y27632 | Inhibitor of ROCK, enhances PPs and supports cluster formation | [124] |
Mature Hepatocyte Differentiation

4.2.3. Functional Evaluation of iPSC-Derived Hepatocytes
| iPSC Source | Protocol | In Vivo/Vitro Efficacy | Stage/Day | Ref. |
|---|---|---|---|---|
| Human iPSCs | Days 1–3: Wnt3A, Activin A. Days 4–5: Activin A. Days 5–8: KSR, DMSO. Days 9–14: HGF, OSM, Hyd | Glycoproteins, glycogen, and lipid production. secretion of albumin, urea, AFP, and A1AT; cytochrome P450 1A2 and 3A4 activities | 3/14 | [10] |
| Human iPSCs | Day 1: Wnt3A, Activin A, BMP4, bFGF. Days 2–3: Activin A, BMP4, bFGF. Days 4–5; KGF, ALK inh. Days 6–9; KGF, BMP2, BMP4, bFGF. Days 10–16/18: Forskolin, ALK inh, EGF, LPA, Dex, S1P, GSK3β inh. Days 17/19–36/38: ALK inh, Forskolin | Glycogen production; secretion of albumin, urea, AFP; cytochrome P450 3A4 activities; ammonia elimination | 6/36–38 | [8] |
| Human iPSCs | Days 1–2: BMP4, Activin A, FGF2. Days 3–4: Activin A. Days 5–9: BMP4, FGF2. Days 10–13: Activin A, FGF10, RA. Days 14–18: EGF, Dex, FFA, Hyd, Nicotinamide, IL-6, TGFβ1, VitC, ITS-X, sDLL-1 | Glycogen, albumin, urea, and A1AT production; cytochrome P450 3A4 activities, intracellular triglyceride content | 4/18 | [72] |
| Human iPSCs | Days 0–3: Activin A. Days 4–7: BMP2, FGF4. Days 8–13: HGF, KGF. Days 14–18: OSM, Dex. Days 19–21: OSM, Dex, N2B27 | Glycogen, albumin, and urea, production; cytochrome P450 activity | 5/21 | [196] |
| Human iPSCs | Days 1–3: B27−, Sodium butyrate, Wnt3a, Activin. Days 4–5/6: B27, Wnt3a, Activin. Days 6/7–9/10: KSR, DMSO, Glutamine, 2ME. Days 9/10–11/12: iPSCs were mixed with MSCs and HUVECs and cultured within OSM, Transferrin, Hyd, VitC, Insulin, GA-1000, Bovine brain extract, hEGF, FBS, HGF, Dex | Cellular polarity and bile acid transport; urea production and glycogen accumulation | 3/12 | [140] |
| Mouse iPSCs | Days 1–5: Activin A, Wnt3a. Days 6–10: BMP4, FGF-2. Days 11–15: HGF. Days 16–20: HGF, OSM, Dex, ITS-X | Urea and albumin production | 4/20 | [141] |
| Human iPSCs | Days 1–3: B27, Activin A, Wnt3a. Days 4–10: DMSO, KSR. Days 11–20: HGF, OSM, Hyd | Urea and albumin production | 3/20 | [143] |
4.3. iPSC-Derived Skeletal Muscle Cells
4.3.1. Development of Skeletal Muscle
4.3.2. Differentiation of iPSCs into Myocytes
Overexpression of Muscle-Specific Transcription Factors
Step-Wise Induction of Skeletal Muscle Cells by Small Molecules

4.3.3. iPSC-Derived Skeletal Muscle Cells and Insulin Resistance
| iPSCs Source | Protocol | In Vivo/Vitro Efficacy | Stage/Day | Ref. |
|---|---|---|---|---|
| Human iPSCs | Days 1–7: GSK3 inh, bFGF, Forskolin. Days 8–9: Serum free media. Days 10–36: Horse serum | Insulin-stimulated glucose uptake, glycogen synthase activity, glycogen accumulation | 3/36 | [160] |
| Human iPSCs | Days 1–5: GSK3 inh. Days 5–19: bFGF, DMSO. Days 20–35: KSR, ITS-X | Glycogen accumulation | 3/35 | [169] |
| Human iPSCs | Days −1–0: DMSO. Days 0–1.5: Insulin, transferrin, FGF2, PI3K inh, BMP4, GSK3 inh. Days 1.5–7: Insulin, Transferrin, FGF2, PI3K inh. Days 8–12: 15% FBS. Days 13–36: horse serum | Spontaneous contractions | 3/36 | [148] |
| Human iPSCs | Days 1: MyoD overexpression. Day 3: Adding G418. Day 4: ROCK inh. Day 5: ROCK inh, Dox. Days 6–10: αMEM, KSR, 2-ME. Days 11–13: horse serum, IGF1, 2-ME, Glutamin | Fusion potential both in vitro and in vivo | 3/13 | [161] |
| Equine iPSCs | Day 1: mTeSR1 media. Days 2–3: ITS-X, GSK3 inh, ALK inh. Days 4–5: ITS-X, bFGF, GSK3 inh, ALK inh. Days 6–7: IGF1, bFGF, HGF, BMP inh. Days 8–12: IGF1, KSR. Days 12–30: IGF1, KSR, bFGF | Intracellular Ca2+ release in response to KCl-induced membrane depolarization | 5/30 | [220] |
| Equine iPSCs | Days 1–14: media containing high glucose, 10% FBS. Day 15: MyoD overexpression. Day 16: washing with PBS and adding puromycin for selection of positive transductants. Puromycin-resistant cells were cultured in high glucose media containing 10% FBS then Dox was added and cells were differentiated for 7 days. | Intracellular Ca2+ release in response to KCl-induced membrane depolarization | ?/? | [220] |
4.4. The iPSC-Derived Adipocyte
4.4.1. Development of Adipocytes
4.4.2. Differentiation of iPSCs into Adipocytes
Formation of Embryoid Bodies
Adipogenic Differentiation
Differentiation of White Adipocytes from iPSCs
Differentiation of Brown Adipocytes from iPSCs
Differentiation of Beige Adipocytes from iPSCs
| iPSC Source | Protocol | Adipocyte | In Vivo/Vitro Efficacy | Stage/Day | Ref. |
|---|---|---|---|---|---|
| Human iPSCs | Days 0–2: Y-27632 and 15%FBS. Days 2–4: 10 µM RA. Days 4–6: 0.1 µM RA. Days 6–7: RA-. Days 7–12: EB plating onto gelatin/matrigel-coated plates. Days 12–20: bFGF. Days 20–30/34: knockout DMEM-F12 containing 10% KSR, Glutamax, IBMX, Dex, Insulin, Indomethacin, and Pioglitazone | White | NR | 3/30–34 | [231] |
| Human iPSCs | Days 0–2: Y-27632 and 15% FBS. Days 2–4: 10 µM RA. Days 4–6: 0.1 µM RA. Days 6–7: RA-. Days 7–12: EB plating onto gelatin/matrigel-coated plates. Days 12–20: bFGF. Days 20–30/34: MEM-α supplemented with 10% FBS, IBMX, Dex, Insulin, Indomethacin, and Roziglitazone | White | NR | 3/30–34 | [231] |
| Human iPSCs | Days 0–2: 20% KSR. Day 2–5: RA. Day 6–12: 20% KSR. Day 12–22: 10% KSR, IBMX, Dex, Insulin, Indomethacin, Pioglitazone on Poly-L-ornithine and fibronectin plate | White | NR | 2/22 | [230] |
| Human and mouse iPSCs | Days 0–3: 10% FBS. Days 3–5: 10% FBS, RA (25 µM). Days 5–7: 10% FBS, RA (50 µM) (floating condition). Days 7–17: 10% FBS, RA (50 µM) (adherent conditions) and overexpression of PRDM16. Days 17–19: 10% FBS, IBMX, Dex, Indomethacin, Insulin, T3, Rosiglitazone. Days 19–27: 10% FBS, Insulin, T3, Rosiglitazone | Brown | ↓ Body weight, serum glucose, LDL, total cholesterol, and triglycerides, and urine glucose in high fat diet-fed mice | 2/27 | [238] |
| Human iPSCs | Days 0–7: 15% FBS. Days 7–12: 10% FBS, 1% GlutaMAX. Days 12–33: KSR, Dex, hPlasmanate, Insulin, Rosiglitazone | White | Insulin-induced phosphorylation of AKT and glucose uptake. Glycerol release in response to forskolin | 3/33 | [176] |
| Human iPSCs | Days 0–2: bFGF. Days 2–5: RA. Days 5–11: Primate ES cell medium. Days 11–14: IBMX, Dex, Insulin, Pioglitazone | White | NR | 2/14 | [233] |
| Human iPSCs | Days 0–7: 15% FBS. Days 7–12: 10% FBS, 1% GlutaMAX. Days 12–28: overexpression of PPARγ2, and adding Dox, KSR, Dex, Insulin, hPlasmanate, Rosiglitazone. Days 28–33: KSR, Dex, hPlasmanate, Insulin, Rosiglitazone | White | Insulin-induced phosphorylation of AKT and glucose uptake. Glycerol release in response to forskolin | 3/33 | [176] |
| Human iPSCs | Days 0–7: 15% FBS. Days 7–12: 10% FBS, 1% GlutaMAX; Days 12–26: overexpression of PPARγ2, C/EBPα, PRDM16, and adding Dox, KSR, Dex, Insulin, hPlasmanate, Rosiglitazone. Days 26–33: KSR, Dex, hPlasmanate, Insulin, Rosiglitazone | Brown | Glycerol release in response to forskolin and isoproterenol. ↑Oxygen consumption and extracellular acidification rate | 3/33 | [176] |
| Human iPSCs | Days 0–2: 20% KSR. Days 2–5: RA. Days 6–8: 20% KSR; Days 8–11: Insulin, Pioglitazone. Days 11–14/16: IBMX, Dex, Insulin, Pioglitazone | White | Insulin-induced phosphorylation of AKT. Glycerol release in response to forskolin | 2/14–16 | [234] |
| Human iPSCs | Days 0–4: GlutaMAX, VitC, BMP4, activin A. Days 4–10: 10% FCS, Insulin, IBMX, Dex, Indomethacin. Days 10–20: 10% FCS, Insulin | Beige | Insulin-induced phosphorylation of AKT | 3/20 | [186] |
| Human iPSCs | Days −2–0: Serum-free MSC medium, TGF-β inh, IL-4. Days 0–3: Insulin, T3, IBMX, Dex, Indomethacin, TGF-β inh, Rosiglitazone. Days 3–12: Insulin, T3, TGF-β inh, Rosiglitazone; | Beige | Insulin-induced phosphorylation of AKT and glucose uptake | 2/12 | [239] |
| Human iPSCs | Days 0–3: 20% KSR. Days 3–5: 20% KSR, RA; Days 6–20: 20% KSR and overexpression of C/EBPβ. Days 20–30: 10% KSR, Insulin, IBMX, Dex, Rosiglitazone | Brown and white | NR | 2/30 | [242] |
5. Future Prospects and Challenges
5.1. Reprogramming of iPSCs
5.2. Immaturity of the iPSC-Derived Insulin/Glucose-Responsive Cells
5.3. Low Efficiency of Engraftment and Safety in Clinical Therapy
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019. [Google Scholar]
- Gheibi, S.; Samsonov, A.P.; Gheibi, S.; Vazquez, A.B.; Kashfi, K. Regulation of carbohydrate metabolism by nitric oxide and hydrogen sulfide: Implications in diabetes. Biochem. Pharmacol. 2020, 176, 113819. [Google Scholar] [CrossRef] [PubMed]
- Schiesser, J.V.; Wells, J.M. Generation of β cells from human pluripotent Stem Cells: Are we there yet? Ann. N. Y. Acad. Science 2014, 1311, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Thakur, G.; Lee, H.-J.; Jeon, R.-H.; Lee, S.-L.; Rho, G.-J. Small Molecule-Induced Pancreatic β-Like Cell Development: Mechanistic Approaches and Available Strategies. Int. J. Mol. Science 2020, 21, 2388. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Pourquie, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef]
- Higuchi, A.; Kumar, S.S.; Ling, Q.-D.; Alarfaj, A.A.; Munusamy, M.A.; Murugan, K.; Hsu, S.-T.; Benelli, G.; Umezawa, A. Polymeric design of cell culture materials that guide the differentiation of human pluripotent Stem Cells. Prog. Polym. Science 2017, 65, 83–126. [Google Scholar] [CrossRef]
- Tran, R.; Moraes, C.; Hoesli, C.A. Controlled clustering enhances PDX1 and NKX6.1 expression in pancreatic endoderm cells derived from pluripotent Stem Cells. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Chen, S.; Wang, J.; Ren, H.; Liu, Y.; Xiang, C.; Li, C.; Lu, S.; Shi, Y.; Deng, H.; Shi, X. Hepatic spheroids derived from human induced pluripotent Stem Cells in bio-artificial liver rescue porcine acute liver failure. Cell Res. 2020, 30, 95–97. [Google Scholar] [CrossRef]
- Legøy, T.A.; Vethe, H.; Abadpour, S.; Strand, B.L.; Scholz, H.; Paulo, J.A.; Ræder, H.; Ghila, L.; Chera, S. Encapsulation boosts islet-cell signature in differentiating human induced pluripotent Stem Cells via integrin signalling. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Calabrese, D.; Roma, G.; Bergling, S.; Carbone, W.; Mele, V.; Nuciforo, S.; Fofana, I.; Campana, B.; Szkolnicka, D.; Hay, D.C.; et al. Liver biopsy derived induced pluripotent Stem Cells provide unlimited supply for the generation of hepatocyte-like cells. PLoS ONE 2019, 14, e0221762. [Google Scholar] [CrossRef]
- Haller, C.; Piccand, J.; De Franceschi, F.; Ohi, Y.; Bhoumik, A.; Boss, C.; De Marchi, U.; Jacot, G.; Metairon, S.; Descombes, P.; et al. Macroencapsulated Human iPSC-Derived Pancreatic Progenitors Protect against STZ-Induced Hyperglycemia in Mice. Stem Cell Rep. 2019, 12, 787–800. [Google Scholar] [CrossRef]
- Halevy, T.; Urbach, A. Comparing ESC and iPSC—Based Models for Human Genetic Disorders. J. Clin. Med. 2014, 3, 1146–1162. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Yabe, S.G.; Fukuda, S.; Nishida, J.; Takeda, F.; Nashiro, K.; Okochi, H. Induction of functional islet-like cells from human iPS cells by suspension culture. Regen. Ther. 2019, 10, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Blelloch, R.; Venere, M.; Yen, J.; Ramalho-Santos, M. Generation of Induced Pluripotent Stem Cells in the Absence of Drug Selection. Cell Stem Cell 2007, 1, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Maherali, N.; Ahfeldt, T.; Rigamonti, A.; Utikal, J.; Cowan, C.; Hochedlinger, K. A High-Efficiency System for the Generation and Study of Human Induced Pluripotent Stem Cells. Cell Stem Cell 2008, 3, 340–345. [Google Scholar] [CrossRef]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent Stem Cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent Stem Cells. Nat. Cell Biol. 2007, 448, 313–317. [Google Scholar] [CrossRef]
- Wernig, M.; Meissner, A.; Cassady, J.P.; Jaenisch, R. c-Myc Is Dispensable for Direct Reprogramming of Mouse Fibroblasts. Cell Stem Cell 2008, 2, 10–12. [Google Scholar] [CrossRef]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of Mouse Induced Pluripotent Stem Cells Without Viral Vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef]
- Sommer, C.A.; Stadtfeld, M.; Murphy, G.J.; Hochedlinger, K.; Kotton, D.N.; Mostoslavsky, G. Induced Pluripotent Stem Cell Generation Using a Single Lentiviral Stem Cell Cassette. Stem Cells 2009, 27, 543–549. [Google Scholar] [CrossRef]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent Stem Cells. Nat. Cell Biol. 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rad, R.; Takeda, J.; Bradley, A. Generation of transgene-free induced pluripotent mouse Stem Cells by the piggyBac transposon. Nat. Methods 2009, 6, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Talluri, T.R.; Anand, T.; Kues, W.A. Transposon-based reprogramming to induced pluripotency. Histol. Histopathol. 2015, 30, 1397–1409. [Google Scholar] [PubMed]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced Pluripotent Stem Cells Generated Without Viral Integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent Stem Cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent Stem Cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef]
- González, F.; Boué, S.; Belmonte, J.C.I. Methods for making induced pluripotent Stem Cells: Reprogramming à la carte. Nat. Rev. Genet. 2011, 12, 231–242. [Google Scholar] [CrossRef]
- Gonzalez, F.; Monasterio, M.B.; Tiscornia, G.; Pulido, N.M.; Vassena, R.; Morera, L.B.; Piza, I.R.; Belmonte, J.C.I. Generation of mouse-induced pluripotent Stem Cells by transient expression of a single nonviral polycistronic vector. Proc. Natl. Acad. Sci. USA 2009, 106, 8918–8922. [Google Scholar] [CrossRef]
- Rim, Y.A.; Nam, Y.; Park, N.; Jung, H.; Lee, K.; Lee, J.; Ju, J.H. Chondrogenic Differentiation from Induced Pluripotent Stem Cells Using Non-Viral Minicircle Vectors. Cells 2020, 9, 582. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human Induced Pluripotent Stem Cells Free of Vector and Transgene Sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.-I.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Steinle, H.; Weber, M.; Behring, A.; Mau-Holzmann, U.; Schlensak, C.; Wendel, H.P.; Avci-Adali, M. Generation of iPSCs by Nonintegrative RNA-Based Reprogramming Techniques: Benefits of Self-Replicating RNA versus Synthetic mRNA. Stem Cells Int. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly Efficient Reprogramming to Pluripotency and Directed Differentiation of Human Cells with Synthetic Modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, N.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of Induced Pluripotent Stem Cells Using Recombinant Proteins. Cell Stem Cell 2009, 4, 581. [Google Scholar] [CrossRef]
- Michiue, H.; Tomizawa, K.; Wei, F.-Y.; Matsushita, M.; Lu, Y.-F.; Ichikawa, T.; Tamiya, T.; Date, I.; Matsui, H. The NH2Terminus of Influenza Virus Hemagglutinin-2 Subunit Peptides Enhances the Antitumor Potency of Polyarginine-mediated p53 Protein Transduction. J. Biol. Chem. 2004, 280, 8285–8289. [Google Scholar] [CrossRef] [PubMed]
- Ichida, J.K.; Blanchard, J.; Lam, K.; Son, E.Y.; Chung, J.E.; Egli, D.; Loh, K.M.; Carter, A.C.; Di Giorgio, F.P.; Koszka, K.; et al. A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell Stem Cell 2009, 5, 491–503. [Google Scholar] [CrossRef]
- Noggle, S.A.; Fung, H.-L.; Gore, A.; Martínez, H.; Satriani, K.C.; Prosser, R.; Oum, K.; Paull, D.; Druckenmiller, S.; Freeby, M.; et al. Human oocytes reprogram somatic cells to a pluripotent state. Nat. Cell Biol. 2011, 478, 70–75. [Google Scholar] [CrossRef]
- Mali, P.; Chou, B.-K.; Yen, J.; Ye, Z.; Zou, J.; Dowey, S.; Brodsky, R.A.; Ohm, J.E.; Yu, W.; Baylin, S.B.; et al. Butyrate Greatly Enhances Derivation of Human Induced Pluripotent Stem Cells by Promoting Epigenetic Remodeling and the Expression of Pluripotency-Associated Genes. Stem Cells 2010, 28, 713–720. [Google Scholar] [CrossRef]
- Zhu, S.; Li, W.; Zhou, H.; Wei, W.; Ambasudhan, R.; Lin, T.; Kim, J.; Zhang, K.; Ding, S. Reprogramming of Human Primary Somatic Cells by OCT4 and Chemical Compounds. Cell Stem Cell 2010, 7, 651–655. [Google Scholar] [CrossRef]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C Enhances the Generation of Mouse and Human Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of pluripotent Stem Cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Vaskova, E.A.; Stekleneva, A.E.; Medvedev, S.P.; Zakian, S.M. “Epigenetic Memory” Phenomenon in Induced Pluripotent Stem Cells. Acta Nat. 2013, 5, 15–21. [Google Scholar] [CrossRef]
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic Memory and Preferential Lineage-Specific Differentiation in Induced Pluripotent Stem Cells Derived from Human Pancreatic Islet Beta Cells. Cell Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent Stem Cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Pictet, R.L.; Clark, W.R.; Williams, R.H.; Rutter, W.J. An ultrastructural analysis of the developing embryonic pancreas. Dev. Biol. 1972, 29, 436–467. [Google Scholar] [CrossRef]
- Jennings, R.E.; Berry, A.A.; Strutt, J.P.; Gerrard, D.T.; Hanley, N.A. Human pancreas development. Development 2015, 142, 3126–3137. [Google Scholar] [CrossRef]
- Jennings, R.E.; Berry, A.A.; Kirkwood-Wilson, R.; Roberts, N.A.; Hearn, T.; Salisbury, R.J.; Blaylock, J.; Hanley, K.P.; Hanley, N.A. Development of the Human Pancreas From Foregut to Endocrine Commitment. Diabetes 2013, 62, 3514–3522. [Google Scholar] [CrossRef]
- Pan, F.C.; Wright, C. Pancreas organogenesis: From bud to plexus to gland. Dev. Dyn. 2011, 240, 530–565. [Google Scholar] [CrossRef]
- Herrera, P.L. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 2000, 127, 2317–2322. [Google Scholar]
- Larsen, H.L.; Grapin-Botton, A. The molecular and morphogenetic basis of pancreas organogenesis. Semin. Cell Dev. Biol. 2017, 66, 51–68. [Google Scholar] [CrossRef]
- Dassaye, R.; Naidoo, S.; Cerf, M.E. Transcription factor regulation of pancreatic organogenesis, differentiation and maturation. Islets 2015, 8, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Vethe, H.; Ghila, L.; Berle, M.; Hoareau, L.; Haaland, Ø.A.; Scholz, H.; Paulo, J.A.; Chera, S.; Ræder, H. The Effect of Wnt Pathway Modulators on Human iPSC-Derived Pancreatic Beta Cell Maturation. Front. Endocrinol. 2019, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Mochida, T.; Ueno, H.; Tsubooka-Yamazoe, N.; Hiyoshi, H.; Ito, R.; Matsumoto, H.; Toyoda, T. Insulin-Deficient Diabetic Condition Upregulates the Insulin-Secreting Capacity of Human Induced Pluripotent Stem Cell-Derived Pancreatic Endocrine Progenitor Cells After Implantation in Mice. Diabetes 2020, 69, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Tam, P.P.L.; Loebel, D.A.F. Gene function in mouse embryogenesis: Get set for gastrulation. Nat. Rev. Genet. 2007, 8, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jiang, W.; Liu, M.; Sui, X.; Yin, X.; Chen, S.; Shi, Y.; Deng, H. Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Res. 2009, 19, 429–438. [Google Scholar] [CrossRef]
- Nostro, M.C.; Sarangi, F.; Ogawa, S.; Holtzinger, A.; Corneo, B.; Li, X.; Micallef, S.J.; Park, I.-H.; Basford, C.; Wheeler, M.B.; et al. Stage-specific signaling through TGFβ family members and WNT regulates patterning and pancreatic specification of human pluripotent Stem Cells. Development 2011, 138, 1445. [Google Scholar] [CrossRef]
- Teo, A.K.K.; Ali, Y.; Wong, K.Y.; Chipperfield, H.; Sadasivam, A.; Poobalan, Y.; Tan, E.K.; Wang, S.T.; Abraham, S.; Tsuneyoshi, N.; et al. Activin and BMP4 Synergistically Promote Formation of Definitive Endoderm in Human Embryonic Stem Cells. Stem Cells 2012, 30, 631–642. [Google Scholar] [CrossRef]
- Ungrin, M.D.; Clarke, G.; Yin, T.; Niebrugge, S.; Nostro, M.C.; Sarangi, F.; Wood, G.; Keller, G.; Zandstra, P.W. Rational bioprocess design for human pluripotent stem cell expansion and endoderm differentiation based on cellular dynamics. Biotechnol. Bioeng. 2012, 109, 853–866. [Google Scholar] [CrossRef]
- Yabe, S.G.; Fukuda, S.; Takeda, F.; Nashiro, K.; Shimoda, M.; Okochi, H. Efficient generation of functional pancreatic β-cells from human induced pluripotent Stem Cells. J. Diabetes 2016, 9, 168–179. [Google Scholar] [CrossRef]
- Wells, J.M.; A Melton, D. Early mouse endoderm is patterned by soluble factors from adjacent germ layers. Development 2000, 127, 1563–1572. [Google Scholar]
- Deutsch, G.; Jung, J.; Zheng, M.; Lóra, J.; Zaret, K.S. A bipotential precursor population for pancreas and liver within the embryonic endoderm. Development 2001, 128, 871–881. [Google Scholar] [PubMed]
- Ameri, J.; Ståhlberg, A.; Pedersen, J.; Johansson, J.K.; Johannesson, M.M.; Artner, I.; Semb, H. FGF2 Specifies hESC-Derived Definitive Endoderm into Foregut/Midgut Cell Lineages in a Concentration-Dependent Manner. Stem Cells 2009, 28, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kelly, O.G.; Chan, M.Y.; Martinson, L.A.; Kadoya, K.; Ostertag, T.M.; Ross, K.G.; Richardson, M.; Carpenter, M.K.; D’Amour, K.A.; Kroon, E.; et al. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic Stem Cells. Nat. Biotechnol. 2011, 29, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Johannesson, M.; Ståhlberg, A.; Ameri, J.; Sand, F.W.; Norrman, K.; Semb, H. FGF4 and Retinoic Acid Direct Differentiation of hESCs into PDX1-Expressing Foregut Endoderm in a Time- and Concentration-Dependent Manner. PLoS ONE 2009, 4, e4794. [Google Scholar] [CrossRef] [PubMed]
- Apelqvist, Å.; Ahlgren, U.; Edlund, H. Sonic hedgehog directs specialised mesoderm differentiation in the intestine and pancreas. Curr. Biol. 1997, 7, 801–804. [Google Scholar] [CrossRef]
- Shahjalal, H.M.; Shiraki, N.; Sakano, D.; Kikawa, K.; Ogaki, S.; Baba, H.; Kume, K.; Kume, S. Generation of insulin-producing β-like cells from human iPS cells in a defined and completely xeno-free culture system. J. Mol. Cell Biol. 2014, 6, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Thatava, T.; Nelson, T.J.; Edukulla, R.; Sakuma, T.; Ohmine, S.; Tonne, J.M.; Yamada, S.; Kudva, Y.C.; Terzic, A.; Ikeda, Y. Indolactam V/GLP-1-mediated differentiation of human iPS cells into glucose-responsive insulin-secreting progeny. Gene Ther. 2011, 18, 283–293. [Google Scholar] [CrossRef]
- Taylor, B.L.; Liu, F.-F.; Sander, M. Nkx6.1 Is Essential for Maintaining the Functional State of Pancreatic Beta Cells. Cell Rep. 2013, 4, 1262–1275. [Google Scholar] [CrossRef]
- Memon, B.; Abdelalim, E.M. Stem Cell Therapy for Diabetes: Beta Cells versus Pancreatic Progenitors. Cells 2020, 9, 283. [Google Scholar] [CrossRef]
- Aigha, I.I.; Memon, B.; Elsayed, A.K.; Abdelalim, E.M. Differentiation of human pluripotent Stem Cells into two distinct NKX6.1 populations of pancreatic progenitors. Stem Cell Res. Ther. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Takeishi, K.; De L’Hortet, A.C.; Wang, Y.; Handa, K.; Guzman-Lepe, J.; Matsubara, K.; Morita, K.; Jang, S.; Haep, N.; Florentino, R.M.; et al. Assembly and Function of a Bioengineered Human Liver for Transplantation Generated Solely from Induced Pluripotent Stem Cells. Cell Rep. 2020, 31, 107711. [Google Scholar] [CrossRef] [PubMed]
- Memon, B.; Karam, M.; Al-Khawaga, S.; Abdelalim, E.M. Enhanced differentiation of human pluripotent Stem Cells into pancreatic progenitors co-expressing PDX1 and NKX6. Stem. Cell Res. Ther. 2018, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Nostro, M.C.; Sarangi, F.; Yang, C.; Holland, A.M.; Elefanty, A.G.; Stanley, E.G.; Greiner, D.L.; Keller, G. Efficient Generation of NKX6-1+ Pancreatic Progenitors from Multiple Human Pluripotent Stem Cell Lines. Stem Cell Rep. 2015, 4, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Memon, B.; Younis, I.; Abubaker, F.; Abdelalim, E.M. PDX1 − /NKX6.1 + progenitors derived from human pluripotent Stem Cells as a novel source of insulin-secreting cells. Diabetes/Metabolism Res. Rev. 2020, 3400. [Google Scholar] [CrossRef] [PubMed]
- Rosado-Olivieri, E.A.; Anderson, K.; Kenty, J.H.; Melton, D.A. YAP inhibition enhances the differentiation of functional stem cell-derived insulin-producing β cells. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.G.; Moroishi, T.; Guan, K.-L. YAP and TAZ: A nexus for Hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent Stem Cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef]
- Kunisada, Y.; Tsubooka-Yamazoe, N.; Shoji, M.; Hosoya, M. Small molecules induce efficient differentiation into insulin-producing cells from human induced pluripotent Stem Cells. Stem Cell Res. 2012, 8, 274–284. [Google Scholar] [CrossRef]
- Miralles, F.; LaMotte, L.; Couton, D.; Joshi, R.L. Interplay between FGF10 and Notch signalling is required for the self-renewal of pancreatic progenitors. Int. J. Dev. Biol. 2006, 50, 17–26. [Google Scholar] [CrossRef]
- Bhushan, A.; Itoh, N.; Kato, S.; Thiery, J.P.; Czernichow, P.; Bellusci, S.; Scharfmann, R. Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development 2001, 128, 5109–5117. [Google Scholar]
- Hart, A.; Papadopoulou, S.; Edlund, H. Fgf10maintains notch activation, stimulates proliferation, and blocks differentiation of pancreatic epithelial cells. Dev. Dyn. 2003, 228, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Pinney, S.E.; Oliverkrasinski, J.M.; Ernst, L.M.; Hughes, N.; Patel, P.; Stoffers, D.A.; Russo, P.; De León, D.D. Neonatal Diabetes and Congenital Malabsorptive Diarrhea Attributable to a Novel Mutation in the Human Neurogenin-3 Gene Coding Sequence. J. Clin. Endocrinol. Metab. 2011, 96, 1960–1965. [Google Scholar] [CrossRef] [PubMed]
- Gradwohl, G.; Dierich, A.; LeMeur, M.; Guillemot, F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. USA 2000, 97, 1607. [Google Scholar] [CrossRef]
- Bliss, C.R.; Sharp, G.W. Glucose-induced insulin release in islets of young rats: Time-dependent potentiation and effects of 2-bromostearate. Am. J. Physiol. Metab. 1992, 263, E890–E896. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Koh, A.; El Khattabi, I.; Li, W.-C.; Toschi, E.; Jermendy, A.; Juhl, K.; Mao, K.; Weir, G.C.; Sharma, A.; et al. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia 2010, 54, 583–593. [Google Scholar] [CrossRef]
- Kataoka, K.; Han, S.I.; Shioda, S.; Hirai, M.; Nishizawa, M.; Handa, H. MafA is a glucose-regulated and pancreatic beta-cell-specific transcriptional activator for the insulin gene. J. Biol. Chem. 2002, 277, 49903–49910. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Brun, T.; Kataoka, K.; Sharma, A.J.; Wollheim, C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 2007, 50, 348–358. [Google Scholar] [CrossRef]
- El Khattabi, I.; Sharma, A. Proper activation of MafA is required for optimal differentiation and maturation of pancreatic β-cells. Best Pr. Res. Clin. Endocrinol. Metab. 2015, 29, 821–831. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of Functional Human Pancreatic β Cells In Vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef]
- Manzar, G.S.; Kim, E.-M.; Zavazava, N. Demethylation of induced pluripotent Stem Cells from type 1 diabetic patients enhances differentiation into functional pancreatic β cells. J. Biol. Chem. 2017, 292, 14066–14079. [Google Scholar] [CrossRef]
- Davis, J.C.; Alves, T.C.; Helman, A.; Chen, J.C.; Kenty, J.H.; Cardone, R.L.; Liu, D.R.; Kibbey, R.G.; Melton, D.A. Glucose Response by Stem Cell-Derived β Cells In Vitro Is Inhibited by a Bottleneck in Glycolysis. Cell Rep. 2020, 31, 107623. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dominguez, J.R.; Donaghey, J.; Rasouli, N.; Kenty, J.H.; Helman, A.; Charlton, J.; Straubhaar, J.R.; Meissner, A.; Melton, D.A. Circadian Entrainment Triggers Maturation of Human In Vitro Islets. Cell Stem Cell 2020, 26, 108–122.e10. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, E.; Wei, Z.; Lin, C.S.; Fang, S.; Ahmadian, M.; Kida, Y.; Tseng, T.; Dai, Y.; Yasuyuki, K.; Liddle, C.; et al. ERRγ Is Required for the Metabolic Maturation of Therapeutically Functional Glucose-Responsive β Cells. Cell Metab. 2016, 23, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Guo, D.; Ruzi, A.; Chen, Y.; Pan, T.; Yang, F.; Li, J.; Xu, K.; Zhou, T.; Qin, D.; et al. Testosterone improves the differentiation efficiency of insulin-producing cells from human induced pluripotent Stem Cells. PLoS ONE 2017, 12, e0179353. [Google Scholar] [CrossRef] [PubMed]
- ElSharkawi, I.; Parambath, D.; Saber-Ayad, M.; Khan, A.A.; El-Serafi, A. Exploring the effect of epigenetic modifiers on developing insulin-secreting cells. Hum. Cell 2019, 33, 1–9. [Google Scholar] [CrossRef]
- Ghazizadeh, Z.; Kao, D.-I.; Amin, S.; Cook, B.; Rao, S.; Zhou, T.; Zhang, T.; Xiang, Z.; Kenyon, R.; Kaymakcalan, O.; et al. ROCKII inhibition promotes the maturation of human pancreatic beta-like cells. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Ameri, J.; Borup, R.; Prawiro, C.; Ramond, C.; Schachter, K.A.; Scharfmann, R.; Semb, H. Efficient Generation of Glucose-Responsive Beta Cells from Isolated GP2 + Human Pancreatic Progenitors. Cell Rep. 2017, 19, 36–49. [Google Scholar] [CrossRef]
- Cogger, K.F.; Sinha, A.; Sarangi, F.; McGaugh, E.C.; Saunders, D.; Dorrell, C.; Mejia-Guerrero, S.; Aghazadeh, Y.; Rourke, J.L.; Screaton, R.A.; et al. Glycoprotein 2 is a specific cell surface marker of human pancreatic progenitors. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Jiang, W.; Sui, X.; Zhang, N.; Liu, M.; Ding, M.; Shi, Y.; Deng, H. CD24: A Novel Surface Marker for PDX1-Positive Pancreatic Progenitors Derived from Human Embryonic Stem Cells. Stem Cells 2011, 29, 609–617. [Google Scholar] [CrossRef]
- Ramond, C.; Glaser, N.; Berthault, C.; Ameri, J.; Kirkegaard, J.S.; Hansson, M.; Honoré, C.; Semb, H.; Scharfmann, R. Reconstructing human pancreatic differentiation by mapping specific cell populations during development. eLife 2017, 6, 6. [Google Scholar] [CrossRef]
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.-R.; Harb, G.; Poh, Y.-C.; Sintov, E.; Gürtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro β-cell differentiation. Nat. Cell Biol. 2019, 569, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Diaz, R.; Caicedo, A. Neural control of the endocrine pancreas. Best Pr. Res. Clin. Endocrinol. Metab. 2014, 28, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; DiIenno, A.; Hollister-Lock, J.; Cahill, C.; Sharma, A.; Weir, G.; Colton, C.; Bonner-Weir, S. MAFA and T3 Drive Maturation of Both Fetal Human Islets and Insulin-Producing Cells Differentiated From hESC. J. Clin. Endocrinol. Metab. 2015, 100, 3651–3659. [Google Scholar] [CrossRef] [PubMed]
- Saber, N.; E Bruin, J.; O’Dwyer, S.; Schuster, H.; Rezania, A.; Kieffer, T.J. Sex Differences in Maturation of Human Embryonic Stem Cell–Derived β Cells in Mice. Endocrinology 2018, 159, 1827–1841. [Google Scholar] [CrossRef]
- Briant, L.J.B.; Reinbothe, T.M.; Spiliotis, I.; Miranda, C.; Rodriguez, B.; Rorsman, P. δ-cells and β-cells are electrically coupled and regulate α-cell activity via somatostatin. J. Physiol. 2018, 596, 197–215. [Google Scholar] [CrossRef]
- Alessandra, G.; Algerta, M.; Paola, M.; Carsten, S.; Cristina, L.; Paolo, M.; Elisa, M.; Gabriella, T.; Carla, P.; Galli, A.; et al. Shaping Pancreatic β-Cell Differentiation and Functioning: The Influence of Mechanotransduction. Cells 2020, 9, 413. [Google Scholar] [CrossRef]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef]
- Ladoux, B.; Mège, R.-M. Mechanobiology of collective cell behaviours. Nat. Rev. Mol. Cell Biol. 2017, 18, 743–757. [Google Scholar] [CrossRef]
- Mui, K.L.; Chen, C.S.; Assoian, R.K. The mechanical regulation of integrin–cadherin crosstalk organizes cells, signaling and forces. J. Cell Sci. 2016, 129, 1093–1100. [Google Scholar] [CrossRef]
- Nyitray, C.E.; Chavez, M.G.; Desai, T.A. Compliant 3D Microenvironment Improves β-Cell Cluster Insulin Expression Through Mechanosensing and β-Catenin Signaling. Tissue Eng. Part A 2014, 20, 1888–1895. [Google Scholar] [CrossRef]
- Ribeiro, D.; Kvist, A.J.; Wittung-Stafshede, P.; Hicks, R.; Forslöw, A. 3D-Models of Insulin-Producing β-Cells: From Primary Islet Cells to Stem Cell-Derived Islets. Stem Cell Rev. Rep. 2017, 14, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Maffioli, E.; Sogne, E.; Moretti, S.; Di Cairano, E.S.; Negri, A.; Nonnis, S.; Norata, G.D.; Bonacina, F.; Borghi, F.; et al. Cluster-assembled zirconia substrates promote long-term differentiation and functioning of human islets of Langerhans. Sci. Rep. 2018, 8, 9979. [Google Scholar] [CrossRef] [PubMed]
- Parnaud, G.; Lavallard, V.; Bédat, B.; Matthey-Doret, D.; Morel, P.; Berney, T.; Bosco, D. Cadherin Engagement Improves Insulin Secretion of Single Human β-Cells. Diabetes 2014, 64, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Charollais, A.; Gjinovci, A.; Huarte, J.; Bauquis, J.; Nadal, A.; Martín, F.; Andreu, E.; Sánchez-Andrés, J.V.; Calabrese, A.; Bosco, D.; et al. Junctional communication of pancreatic β cells contributes to the control of insulin secretion and glucose tolerance. J. Clin. Investig. 2000, 106, 235–243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Di Cairano, E.S.; Moretti, S.; Marciani, P.; Sacchi, V.F.; Castagna, M.; Davalli, A.M.; Folli, F.; Perego, C. Neurotransmitters and Neuropeptides: New Players in the Control of Islet of Langerhans’ Cell Mass and Function. J. Cell. Physiol. 2016, 231, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Arous, C.; Wehrle-Haller, B. Role and impact of the extracellular matrix on integrin-mediated pancreatic β-cell functions. Biol. Cell 2017, 109, 223–237. [Google Scholar] [CrossRef]
- Caicedo, A. Paracrine and autocrine interactions in the human islet: More than meets the eye. Semin. Cell Dev. Biol. 2013, 24, 11–21. [Google Scholar] [CrossRef]
- Barker, C.J.; Leibiger, I.B.; Berggren, P.-O. The pancreatic islet as a signaling hub. Adv. Biol. Regul. 2013, 53, 156–163. [Google Scholar] [CrossRef]
- Lammert, E.; Cleaver, O.; Melton, D. Role of endothelial cells in early pancreas and liver development. Mech. Dev. 2003, 120, 59–64. [Google Scholar] [CrossRef]
- Athirasala, A.; Hirsch, N.; Buxboim, A. Nuclear mechanotransduction: Sensing the force from within. Curr. Opin. Cell Biol. 2017, 46, 119–127. [Google Scholar] [CrossRef]
- Mamidi, A.; Prawiro, C.; Seymour, P.A.; De Lichtenberg, K.H.; Jackson, A.; Serup, P.; Semb, H. Mechanosignalling via integrins directs fate decisions of pancreatic progenitors. Nat. Cell Biol. 2018, 564, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells. Nat. Cell Biol. 2019, 21, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Velazco-Cruz, L.; Song, J.; Maxwell, K.G.; Goedegebuure, M.M.; Augsornworawat, P.; Hogrebe, N.J.; Millman, J.R. Acquisition of Dynamic Function in Human Stem Cell-Derived beta Cells. Stem Cell Rep. 2019, 12, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Hogrebe, N.J.; Augsornworawat, P.; Maxwell, K.G.; Velazco-Cruz, L.; Millman, J.R. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent Stem Cells. Nat. Biotechnol. 2020, 38, 460–470. [Google Scholar] [CrossRef]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef]
- McCarthy, M.I. Genomics, Type 2 Diabetes, and Obesity. N. Engl. J. Med. 2010, 363, 2339–2350. [Google Scholar] [CrossRef]
- Maehr, R.; Chen, S.; Snitow, M.; Ludwig, T.; Yagasaki, L.; Goland, R.; Leibel, R.L.; Melton, D.A. Generation of pluripotent Stem Cells from patients with type 1 diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 15768–15773. [Google Scholar] [CrossRef]
- Ohmine, S.; Squillace, K.A.; Hartjes, K.A.; Deeds, M.C.; Armstrong, A.S.; Thatava, T.; Sakuma, T.; Terzic, A.; Kudva, Y.; Ikeda, Y. Reprogrammed keratinocytes from elderly type 2 diabetes patients suppress senescence genes to acquire induced pluripotency. Aging 2012, 4, 60–73. [Google Scholar] [CrossRef]
- Zhao, T.; Zhang, Z.-N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent Stem Cells. Nat. Cell Biol. 2011, 474, 212–215. [Google Scholar] [CrossRef]
- Tang, C.; Weissman, I.L.; Drukker, M. Immunogenicity of in vitro maintained and matured populations: Potential barriers to engraftment of human pluripotent stem cell derivatives. In Embryonic Stem Cell Immunobiology. Methods in Molecular Biology; Zavazava, N., Ed.; Springer Science and Business Media LLC: Totowa, NJ, USA, 2013; Volume 1029, pp. 17–31. [Google Scholar] [CrossRef]
- Millman, J.R.; Xie, C.; Van Dervort, A.; Gürtler, M.; Pagliuca, F.W.; Melton, D.A. Generation of stem cell-derived β-cells from patients with type 1 diabetes. Nat. Commun. 2016, 7, 11463. [Google Scholar] [CrossRef]
- Balboa, D.; Saarimäki-Vire, J.; Borshagovski, D.; Survila, M.; Lindholm, P.; Galli, E.; Eurola, S.; Ustinov, J.; Grym, H.; Huopio, H.; et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. eLife 2018, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.C.; Warram, J.H.; Rosner, B.; Rich, S.S.; Soeldner, J.S.; Krolewski, A.S. Familial clustering of insulin sensitivity. Diabetes 1992, 41, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Shulman, R.G. Nuclear Magnetic Resonance Studies of Glucose Metabolism in Non-Insulin-Dependent Diabetes Mellitus Subjects. Mol. Med. 1996, 2, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Monga, S.P. Cellular and Molecular Basis of Liver Development. Compr. Physiol. 2013, 3, 799–815. [Google Scholar] [CrossRef] [PubMed]
- Ober, E.A.; Lemaigre, F.P. Development of the liver: Insights into organ and tissue morphogenesis. J. Hepatol. 2018, 68, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Nimgaonkar, I.; Chu, V.; Xia, Y.; Hu, Z.; Liang, T.J. Hepatic differentiation of human pluripotent Stem Cells in miniaturized format suitable for high-throughput screen. Stem Cell Res. 2016, 16, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Varghese, D.S.; Alawathugoda, T.T.; Ansari, S.A. Fine Tuning of Hepatocyte Differentiation from Human Embryonic Stem Cells: Growth Factor vs. Small Molecule-Based Approaches. Stem Cells Int. 2019, 2019, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Aihara, E.; Watson, C.; Mourya, R.; Mizuochi, T.; Shivakumar, P.; Phelan, K.; Mayhew, C.; Helmrath, M.; Takebe, T.; et al. Paracrine signals regulate human liver organoid maturation from induced pluripotent Stem Cells. Development 2017, 144, 1056–1064. [Google Scholar] [CrossRef]
- Balasiddaiah, A.; Moreno, D.; Guembe, L.; Prieto, J.; Aldabe, R. Hepatic differentiation of mouse iPS cells and analysis of liver engraftment potential of multistage iPS progeny. J. Physiol. Biochem. 2013, 69, 835–845. [Google Scholar] [CrossRef]
- Goulart, E.; De Caires-Junior, L.C.; Telles-Silva, K.A.; Araujo, B.H.S.; Rocco, S.A.; Sforca, M.; De Sousa, I.L.; Kobayashi, G.S.; Musso, C.M.; Assoni, A.F.; et al. 3D bioprinting of liver spheroids derived from human induced pluripotent Stem Cells sustain liver function and viability in vitro. Biofabrication 2019, 12, 015010. [Google Scholar] [CrossRef]
- Goulart, E.; De Caires-Junior, L.C.; Telles-Silva, K.A.; Araujo, B.H.S.; Kobayashi, G.S.; Musso, C.M.; Assoni, A.F.; Oliveira, D.; Caldini, E.; Gerstenhaber, J.A.; et al. Adult and iPS-derived non-parenchymal cells regulate liver organoid development through differential modulation of Wnt and TGF-β. Stem Cell Res. Ther. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.B.; D’Amour, K.A.; Jones, K.L.; Krishnamoorthy, M.; Kulik, M.J.; Reynolds, D.M.; Sheppard, A.M.; Liu, H.; Xu, Y.; Baetge, E.E.; et al. Activin A Efficiently Specifies Definitive Endoderm from Human Embryonic Stem Cells Only When Phosphatidylinositol 3-Kinase Signaling Is Suppressed. Stem Cells 2007, 25, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Massumi, M.; Pourasgari, F.; Nalla, A.; Batchuluun, B.; Nagy, K.; Neely, E.; Gull, R.; Nagy, A.; Wheeler, M.B. An Abbreviated Protocol for In Vitro Generation of Functional Human Embryonic Stem Cell-Derived Beta-Like Cells. PLoS ONE 2016, 11, e0164457. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, H.; Luu, N.-T.; Alwahsh, S.M.; Ginai, M.; Alhaque, S.; Dong, H.; Tomaz, R.A.; Vernay, B.; Vigneswara, V.; Hallett, J.M.; et al. 3D human liver tissue from pluripotent Stem Cells displays stable phenotype in vitro and supports compromised liver function in vivo. Arch. Toxicol. 2018, 92, 3117–3129. [Google Scholar] [CrossRef]
- Cho, Y.M.; Lim, J.M.; Yoo, D.H.; Kim, J.H.; Chung, S.S.; Park, S.G.; Kim, T.H.; Oh, S.K.; Choi, Y.M.; Moon, S.Y.; et al. Betacellulin and nicotinamide sustain PDX1 expression and induce pancreatic beta-cell differentiation in human embryonic Stem Cells. Biochem. Biophys. Res. Commun. 2008, 366, 129–134. [Google Scholar] [CrossRef]
- Swartz, E.W.; Baek, J.; Pribadi, M.; Wojta, K.J.; Almeida, S.; Karydas, A.; Gao, F.-B.; Miller, B.L.; Coppola, G. A Novel Protocol for Directed Differentiation of C9orf72-Associated Human Induced Pluripotent Stem Cells Into Contractile Skeletal Myotubes. Stem Cells Transl. Med. 2016, 5, 1461–1472. [Google Scholar] [CrossRef]
- Takeuchi, H.; Nakatsuji, N.; Suemori, H. Endodermal differentiation of human pluripotent Stem Cells to insulin-producing cells in 3D culture. Sci. Rep. 2014, 4, 4488. [Google Scholar] [CrossRef]
- Kim, S.K.; Melton, U.A. Pancreas development is promoted by cyclopamine, a Hedgehog signaling inhibitor. Proc. Natl. Acad. Sci. USA 1998, 95, 13036–13041. [Google Scholar] [CrossRef]
- D’Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of pancreatic hormone–expressing endocrine cells from human embryonic Stem Cells. Nat. Biotechnol. 2006, 24, 1392–1401. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, H.; Ko, U.H.; Oh, Y.; Lim, A.; Sohn, J.-W.; Shin, J.H.; Kim, H.; Han, Y.-M. Islet-like organoids derived from human pluripotent Stem Cells efficiently function in the glucose responsiveness in vitro and in vivo. Sci. Rep. 2016, 6, 35145. [Google Scholar] [CrossRef]
- Scott, M.A.; Nguyen, V.T.; Levi, B.; James, A.W. Current Methods of Adipogenic Differentiation of Mesenchymal Stem Cells. Stem Cells Dev. 2011, 20, 1793–1804. [Google Scholar] [CrossRef] [PubMed]
- Czysz, K.; Minger, S.; Thomas, N. DMSO Efficiently Down Regulates Pluripotency Genes in Human Embryonic Stem Cells during Definitive Endoderm Derivation and Increases the Proficiency of Hepatic Differentiation. PLoS ONE 2015, 10, e0117689. [Google Scholar] [CrossRef] [PubMed]
- Siller, R.; Greenhough, S.; Naumovska, E.; Sullivan, G.J. Small-Molecule-Driven Hepatocyte Differentiation of Human Pluripotent Stem Cells. Stem Cell Rep. 2015, 4, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, X.; Pineyro, M.A.; Egan, J.M. Glucagon-like peptide 1 and exendin-4 convert pancreatic AR42J cells into glucagon- and insulin-producing cells. Diabetes 1999, 48, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Korostylev, A.; Mahaddalkar, P.U.; Keminer, O.; Hadian, K.; Schorpp, K.; Gribbon, P.; Lickert, H. A high-content small molecule screen identifies novel inducers of definitive endoderm. Mol. Metab. 2017, 6, 640–650. [Google Scholar] [CrossRef]
- McGaugh, E.C.; Nostro, M.C. Efficient Differentiation of Pluripotent Stem Cells to NKX6-1+ Pancreatic Progenitors. J. Vis. Exp. 2017, 2017. [Google Scholar] [CrossRef]
- Shim, J.-H.; Kim, J.; Han, J.; An, S.Y.; Jang, Y.J.; Son, J.; Woo, N.-H.; Kim, S.-K.; Kim, J.S. Pancreatic Islet-Like Three-Dimensional Aggregates Derived from Human Embryonic Stem Cells Ameliorate Hyperglycemia in Streptozotocin-Induced Diabetic Mice. Cell Transplant. 2015, 24, 2155–2168. [Google Scholar] [CrossRef]
- Iovino, S.; Burkart, A.M.; Warren, L.; Patti, M.E.; Kahn, C.R. Myotubes derived from human-induced pluripotent Stem Cells mirror in vivo insulin resistance. Proc. Natl. Acad. Sci. USA 2016, 113, 1889–1894. [Google Scholar] [CrossRef]
- Tanaka, A.; Woltjen, K.; Miyake, K.; Hotta, A.; Ikeya, M.; Yamamoto, T.; Nishino, T.; Shoji, E.; Sehara-Fujisawa, A.; Manabe, Y.; et al. Efficient and reproducible myogenic differentiation from human iPS cells: Prospects for modeling Miyoshi Myopathy in vitro. PLoS ONE 2013, 8, e61540. [Google Scholar] [CrossRef]
- Sullivan, G.J.; Hay, D.C.; Park, I.-H.; Fletcher, J.; Hannoun, Z.; Payne, C.M.; Dalgetty, D.; Black, J.R.; Ross, J.A.; Samuel, K.; et al. Generation of functional human hepatic endoderm from human induced pluripotent Stem Cells. Hepatology 2010, 51, 329–335. [Google Scholar] [CrossRef]
- Du, C.; Feng, Y.; Qiu, D.; Xu, Y.; Pang, M.; Cai, N.; Xiang, A.P.; Zhang, Q. Highly efficient and expedited hepatic differentiation from human pluripotent Stem Cells by pure small-molecule cocktails. Stem Cell Res. Ther. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; You, L.; Cui, X.; Li, Y.; Wang, X.; Xu, P.; Zhu, L.; Wen, J.; Pang, L.; Guo, X.; et al. Evaluation and optimization of differentiation conditions for human primary brown adipocytes. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Borowiak, M.; Maehr, R.; Chen, S.; Chen, A.E.; Tang, W.; Fox, J.L.; Schreiber, S.L.; Melton, D.A. Small Molecules Efficiently Direct Endodermal Differentiation of Mouse and Human Embryonic Stem Cells. Cell Stem Cell 2009, 4, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Al Tanoury, Z.; Hestin, M.; Gobert, B.; Aivio, S.; Hick, A.; Cherrier, T.; Nesmith, A.P.; Parker, K.K.; Pourquié, O. Generation of human muscle fibers and satellite-like cells from human pluripotent Stem Cells in vitro. Nat. Protoc. 2016, 11, 1833–1850. [Google Scholar] [CrossRef]
- Chen, S.; Borowiak, M.; Fox, J.L.; Maehr, R.; Osafune, K.; Davidow, L.S.; Lam, K.; Peng, L.F.; Schreiber, S.L.; Rubin, L.L.; et al. A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat. Chem. Biol. 2009, 5, 258–265. [Google Scholar] [CrossRef]
- Wang, Q.; Donelan, W.; Ye, H.; Jin, Y.; Lin, Y.; Wu, X.; Wang, Y.; Xi, Y. Real-time observation of pancreatic beta cell differentiation from human induced pluripotent Stem Cells. Am. J. Transl. Res. 2019, 11, 3490–3504. [Google Scholar]
- Van Der Wal, E.; Herrero-Hernandez, P.; Wan, R.; Broeders, M.; Groen, S.L.; Van Gestel, T.J.; Van Ijcken, W.F.; Cheung, T.H.; Van Der Ploeg, A.T.; Schaaf, G.J.; et al. Large-Scale Expansion of Human iPSC-Derived Skeletal Muscle Cells for Disease Modeling and Cell-Based Therapeutic Strategies. Stem Cell Rep. 2018, 10, 1975–1990. [Google Scholar] [CrossRef]
- A Russ, H.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef]
- Tan, M.; Jiang, L.; Li, Y.; Jiang, W. Dual Inhibition of BMP and WNT Signals Promotes Pancreatic Differentiation from Human Pluripotent Stem Cells. Stem Cells Int. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- Zhu, Z.; Chew-Li, S.; Lee, K.; Rosen, B.P.; González, F.; Soh, C.-L.; Huangfu, D. Genome Editing of Lineage Determinants in Human Pluripotent Stem Cells Reveals Mechanisms of Pancreatic Development and Diabetes. Cell Stem Cell 2016, 18, 755–768. [Google Scholar] [CrossRef]
- Tomizawa, M.; Shinozaki, F.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Ishige, N. Improved Survival and Initiation of Differentiation of Human Induced Pluripotent Stem Cells to Hepatocyte-Like Cells upon Culture in William’s E Medium followed by Hepatocyte Differentiation Inducer Treatment. PLoS ONE 2016, 11, e0153435. [Google Scholar] [CrossRef] [PubMed]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent Stem Cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Sandouk, T.; Reda, D.; Hofmann, C. Antidiabetic agent pioglitazone enhances adipocyte differentiation of 3T3-F442A cells. Am. J. Physiol. Physiol. 1993, 264, C1600–C1608. [Google Scholar] [CrossRef] [PubMed]
- Ahfeldt, T.; Schinzel, R.T.; Lee, Y.-K.; Hendrickson, D.; Kaplan, A.; Lum, D.H.; Camahort, R.; Xia, F.; Shay, J.; Rhee, E.P.; et al. Programming human pluripotent Stem Cells into white and brown adipocytes. Nat. Cell Biol. 2012, 14, 209–219. [Google Scholar] [CrossRef]
- Pezzolla, D.; López-Beas, J.; Lachaud, C.C.; Domínguez-Rodríguez, A.; Smani, T.; Hmadcha, A.; Soria, B. Resveratrol ameliorates the maturation process of beta-cell-like cells obtained from an optimized differentiation protocol of human embryonic Stem Cells. PLoS ONE 2015, 10, e0119904. [Google Scholar] [CrossRef]
- Lorberbaum, D.S.; Kishore, S.; Rosselot, C.; Sarbaugh, D.; Brooks, E.P.; Aragon, E.; Xuan, S.; Simon, O.; Ghosh, D.; Mendelsohn, C.; et al. Retinoic acid signaling within pancreatic endocrine progenitors regulates mouse and human beta cell specification. Development 2020, 147. [Google Scholar] [CrossRef]
- Öström, M.; Loffler, K.A.; Edfalk, S.; Selander, L.; Dahl, U.; Ricordi, C.; Jeon, J.; Correa-Medina, M.; Diez, J.; Edlund, H. Retinoic acid promotes the generation of pancreatic endocrine progenitor cells and their further differentiation into beta-cells. PLoS ONE 2008, 3, e2841. [Google Scholar] [CrossRef]
- Villarroya, J.; Giralt, M.; Iglesias, R. Retinoids and adipose tissues: Metabolism, cell differentiation and gene expression. Int. J. Obes. 1999, 23, 1–6. [Google Scholar] [CrossRef]
- Fayyad, A.M.; Khan, A.A.; Abdallah, S.H.; Alomran, S.S.; Bajou, K.; Khattak, M.N.K. Rosiglitazone Enhances Browning Adipocytes in Association with MAPK and PI3-K Pathways During the Differentiation of Telomerase-Transformed Mesenchymal Stromal Cells into Adipocytes. Int. J. Mol. Sci. 2019, 20, 1618. [Google Scholar] [CrossRef]
- Ren, M.; Yan, L.; Shang, C.; Cao, J.; Min, J.; Cheng, H.; Lu, L.-H. Effects of sodium butyrate on the differentiation of pancreatic and hepatic progenitor cells from mouse embryonic Stem Cells. J. Cell. Biochem. 2009, 109, 236–244. [Google Scholar] [CrossRef]
- Kondo, Y.; Toyoda, T.; Ito, R.; Funato, M.; Hosokawa, Y.; Matsui, S.; Sudo, T.; Nakamura, M.; Okada, C.; Zhuang, X.; et al. Identification of a small molecule that facilitates the differentiation of human iPSCs/ESCs and mouse embryonic pancreatic explants into pancreatic endocrine cells. Diabetologia 2017, 60, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wurdak, H.; Wang, J.; Lyssiotis, C.A.; Peters, E.C.; Cho, C.Y.; Wu, X.; Schultz, P.G. A Small Molecule Primes Embryonic Stem Cells for Differentiation. Cell Stem Cell 2009, 4, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Phadnis, S.; Nair, P.D.; Bhonde, R.R. Generation of Pancreatic Hormone-Expressing Islet-Like Cell Aggregates from Murine Adipose Tissue-Derived Stem Cells. Stem Cells 2009, 27, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Guénantin, A.-C.; Briand, N.; Capel, E.; Dumont, F.; Morichon, R.; Provost, C.; Stillitano, F.; Jeziorowska, R.; Siffroi, J.-P.; Hajjar, R.J.; et al. Functional Human Beige Adipocytes From Induced Pluripotent Stem Cells. Diabetes 2017, 66, 1470–1478. [Google Scholar] [CrossRef]
- Ang, L.T.; Tan, A.K.Y.; Autio, M.I.; Goh, S.H.; Choo, S.H.; Lee, K.L.; Tan, J.; Pan, B.; Lee, J.J.H.; Lum, J.J.; et al. A Roadmap for Human Liver Differentiation from Pluripotent Stem Cells. Cell Rep. 2018, 22, 2190–2205. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-H.; Chang, C.-C.; Huang, H.-C.; Chen, Y.-J.; Tsai, P.-H.; Jeng, S.-Y.; Hung, S.-I.; Hsieh, J.-H.; Huang, H.-S.; Chiou, S.-H.; et al. Investigation of Hepatoprotective Activity of Induced Pluripotent Stem Cells in the Mouse Model of Liver Injury. J. Biomed. Biotechnol. 2011, 2011, 1–11. [Google Scholar] [CrossRef]
- Baxter, M.; Withey, S.; Harrison, S.; Segeritz, C.-P.; Zhang, F.; Atkinson-Dell, R.; Rowena, S.-Y.; Gerrard, D.T.; Sison-Young, R.; Jenkins, R.; et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 2015, 62, 581–589. [Google Scholar] [CrossRef]
- Gao, X.; Liu, Y. A transcriptomic study suggesting human iPSC-derived hepatocytes potentially offer a better in vitro model of hepatotoxicity than most hepatoma cell lines. Cell Biol. Toxicol. 2017, 33, 407–421. [Google Scholar] [CrossRef]
- Grant, R.; Hay, D.C.; Callanan, A. A Drug-Induced Hybrid Electrospun Poly-Capro-Lactone: Cell-Derived Extracellular Matrix Scaffold for Liver Tissue Engineering. Tissue Eng. Part A 2017, 23, 650–662. [Google Scholar] [CrossRef]
- Grant, R.; Hallett, J.; Forbes, S.; Hay, D.; Callanan, A. Blended electrospinning with human liver extracellular matrix for engineering new hepatic microenvironments. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Huse, S.M.; Gruppuso, P.A.; Boekelheide, K.; Sanders, J.A. Patterns of gene expression and DNA methylation in human fetal and adult liver. BMC Genom. 2015, 16, 981. [Google Scholar] [CrossRef]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent Stem Cells. J. Clin. Investig. 2010, 120, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rashid, S.T.; Strick-Marchand, H.; Varela, I.; Liu, P.-Q.; Paschon, D.E.; Miranda, E.; Ordóñez, A.; Hannan, N.R.F.; Rouhani, F.J.; et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent Stem Cells. Nat. Cell Biol. 2011, 478, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Cai, J.; Liu, Y.; Zhao, D.; Yong, J.; Duo, S.; Song, X.; Guo, Y.; Zhao, Y.; Qin, H.; et al. Efficient generation of hepatocyte-like cells from human induced pluripotent Stem Cells. Cell Res. 2009, 19, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Grefte, S.; Kuijpers-Jagtman, A.M.; Torensma, R.; Hoff, J.W.V.D. Skeletal Muscle Development and Regeneration. Stem Cells Dev. 2007, 16, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, P.; Gruss, P. Pax: Genes for mice and men. Pharmacol. Ther. 1994, 61, 205–226. [Google Scholar] [CrossRef]
- Buckingham, M.; Bajard, L.; Chang, T.; Daubas, P.; Hadchouel, J.; Meilhac, S.; Montarras, D.; Rocancourt, D.; Relaix, F. The formation of skeletal muscle: From somite to limb. J. Anat. 2003, 202, 59–68. [Google Scholar] [CrossRef]
- Weintraub, H.; Tapscott, S.J.; Davis, R.L.; Thayer, M.J.; Adam, M.A.; Lassar, A.B.; Miller, A.D. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc. Natl. Acad. Sci. USA 1989, 86, 5434–5438. [Google Scholar] [CrossRef]
- Darabi, R.; Arpke, R.W.; Irion, S.; Dimos, J.T.; Grskovic, M.; Kyba, M.; Perlingeiro, R.C.R. Human ES- and iPS-Derived Myogenic Progenitors Restore DYSTROPHIN and Improve Contractility upon Transplantation in Dystrophic Mice. Cell Stem Cell 2012, 10, 610–619. [Google Scholar] [CrossRef]
- Borchin, B.; Chen, J.; Barberi, T. Derivation and FACS-mediated purification of PAX3+/PAX7+ skeletal muscle precursors from human pluripotent Stem Cells. Stem Cell Rep. 2013, 1, 620–631. [Google Scholar] [CrossRef]
- De La Serna, I.L.; Roy, K.; Carlson, K.A.; Imbalzano, A.N. MyoD Can Induce Cell Cycle Arrest but Not Muscle Differentiation in the Presence of Dominant Negative SWI/SNF Chromatin Remodeling Enzymes. J. Biol. Chem. 2001, 276, 41486–41491. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building Muscle: Molecular Regulation of Myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.; Metz, J.; Liu, J.; Carpenedo, R.L.; Demers, S.-P.; Stanford, W.L.; Skerjanc, I.S. Derivation and Expansion of PAX7-Positive Muscle Progenitors from Human and Mouse Embryonic Stem Cells. Stem Cell Rep. 2014, 3, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Tabebordbar, M.; Iovino, S.; Ciarlo, C.; Liu, J.; Castiglioni, A.; Price, E.; Liu, M.; Barton, E.R.; Kahn, C.R.; et al. A Zebrafish Embryo Culture System Defines Factors that Promote Vertebrate Myogenesis across Species. Cell 2013, 155, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Oginuma, M.; Al Tanoury, Z.; Gobert, B.; Sumara, O.; Hick, A.; Bousson, F.; Zidouni, Y.; Mursch, C.; Moncuquet, P.; et al. Differentiation of pluripotent Stem Cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol. 2015, 33, 962–969. [Google Scholar] [CrossRef]
- Caron, L.; Kher, D.; Lee, K.L.; McKernan, R.; Dumevska, B.; Hidalgo, A.; Leslie, C.; Yang, H.; Main, H.; Ferri, G.; et al. A Human Pluripotent Stem Cell Model of Facioscapulohumeral Muscular Dystrophy-Affected Skeletal Muscles. Stem Cells Transl. Med. 2016, 5, 1145–1161. [Google Scholar] [CrossRef]
- Chetty, S.; Pagliuca, F.W.; Honore, C.; Kweudjeu, A.; Rezania, A.; Melton, D.A. A simple tool to improve pluripotent stem cell differentiation. Nat. Methods 2013, 10, 553–556. [Google Scholar] [CrossRef]
- Aulehla, A.; Pourquié, O. Signaling Gradients during Paraxial Mesoderm Development. Cold Spring Harb. Perspect. Biol. 2009, 2, a000869. [Google Scholar] [CrossRef]
- Balhara, B.; Burkart, A.; Topcu, V.; Lee, Y.-K.; Cowan, C.; Kahn, C.R.; Patti, M.-E. Severe Insulin Resistance Alters Metabolism in Mesenchymal Progenitor Cells. Endocrinology 2015, 156, 2039–2048. [Google Scholar] [CrossRef]
- Wang, L.; Schulz, T.C.; Sherrer, E.S.; Dauphin, D.S.; Shin, S.; Nelson, A.M.; Ware, C.B.; Zhan, M.; Song, C.-Z.; Chen, X.; et al. Self-renewal of human embryonic Stem Cells requires insulin-like growth factor-1 receptor and ERBB2 receptor signaling. Blood 2007, 110, 4111–4119. [Google Scholar] [CrossRef]
- Burkart, A.M.; Tan, K.; Warren, L.; Iovino, S.; Hughes, K.J.; Kahn, C.R.; Patti, M.-E. Insulin Resistance in Human iPS Cells Reduces Mitochondrial Size and Function. Sci. Rep. 2016, 6, 22788. [Google Scholar] [CrossRef] [PubMed]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetology 2005, 48, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Pravenec, M.; Hyakukoku, M.; Houstek, J.; Zidek, V.; Landa, V.; Mlejnek, P.; Miksik, I.; Dudová-Mothejzikova, K.; Pecina, P.; Vrbacký, M.; et al. Direct linkage of mitochondrial genome variation to risk factors for type 2 diabetes in conplastic strains. Genome Res. 2007, 17, 1319–1326. [Google Scholar] [CrossRef]
- Crawford, S.; Hoogeveen, R.C.; Brancati, F.L.; Astor, B.C.; Ballantyne, C.M.; Schmidt, M.I.; Young, J.H. Association of blood lactate with type 2 diabetes: The Atherosclerosis Risk in Communities Carotid MRI Study. Int. J. Epidemiol. 2010, 39, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, J.; Newby, F.; Gebhart, S.; DiGirolamo, M. Insulin resistance in obesity is associated with elevated basal lactate levels and diminished lactate appearance following intravenous glucose and insulin. Metabolism 1992, 41, 22–27. [Google Scholar] [CrossRef]
- Ørtenblad, N.; Mogensen, M.; Petersen, I.; Højlund, K.; Levin, K.; Sahlin, K.; Beck-Nielsen, H.; Gaster, M. Reduced insulin-mediated citrate synthase activity in cultured skeletal muscle cells from patients with type 2 diabetes: Evidence for an intrinsic oxidative enzyme defect. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2005, 1741, 206–214. [Google Scholar] [CrossRef]
- Christe, M.; Hirzel, E.; Lindinger, A.; Kern, B.; Von Flüe, M.; Peterli, R.; Peters, T.; Eberle, A.N.; Lindinger, P.W. Obesity Affects Mitochondrial Citrate Synthase in Human Omental Adipose Tissue. ISRN Obes. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Amilon, K.R.; Cortes-Araya, Y.; Moore, B.; Lee, S.; Lillico, S.G.; Breton, A.; Esteves, C.L.; Donadeu, F.X. Generation of Functional Myocytes from Equine Induced Pluripotent Stem Cells. Cell. Reprogramm. 2018, 20, 275–281. [Google Scholar] [CrossRef]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.-H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige Adipocytes Are a Distinct Type of Thermogenic Fat Cell in Mouse and Human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef]
- Cinti, S. The adipose organ at a glance. Dis. Model. Mech. 2012, 5, 588–594. [Google Scholar] [CrossRef]
- Ema, X.; Elee, P.; Chisholm, D.J.; James, D.E. Control of Adipocyte Differentiation in Different Fat Depots; Implications for Pathophysiology or Therapy. Front. Endocrinol. 2015, 6, 1. [Google Scholar] [CrossRef]
- Moseti, D.; Regassa, A.; Kim, W.-K. Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules. Int. J. Mol. Sci. 2016, 17, 124. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tang, J.; Gao, X.; Ruan, H.-B. Inhibition of PPAR Gamma, Adipogenesis, and Insulin Sensitivity by MAGED1. Diabetes 2018, 67, 1750. [Google Scholar] [CrossRef]
- Linhart, H.G.; Ishimura-Oka, K.; DeMayo, F.; Kibe, T.; Repka, D.; Poindexter, B.; Bick, R.J.; Darlington, G.J. C/EBP is required for differentiation of white, but not brown, adipose tissue. Proc. Natl. Acad. Sci. USA 2001, 98, 12532–12537. [Google Scholar] [CrossRef]
- Supruniuk, E.; Mikłosz, A.; Chabowski, A. The Implication of PGC-1α on Fatty Acid Transport across Plasma and Mitochondrial Membranes in the Insulin Sensitive Tissues. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Seale, P.; Conroe, H.M.; Estall, J.; Kajimura, S.; Frontini, A.; Ishibashi, J.; Cohen, P.; Cinti, S.; Spiegelman, B.M. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Investig. 2011, 121, 96–105. [Google Scholar] [CrossRef]
- Cohen, P.; Levy, J.D.; Zhang, Y.; Frontini, A.; Kolodin, D.P.; Svensson, K.J.; Lo, J.C.; Zeng, X.; Ye, L.; Khandekar, M.J.; et al. Ablation of PRDM16 and Beige Adipose Causes Metabolic Dysfunction and a Subcutaneous to Visceral Fat Switch. Cell 2014, 156, 304–316. [Google Scholar] [CrossRef]
- Taura, D.; Noguchi, M.; Sone, M.; Hosoda, K.; Mori, E.; Okada, Y.; Takahashi, K.; Homma, K.; Oyamada, N.; Inuzuka, M.; et al. Adipogenic differentiation of human induced pluripotent Stem Cells: Comparison with that of human embryonic Stem Cells. FEBS Lett. 2009, 583, 1029–1033. [Google Scholar] [CrossRef]
- Karam, M.; Younis, I.; Elareer, N.R.; Nasser, S.; Abdelalim, E.M. Scalable Generation of Mesenchymal Stem Cells and Adipocytes from Human Pluripotent Stem Cells. Cells 2020, 9, 710. [Google Scholar] [CrossRef]
- Pettinato, G.; Wen, X.; Zhang, N. Formation of Well-defined Embryoid Bodies from Dissociated Human Induced Pluripotent Stem Cells using Microfabricated Cell-repellent Microwell Arrays. Sci. Rep. 2014, 4, 7402. [Google Scholar] [CrossRef]
- Mori, E.; Fujikura, J.; Noguchi, M.; Nakao, K.; Matsubara, M.; Sone, M.; Taura, D.; Kusakabe, T.; Ebihara, K.; Tanaka, T.; et al. Impaired adipogenic capacity in induced pluripotent Stem Cells from lipodystrophic patients with BSCL2 mutations. Metabolism 2016, 65, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Hosoda, K.; Nakane, M.; Mori, E.; Nakao, K.; Taura, D.; Yamamoto, Y.; Kusakabe, T.; Sone, M.; Sakurai, H.; et al. In Vitro Characterization and Engraftment of Adipocytes Derived from Human Induced Pluripotent Stem Cells and Embryonic Stem Cells. Stem Cells Dev. 2013, 22, 2895–2905. [Google Scholar] [CrossRef] [PubMed]
- Marquez, M.P.; Alencastro, F.; Madrigal, A.; Jimenez, J.L.; Blanco, G.; Gureghian, A.; Keagy, L.; Lee, C.; Liu, R.; Tan, L.; et al. The Role of Cellular Proliferation in Adipogenic Differentiation of Human Adipose Tissue-Derived Mesenchymal Stem Cells. Stem Cells Dev. 2017, 26, 1578–1595. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; He, J.; Jiang, H.; Zu, L.; Zhai, W.; Pu, S.; Xu, G. Direct effect of glucocorticoids on lipolysis in adipocytes. Mol. Endocrinol. 2009, 23, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; I Barnes, R.; Garg, A. Genetic basis of congenital generalized lipodystrophy. Int. J. Obes. 2003, 28, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Kishida, T.; Ejima, A.; Yamamoto, K.; Tanaka, S.; Yamamoto, T.; Mazda, O. Reprogrammed Functional Brown Adipocytes Ameliorate Insulin Resistance and Dyslipidemia in Diet-Induced Obesity and Type 2 Diabetes. Stem Cell Rep. 2015, 5, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Guntur, A.R.; Nguyen, D.C.; Fakory, S.S.; Doucette, C.C.; Leech, C.; Lotana, H.; Kelley, M.; Kohli, J.; Martino, J.; et al. A Renewable Source of Human Beige Adipocytes for Development of Therapies to Treat Metabolic Syndrome. Cell Rep. 2018, 25, 3215–3228.e9. [Google Scholar] [CrossRef]
- Lizcano, F.; Vargas, D.; Gómez, Á.; Torrado, A. Human ADMC-Derived Adipocyte Thermogenic Capacity Is Regulated by IL-4 Receptor. Stem Cells Int. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Jiang, Y.; Berry, D.C.; Graff, J.M. Distinct cellular and molecular mechanisms for β3 adrenergic receptor-induced beige adipocyte formation. eLife 2017, 6, e30329. [Google Scholar] [CrossRef]
- Mohsen-Kanson, T.; Hafner, A.-L.; Wdziekonski, B.; Takashima, Y.; Villageois, P.; Carrière, A.; Svensson, M.; Bagnis, C.; Chignon-Sicard, B.; Svensson, P.-A.; et al. Differentiation of Human Induced Pluripotent Stem Cells into Brown and White Adipocytes: Role of Pax3. Stem Cells 2014, 32, 1459–1467. [Google Scholar] [CrossRef]
- Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the safety of induced pluripotent stem cell lines. Nat. Biotechnol. 2009, 27, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, M.; Aoi, T.; Okita, K.; Takahashi, R.; Inoue, H.; Takayama, N.; Endo, H.; Eto, K.; Toguchida, J.; Uemoto, S.; et al. Donor-dependent variations in hepatic differentiation from human-induced pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12538–12543. [Google Scholar] [CrossRef] [PubMed]
- Suhr, S.T.; Chang, E.A.; Tjong, J.; Alcasid, N.; Perkins, G.A.; Goissis, M.D.; Ellisman, M.H.; Perez, G.I.; Cibelli, J.B. Mitochondrial Rejuvenation After Induced Pluripotency. PLoS ONE 2010, 5, e14095. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent Stem Cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef]
- Helman, A.; Cangelosi, A.L.; Davis, J.C.; Pham, Q.; Rothman, A.; Faust, A.L.; Straubhaar, J.R.; Sabatini, D.M.; Melton, D.A. A Nutrient-Sensing Transition at Birth Triggers Glucose-Responsive Insulin Secretion. Cell Metab. 2020, 31, 1004–1016.e5. [Google Scholar] [CrossRef]
- Jaafar, R.; Tran, S.; Shah, A.N.; Sun, G.; Valdearcos, M.; Marchetti, P.; Masini, M.; Swisa, A.; Giacometti, S.; Bernal-Mizrachi, E.; et al. mTORC1-to-AMPK switching underlies β cell metabolic plasticity during maturation and diabetes. J. Clin. Investig. 2019, 129, 4124–4137. [Google Scholar] [CrossRef]
- Higuchi, A.; Ling, Q.-D.; Kumar, S.S.; A Munusamy, M.; A Alarfaj, A.; Chang, Y.; Kao, S.-H.; Lin, K.-C.; Wang, H.-C.; Umezawa, A. Generation of pluripotent Stem Cells without the use of genetic material. Lab. Investig. 2014, 95, 26–42. [Google Scholar] [CrossRef]
- Higuchi, A.; Ling, Q.-D.; Kumar, S.S.; Munusamy, M.; Alarfajj, A.A.; Umezawa, A.; Wu, G.-J. Design of polymeric materials for culturing human pluripotent Stem Cells: Progress toward feeder-free and xeno-free culturing. Prog. Polym. Sci. 2014, 39, 1348–1374. [Google Scholar] [CrossRef]
- Agulnick, A.D.; Ambruzs, D.M.; Moorman, M.A.; Bhoumik, A.; Cesario, R.M.; Payne, J.K.; Kelly, J.R.; Haakmeester, C.; Srijemac, R.; Wilson, A.Z.; et al. Insulin-Producing Endocrine Cells Differentiated In Vitro From Human Embryonic Stem Cells Function in Macroencapsulation Devices In Vivo. Stem Cells Transl. Med. 2015, 4, 1214–1222. [Google Scholar] [CrossRef]
- Wan, J.; Huang, Y.; Zhou, P.; Guo, Y.; Wu, C.; Zhu, S.; Wang, Y.; Wang, L.; Lu, Y.; Wang, Z. Culture of iPSCs Derived Pancreatic β-Like Cells In Vitro Using Decellularized Pancreatic Scaffolds: A Preliminary Trial. BioMed Res. Int. 2017, 2017, 4276928. [Google Scholar] [CrossRef]
- Xu, H.; Wang, B.; Ono, M.; Kagita, A.; Fujii, K.; Sasakawa, N.; Ueda, T.; Gee, P.; Nishikawa, M.; Nomura, M.; et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell 2019, 24, 566–578.e7. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, M.; Duan, S.; Franco, P.J.; Kenty, J.H.-R.; Hedrick, P.; Xia, Y.; Allen, A.; Ferreira, L.M.R.; Strominger, J.L.; et al. Generation of hypoimmunogenic human pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 10441–10446. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gheibi, S.; Singh, T.; da Cunha, J.P.M.C.M.; Fex, M.; Mulder, H. Insulin/Glucose-Responsive Cells Derived from Induced Pluripotent Stem Cells: Disease Modeling and Treatment of Diabetes. Cells 2020, 9, 2465. https://doi.org/10.3390/cells9112465
Gheibi S, Singh T, da Cunha JPMCM, Fex M, Mulder H. Insulin/Glucose-Responsive Cells Derived from Induced Pluripotent Stem Cells: Disease Modeling and Treatment of Diabetes. Cells. 2020; 9(11):2465. https://doi.org/10.3390/cells9112465
Chicago/Turabian StyleGheibi, Sevda, Tania Singh, Joao Paulo M. C. M. da Cunha, Malin Fex, and Hindrik Mulder. 2020. "Insulin/Glucose-Responsive Cells Derived from Induced Pluripotent Stem Cells: Disease Modeling and Treatment of Diabetes" Cells 9, no. 11: 2465. https://doi.org/10.3390/cells9112465
APA StyleGheibi, S., Singh, T., da Cunha, J. P. M. C. M., Fex, M., & Mulder, H. (2020). Insulin/Glucose-Responsive Cells Derived from Induced Pluripotent Stem Cells: Disease Modeling and Treatment of Diabetes. Cells, 9(11), 2465. https://doi.org/10.3390/cells9112465