Reading between the (Genetic) Lines: How Epigenetics is Unlocking Novel Therapies for Type 1 Diabetes


Abstract
1. Introduction
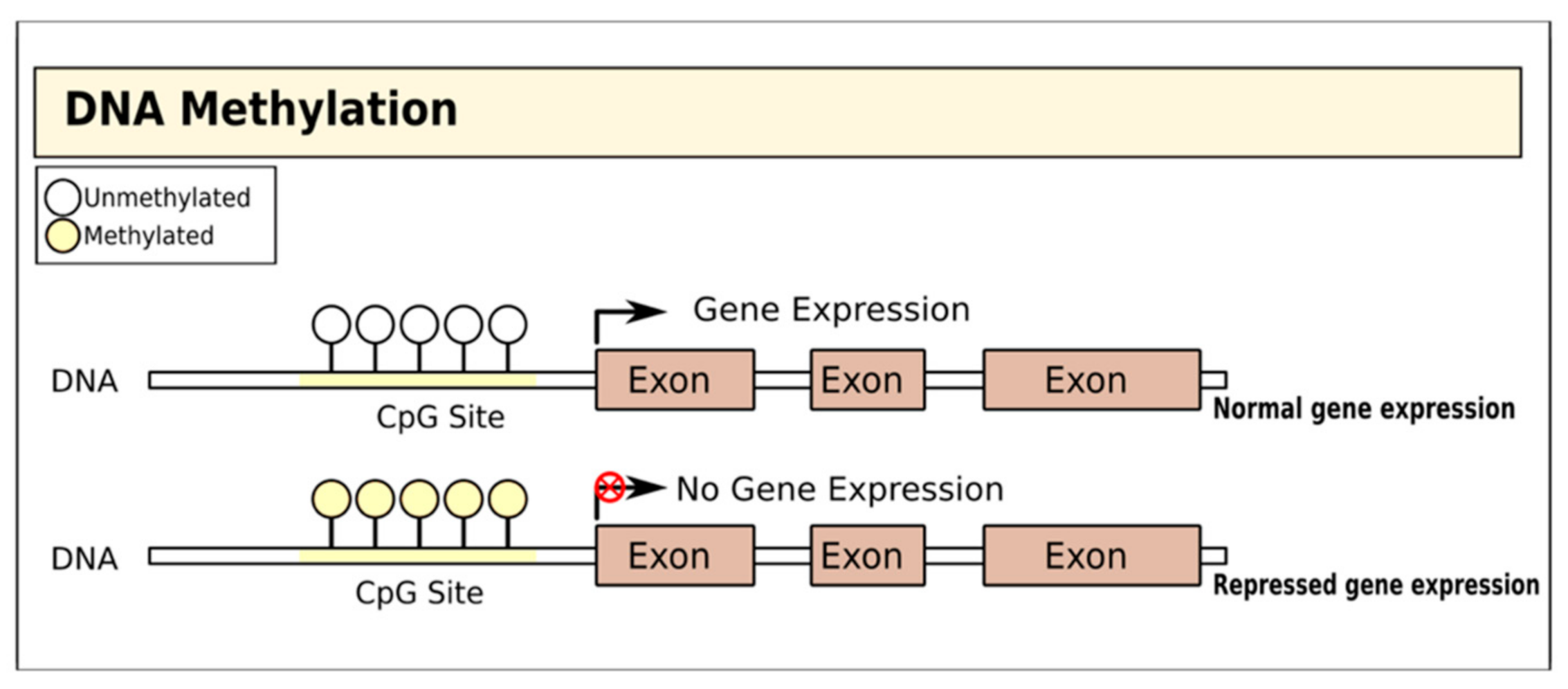
2. Therapies Targeting DNA Methylation in T1D
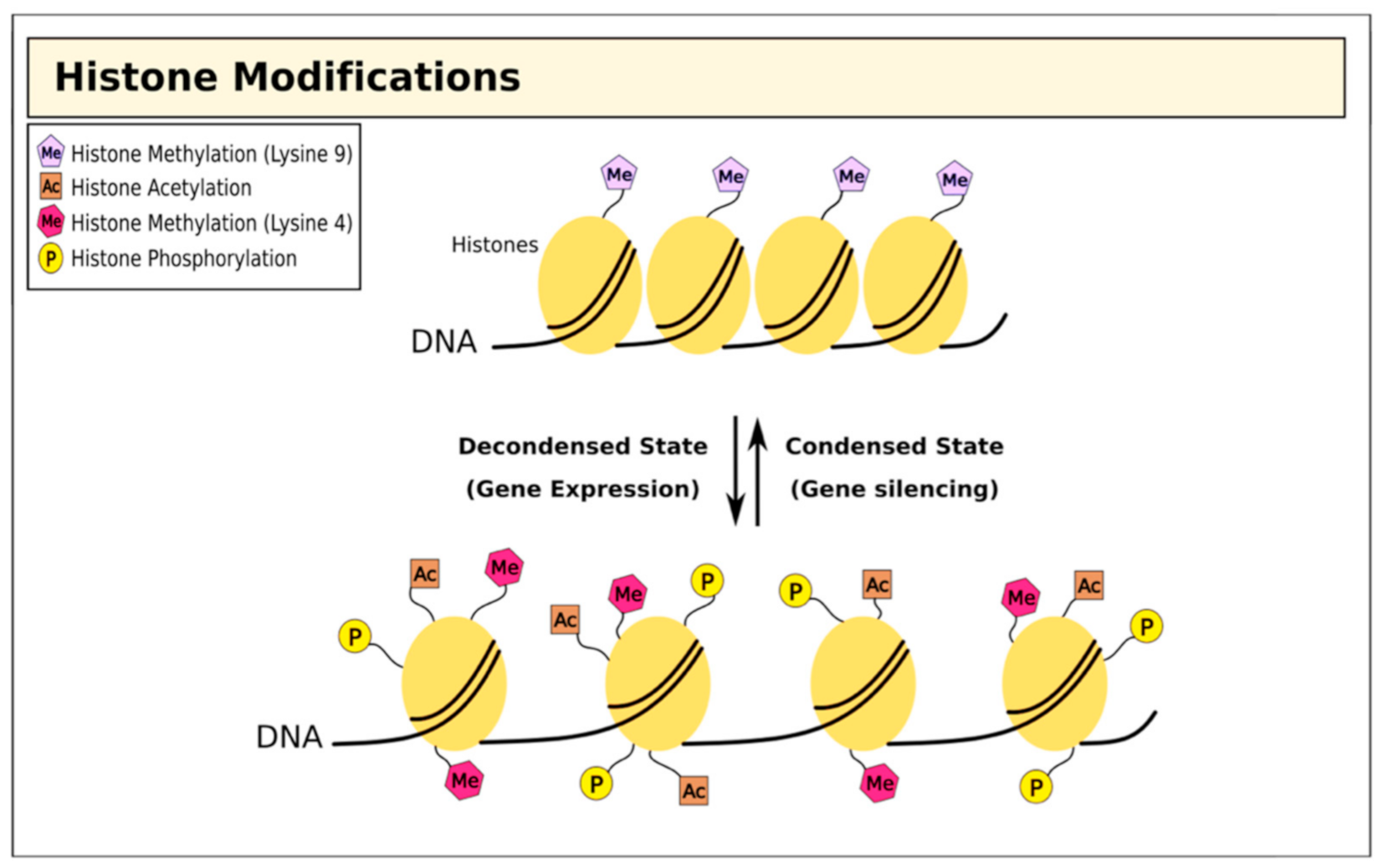
3. Therapies Targeting Histone Modifications in T1D
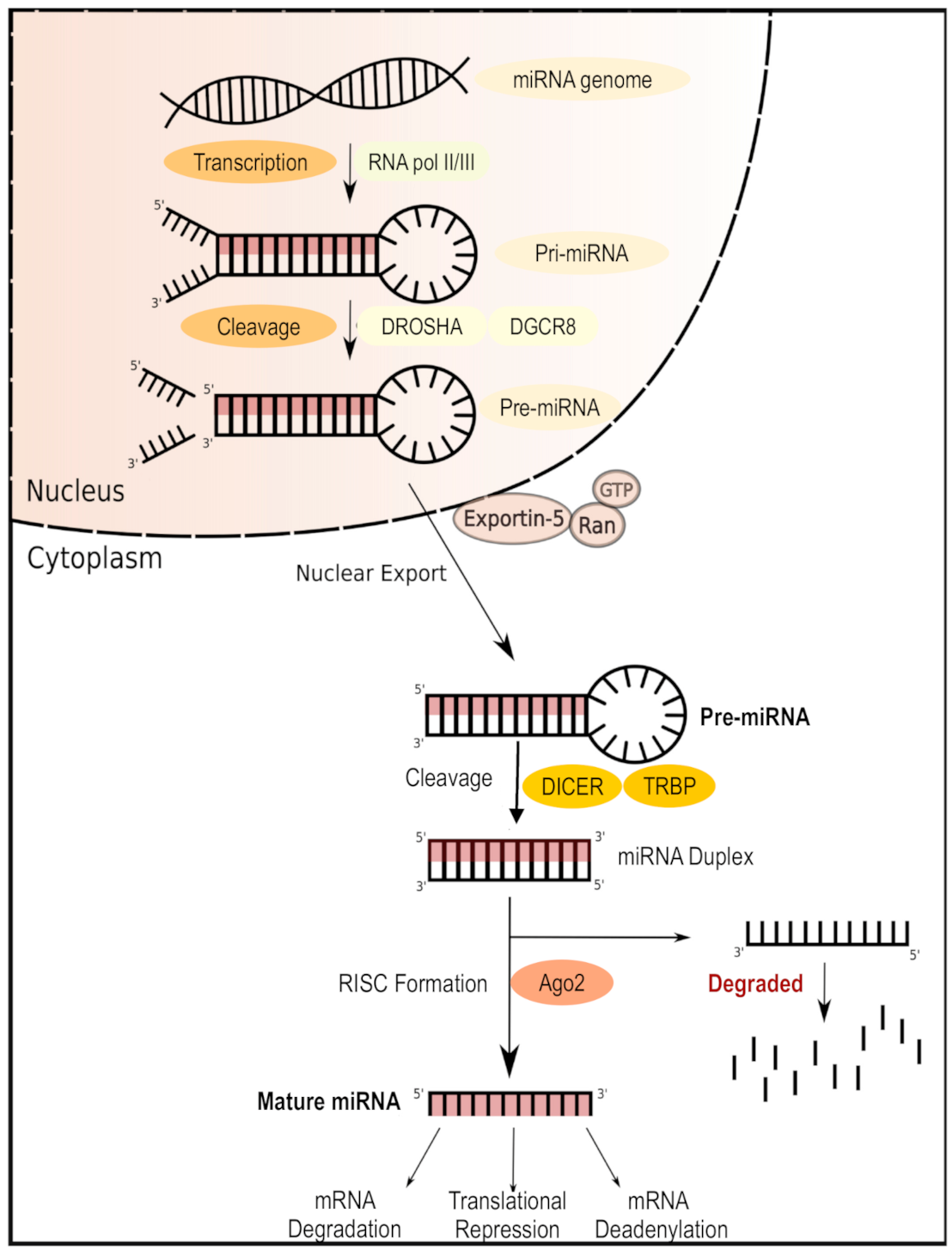
4. Therapies Targeting MicroRNAs in T1D

{kind=link}
{kind=link}
{kind=link}
| miRNA | Target Gene/Pathway Related to T1D | Regulation T1D Patients/Controls | Tissue/Cell | Reference |
|---|---|---|---|---|
| miR-101-3p | c-MET proto-oncogene, receptor tyrosine kinase (c-Met-HGF), ephrin receptor, and signal transducer and activator of transcription 3 (STAT3) pathways linked to insulin secretion and β cells survival | Upregulated | Serum | [65] |
| miR-125b-5p and miR-365a-3p | variation of hyperglycemia, measured (HbA1c) | Upregulated | Plasma | [66] |
| miR-5190 and miR-770-5p | variation of hyperglycemia, measured (HbA1c) | Downregulated | Plasma | [66] |
| miR-100-5p | Wnt signaling pathway | Downregulated | Serum | [54] |
| miR-150-5p | transcription factor cMyb and IFN-r | Downregulated | PBMCs | [67] |
| miR-154-3p | glycosaminoglycan biosynthesis | Downregulated | Serum | [54] |
| miR-24-3p | MAPK and Wnt signaling pathways | Upregulated | Serum | [54] |
| miR-25-3p | suppressor of cytokine Ssgnaling 5 (SOCS5), BTG anti-proliferation factor 2 (BTG2), PTEN/Akt pathway, and Notch signaling pathway | Upregulated | Serum | [54] |
| miR-324-3p | GLI family zinc finger 3 (GLI3), Wnt family member 2B (WNT2B), MAPK pathway, Wnt/β-catenin signaling pathway | Upregulated | Serum | [54] |
| miR-377-3p | Lysine degradation and ubiquitin-mediated proteolysis pathways | Upregulated | Serum | [54] |
| miR-378e | Insulin-like growth factor receptor and lipid metabolism pathways, Adiponectin expression pathway | Downregulated | Plasma-derived exosome | [68] |
| miR-424-5p | caudal type homeobox 2 (CDX2) transcriptional factor pathway | Downregulated | PBMCs | [67] |
| miR-454-3p | nuclear factor of activated T cells 2 (NFATc2) and vitamin D-dependent calcium-binding protein calbindin 1 (CALB1), Wnt/β-catenin pathways | Upregulated | Serum | [54] |
| miR-490-5p | Calcium-binding protein 5 (CABP5), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), nuclear factor of activated T cells 5 (NFAT5) and TGF-beta-signaling pathways | Downregulated | Serum | [54] |
| miR-574-3p | Mothers against decapentaplegic homolog 4(SMAD4) signaling pathway | Downregulated | Plasma-derived exosome | [68] |
| miR-23a-3p | Death protein 5 (DP5), p53-upregulated modulator of apoptosis (PUMA), BCL2-associated X, apoptosis regulator (BAX), and BIM protein encoded by the BCL2L11 gene | Downregulated | Pancreatic cells | [56] |
| miR-23b-3p | DP5 and PUMA | Downregulated | Pancreatic cells | [56] |
| miR-149-5p | DP5, PUMA, and BAX | Downregulated | Pancreatic cells | [56] |
| miR-98 | TNF-related apoptosis-inducing ligand (TRAIL), TRAIL-receptor 2, FAS cell surface death receptor, and FASLG (Ligand) | Upregulated | T cells | [57] |
| miR-146a | TNF receptor-associated factor 6 (TRAF6), NUMB endocytic adaptor protein, syntaxin 3 (STX3) and BAF chromatin-remodeling complex subunit(BCL11A) | Downregulated | PBMCs | [67] |
| miR-590-5p | TRAIL, TRAIL-R2, FAS, and FASLG | Upregulated | T cells | [57] |
| miR-23b | TRAIL, TRAIL-R2, FAS, and FASLG | Upregulated | T cells | [57] |
5. Targeting the Drivers of Epigenetic Modification in T1D
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Patterson, C.C.; Karuranga, S.; Salpea, P.; Saeedi, P.; Dahlquist, G.; Soltesz, G.; Ogle, G.D. Worldwide estimates of incidence prevalence mortality of type 1 diabetes in children adolescents: Results from the International Diabetes Federation Diabetes Atlas 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107842. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F. Type 1 diabetes genome-wide association studies: Not to be lost in translation. Clin. Transl. Immunol. 2017, 6, e162. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Cudworth, A.G.; Woodrow, J.C. HL-A antigens and diabetes mellitus. Lancet 1974, 304, 1153. [Google Scholar] [CrossRef]
- Rojas, J.; Bermudez, V.; Palmar, J.; Martinez, M.S.; Olivar, L.C.; Nava, M.; Tomey, D.; Rojas, M.; Salazar, J.; Garicano, C.; et al. Pancreatic Beta Cell Death: Novel Potential Mechanisms in Diabetes Therapy. J. Diabetes Res. 2018, 2018, 9601801. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Akolkar, B.; Concannon, P.; Erlich, H.A.; Julier, C.; Morahan, G.; Nierras, C.R.; Todd, J.A.; Rich, S.S.; Nerup, J. Genetics of Type 1 Diabetes: What’s next? Diabetes 2010, 59, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Ilonen, J.; Reijonen, H.; Herva, E.; Sjoroos, M.; Iitia, A.; Lovgren, T.; Veijola, R.; Knip, M.; Akerblom, H.K. Rapid HLA-DQB1 genotyping for four alleles in the assessment of risk for IDDM in the Finnish population. Diabetes Care 1996, 19, 795–800. [Google Scholar] [CrossRef]
- Steck, A.K.; Rewers, M.J. Genetics of type 1 diabetes. Clin. Chem. 2011, 57, 176–185. [Google Scholar] [CrossRef]
- Castillo-Fernandez, J.E.; Spector, T.D.; Bell, J.T. Epigenetics of discordant monozygotic twins: Implications for disease. Genome med. 2014, 6, 60. [Google Scholar] [CrossRef]
- Egro, F.M. Why is type 1 diabetes increasing? J. Mol. Endocrinol. 2013, 51, R1–R13. [Google Scholar] [CrossRef]
- Vehik, K.; Dabelea, D. The changing epidemiology of type 1 diabetes: Why is it going through the roof? Diabetes Metab. Res. Rev. 2011, 27, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.M.; Aitken, R.J.; Wilson, I.; Williams, A.J.; Bingley, P.J. Early onset of diabetes in the proband is the major determinant of risk in HLA DR3-DQ2/DR4-DQ8 siblings. Diabetes 2014, 63, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Cerna, M. Epigenetic Regulation in Etiology of Type 1 Diabetes Mellitus. Int. J. Mol. Sci 2019, 21, 36. [Google Scholar] [CrossRef] [PubMed]
- Jerram, S.T.; Dang, M.N.; Leslie, R.D. The Role of Epigenetics in Type 1 Diabetes. Curr. Diab. Rep. 2017, 17, 89. [Google Scholar] [CrossRef]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Pinney, S.E. DNA methylation and its role in the pathogenesis of diabetes. Pediatr. Diabetes 2017, 18, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Beyan, H.; Down, T.A.; Hawa, M.I.; Maslau, S.; Aden, D.; Daunay, A.; Busato, F.; Mein, C.A.; Manfras, B.; et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 2011, 7, e1002300. [Google Scholar] [CrossRef]
- Stefan, M.; Zhang, W.; Concepcion, E.; Yi, Z.; Tomer, Y. DNA methylation profiles in type 1 diabetes twins point to strong epigenetic effects on etiology. J. Autoimmun. 2014, 50, 33–37. [Google Scholar] [CrossRef]
- Paul, D.S.; Teschendorff, A.E.; Dang, M.A.N.; Lowe, R.; Hawa, M.I.; Ecker, S.; Beyan, H.; Cunningham, S.; Fouts, A.R.; Ramelius, A.; et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016, 7, 13555. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.H.; Volkov, P.; Bacos, K.; Dayeh, T.; Hall, E.; Nilsson, E.A.; Ladenvall, C.; Rönn, T.; Ling, C. Genome-Wide Associations between Genetic and Epigenetic Variation Influence mRNA Expression and Insulin Secretion in Human Pancreatic Islets. PLoS Genet. 2014, 10, e1004735. [Google Scholar] [CrossRef]
- Ye, J.; Richardson, T.G.; McArdle, W.L.; Relton, C.L.; Gillespie, K.M.; Suderman, M.; Hemani, G. Identification of loci where DNA methylation potentially mediates genetic risk of type 1 diabetes. J. Autoimmun. 2018, 93, 66–75. [Google Scholar] [CrossRef]
- Johnson, R.K.; Vanderlinden, L.A.; Dong, F.; Carry, P.M.; Seifert, J.; Waugh, K.; Shorrosh, H.; Fingerlin, T.; Frohnert, B.I.; Yang, I.V.; et al. Longitudinal DNA methylation differences precede type 1 diabetes. Sci. Rep. 2020, 10, 3721. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, B.; Belaich, S.; Longue, J.; Vandewalle, B.; Oberholzer, J.; Gmyr, V.; Pattou, F.; Kerr-Conte, J. 5′-AZA induces Ngn3 expression and endocrine differentiation in the PANC-1 human ductal cell line. Biochem. Biophys. Res. Commun. 2010, 391, 305–309. [Google Scholar] [CrossRef]
- Chakravarthy, H.; Gu, X.; Enge, M.; Dai, X.; Wang, Y.; Damond, N.; Downie, C.; Liu, K.; Wang, J.; Xing, Y.; et al. Converting Adult Pancreatic Islet alpha Cells into beta Cells by Targeting Both Dnmt1 and Arx. Cell Metab. 2017, 25, 622–634. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Petrie, J.R.; Chaturvedi, N.; Ford, I.; Hramiak, I.; Hughes, A.D.; Jenkins, A.J.; Klein, B.E.; Klein, R.; Ooi, T.C.; Rossing, P.; et al. Metformin in adults with type 1 diabetes: Design and methods of REducing with MetfOrmin Vascular Adverse Lesions (REMOVAL): An. international multicentre trial. Diabetes Obes. Metab. 2017, 19, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Das, A.; Chen, J.; Wu, P.; Li, X.; Fang, Z. Metformin in patients with and without diabetes: A paradigm shift in cardiovascular disease management. Cardiovasc. Diabetol. 2019, 18, 54. [Google Scholar] [CrossRef]
- Livingstone, R.; Boyle, J.G.; Petrie, J.R. & REMOVAL Study Team. A new perspective on metformin therapy in type 1 diabetes. Diabetologia 2017, 60, 1594–1600. [Google Scholar]
- García-Calzón, S.; Perfilyev, A.; Männistö, V.; de Mello, V.D.; Nilsson, E.; Pihlajamäki, J.; Ling, C. Diabetes medication associates with DNA methylation of metformin transporter genes in the human liver. Clin. Epigenetics 2017, 9, 102. [Google Scholar] [CrossRef]
- El-Hadidy, W.F.; Mohamed, A.R.; Mannaa, H.F. Possible protective effect of procainamide as an epigenetic modifying agent in experimentally induced type 2 diabetes mellitus in rats. Alexandria J. Med. 2015, 51, 65–71. [Google Scholar] [CrossRef][Green Version]
- Sato, T.; Issa, J.-P.J.; Kropf, P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026948. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Chen, Z.; Genuth, S.; Paterson, A.; Zhang, L.; Wu, X.; Li, S.M.; Cleary, P.; Riggs, A.; Harlan, D.M.; et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 2014, 63, 1748–1762. [Google Scholar] [CrossRef] [PubMed]
- Puthanmadhom Narayanan, S.; Lee, J.-H.; Bhagwate, A.; Kuwelker, S.; Yan, H.; Ordog, T.; Bharucha, A.E. Epigenetic Alterations Are Associated with Gastric Emptying Disturbances in Diabetes Mellitus. Clin. Transl. Gastroenterol. 2020, 11, e00136. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Cao, J.; Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol. 2012, 2, 26. [Google Scholar] [CrossRef]
- Wang, Z.; Xie, Z.; Lu, Q.; Chang, C.; Zhou, Z. Beyond Genetics: What Causes Type 1 Diabetes. Clin. Rev. Allergy Immunol. 2017, 52, 273–286. [Google Scholar] [CrossRef]
- Miao, F.; Smith, D.D.; Zhang, L.; Min, A.; Feng, W.; Natarajan, R. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: An. epigenetic study in diabetes. Diabetes 2008, 57, 3189–3198. [Google Scholar] [CrossRef]
- Miao, F.; Chen, Z.; Zhang, L.; Liu, Z.; Wu, X.; Yuan, Y.C.; Natarajan, R. Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. J. Biol. Chem. 2012, 287, 16335–16345. [Google Scholar] [CrossRef] [PubMed]
- Caramori, M.L.; Kim, Y.; Moore, J.H.; Rich, S.S.; Mychaleckyj, J.C.; Kikyo, N.; Mauer, M. Gene expression differences in skin fibroblasts in identical twins discordant for type 1 diabetes. Diabetes 2012, 61, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Lundh, M.; Christensen, D.P.; Nielsen, M.D.; Richardson, S.J.; Dahllof, M.S.; Skovgaard, T.; Berthelsen, J.; Dinarello, C.A.; Stevenazzi, A.; Mascagni, P.; et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia 2012, 55, 2421–2431. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.P.; Gysemans, C.; Lundh, M.; Dahllöf, M.S.; Noesgaard, D.; Schmidt, S.F.; Mandrup, S.; Birkbak, N.; Workman, C.T.; Piemonti, L.; et al. Lysine deacetylase inhibition prevents diabetes by chromatin-independent immunoregulation and β-cell protection. Proc. Natl. Acad. Sci. USA 2014, 111, 1055–1059. [Google Scholar] [CrossRef]
- Khan, S.; Jena, G.B. Protective role of sodium butyrate, a HDAC inhibitor on beta-cell proliferation, function and glucose homeostasis through modulation of p38/ERK MAPK and apoptotic pathways: Study in juvenile diabetic rat. Chem. Biol. Interact. 2014, 213, 1–12. [Google Scholar] [CrossRef]
- Khan, S.; Jena, G. Valproic Acid Improves Glucose Homeostasis by Increasing Beta-Cell Proliferation, Function, and Reducing its Apoptosis through HDAC Inhibition in Juvenile Diabetic Rat. J. Biochem. Mol. Toxicol. 2016, 30, 438–446. [Google Scholar] [CrossRef]
- Grammatiki, M.; Rapti, E.; Karras, S.; Ajjan, R.A.; Kotsa, K. Vitamin D and diabetes mellitus: Causal or casual association? Rev. Endocr. Metab. Disord. 2017, 18, 227–241. [Google Scholar] [CrossRef]
- Martín-Subero, J.I.; Esteller, M. Profiling Epigenetic Alterations in Disease, in Epigenetic Contributions in Autoimmune Disease. In Advances in Experimental Medicine and Biology; Ballestar, E., Ed.; Springer US: Boston, MA, USA, 2011; pp. 162–177. [Google Scholar]
- Xu, C.-R.; Li, L.-C.; Donahue, G.; Ying, L.; Zhang, Y.-W.; Gadue, P.; Zaret, K.S. Dynamics of genomic H3K27me3 domains and role of EZH2 during pancreatic endocrine specification. EMBO J. 2014, 33, 2157–2170. [Google Scholar] [CrossRef]
- Fontcuberta-PiSunyer, M.; Cervantes, S.; Miquel, E.; Mora-Castilla, S.; Laurent, L.C.; Raya, A.; Gomis, R.; Gasa, R. Modulation of the endocrine transcriptional program by targeting histone modifiers of the H3K27me3 mark. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 473–480. [Google Scholar] [CrossRef]
- Katz, L.S.; Geras-Raaka, E.; Gershengorn, M.C. Reprogramming adult human dermal fibroblasts to islet-like cells by epigenetic modification coupled to transcription factor modulation. Stem Cells Dev. 2013, 22, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Marino, E.; Richards, J.L.; McLeod, K.H.; Stanley, D.; Yap, Y.A.; Knight, J.; McKenzie, C.; Kranich, J.; Oliveira, A.C.; Rossello, F.J.; et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 2017, 18, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, M.V.; Parekh, V.S.; Hardikar, A.A. New pancreas from old: Microregulators of pancreas regeneration. Trends Endocrinol. Metab. 2007, 18, 393–400. [Google Scholar] [CrossRef]
- Erener, S.; Marwaha, A.; Tan, R.; Panagiotopoulos, C.; Kieffer, T.J. Profiling of circulating microRNAs in children with recent onset of type 1 diabetes. JCI Insight 2017, 2, e89656. [Google Scholar] [CrossRef]
- Kim, K.W.; Ho, A.; Alshabee-Akil, A.; Hardikar, A.A.; Kay, T.W.; Rawlinson, W.D.; Craig, M.E. Coxsackievirus B5 Infection Induces Dysregulation of microRNAs Predicted to Target. Known Type 1 Diabetes Risk Genes in Human Pancreatic Islets. Diabetes 2016, 65, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Grieco, F.A.; Sebastiani, G.; Juan-Mateu, J.; Villate, O.; Marroqui, L.; Ladriere, L.; Tugay, K.; Regazzi, R.; Bugliani, M.; Marchetti, P.; et al. MicroRNAs miR-23a-3p, miR-23b-3p, and miR-149–5p Regulate the Expression of Proapoptotic BH3-Only Proteins DP5 and PUMA in Human Pancreatic beta-Cells. Diabetes 2017, 66, 100–112. [Google Scholar] [CrossRef]
- de Jong, V.M.; van der Slik, A.R.; Laban, S.; Slot, R.v.; Koeleman, B.P.; Zaldumbide, A.; Roep, B.O. Survival of autoreactive T lymphocytes by microRNA-mediated regulation of apoptosis through TRAIL and Fas in type 1 diabetes. Genes Immun. 2016, 17, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Osmai, M.; Osmai, Y.; Bang-Berthelsen, C.H.; Pallesen, E.M.H.; Vestergaard, A.L.; Novotny, G.W.; Pociot, F.; Mandrup-Poulsen, T. MicroRNAs as regulators of beta-cell function and dysfunction. Diabetes Metab. Res. Rev. 2016, 32, 334–349. [Google Scholar] [CrossRef] [PubMed]
- Roggli, E.; Britan, A.; Gattesco, S.; Lin-Marq, N.; Abderrahmani, A.; Meda, P.; Regazzi, R. Involvement of microRNAs in the cytotoxic effects exerted by proinflammatory cytokines on pancreatic beta-cells. Diabetes 2010, 59, 978–986. [Google Scholar] [CrossRef]
- Lovis, P.; Roggli, E.; Laybutt, D.R.; Gattesco, S.; Yang, J.Y.; Widmann, C.; Abderrahmani, A.; Regazzi, R. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes 2008, 57, 2728–2736. [Google Scholar] [CrossRef]
- Pileggi, A.; Klein, D.; Fotino, C.; Bravo-Egana, V.; Rosero, S.; Doni, M.; Podetta, M.; Ricordi, C.; Molano, R.D.; Pastori, R.L. MicroRNAs in islet immunobiology and transplantation. Immunol. Res. 2013, 57, 185–196. [Google Scholar] [CrossRef]
- Wang, P.; Liu, Q.; Zhao, H.; Bishop, J.O.; Zhou, G.; Olson, L.K.; Moore, A. miR-216a-targeting theranostic nanoparticles promote proliferation of insulin-secreting cells in type 1 diabetes animal model. Sci. Rep. 2020, 10, 5302. [Google Scholar] [CrossRef]
- Chen, H.; Lan, H.-Y.; Roukos, D.H.; Cho, W.C. Application of microRNAs in diabetes mellitus. J. Endocrinol. 2014, 222, R1–R10. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Chuang, C.-C.; Zuo, L. Potential roles of microRNAs and ROS in colorectal cancer: Diagnostic biomarkers and therapeutic targets. Oncotarget 2017, 8, 17328–17346. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.S.; Neto, E.C.; Fukui, R.T.; Ferreira, L.R.P.; Silva, M.E.R. Increased Expression of Circulating microRNA 101–3p in Type 1 Diabetes Patients: New Insights into miRNA-Regulated Pathophysiological Pathways for Type 1 Diabetes. Front. Immunol. 2019, 10, 1637. [Google Scholar] [CrossRef]
- Satake, E.; Pezzolesi, M.G.; Dom, Z.I.M.; Smiles, A.M.; Niewczas, M.A.; Krolewski, A.S. Circulating miRNA Profiles Associated with Hyperglycemia in Patients with Type 1 Diabetes. Diabetes 2018, 67, 1013–1023. [Google Scholar] [CrossRef]
- Wang, G.; Gu, Y.; Xu, N.; Zhang, M.; Yang, T. Decreased expression of miR-150, miR146a and miR424 in type 1 diabetic patients: Association with ongoing islet autoimmunity. Biochem. Biophys. Res. Commun. 2018, 498, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Contreras, M.; Shah, S.H.; Tamayo, A.; Robbins, P.D.; Golberg, R.B.; Mendez, A.J.; Ricordi, C. Plasma-derived exosome characterization reveals a distinct microRNA signature in long duration Type 1 diabetes. Sci. Rep. 2017, 7, 5998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Long, H.; Chang, C.; Zhao, M.; Lu, Q. Crosstalk between metabolism and epigenetic modifications in autoimmune diseases: A comprehensive overview. Cell Mol. Life Sci. 2018, 75, 3353–3369. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Bushell, M.; Zhang, C.-Y.; Dashwood, R.; James, W.; Harris, C.; Baltimore, D. The role of microRNA in nutritional control. J. Intern. Med. 2015, 278, 99–109. [Google Scholar]
- Guay, C.; Regazzi, R. Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat. Rev. Endocrinol. 2013, 9, 513. [Google Scholar] [CrossRef]
- Kilberg, M.S.; Pan, Y.X.; Chen, H.; Leung-Pineda, V. Nutritional control of gene expression: How mammalian cells respond to amino acid limitation. Annu. Rev. Nutr. 2005, 25, 59–85. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, R.; Zwergel, C.; Artico, M.; Taurone, S.; Ralli, M.; Greco, A.; Mai, A. The emerging role of epigenetics in human autoimmune disorders. Clin. Epigenetics 2019, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Stover, P.J.; James, W.P.T.; Krook, A.; Garza, C. Emerging concepts on the role of epigenetics in the relationships between nutrition and health. J. Intern. Med. 2018, 284, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Tiziani, S.; Firestein, G.S. Metabolomics in rheumatic diseases: Desperately seeking biomarkers. Nat. Rev. Rheumatol. 2016, 12, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Heiden, M.G.V.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Jerónimo, C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef]
- Wang, Y.-P.; Lei, Q.-Y. Metabolic recoding of epigenetics in cancer. Cancer Commu. 2018, 38, 25. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akil, A.-S.A.-S.; Jerman, L.F.; Yassin, E.; Padmajeya, S.S.; Al-Kurbi, A.; Fakhro, K.A. Reading between the (Genetic) Lines: How Epigenetics is Unlocking Novel Therapies for Type 1 Diabetes. Cells 2020, 9, 2403. https://doi.org/10.3390/cells9112403
Akil A-SA-S, Jerman LF, Yassin E, Padmajeya SS, Al-Kurbi A, Fakhro KA. Reading between the (Genetic) Lines: How Epigenetics is Unlocking Novel Therapies for Type 1 Diabetes. Cells. 2020; 9(11):2403. https://doi.org/10.3390/cells9112403
Chicago/Turabian StyleAkil, Ammira-Sarah AL-Shabeeb, Laila F. Jerman, Esraa Yassin, Sujitha S. Padmajeya, Alya Al-Kurbi, and Khalid A. Fakhro. 2020. "Reading between the (Genetic) Lines: How Epigenetics is Unlocking Novel Therapies for Type 1 Diabetes" Cells 9, no. 11: 2403. https://doi.org/10.3390/cells9112403
APA StyleAkil, A.-S. A.-S., Jerman, L. F., Yassin, E., Padmajeya, S. S., Al-Kurbi, A., & Fakhro, K. A. (2020). Reading between the (Genetic) Lines: How Epigenetics is Unlocking Novel Therapies for Type 1 Diabetes. Cells, 9(11), 2403. https://doi.org/10.3390/cells9112403