Pancreatic Fibroblast Heterogeneity: From Development to Cancer

Abstract
1. Introduction
2. Mesenchyme Function and Heterogeneity during Pancreas Development
3. Fibroblast Heterogeneity in the Healthy Pancreas
4. Fibroblast Heterogeneity in Pancreatic Injury and Disease
5. Fibroblast Heterogeneity in Pancreatic Cancer
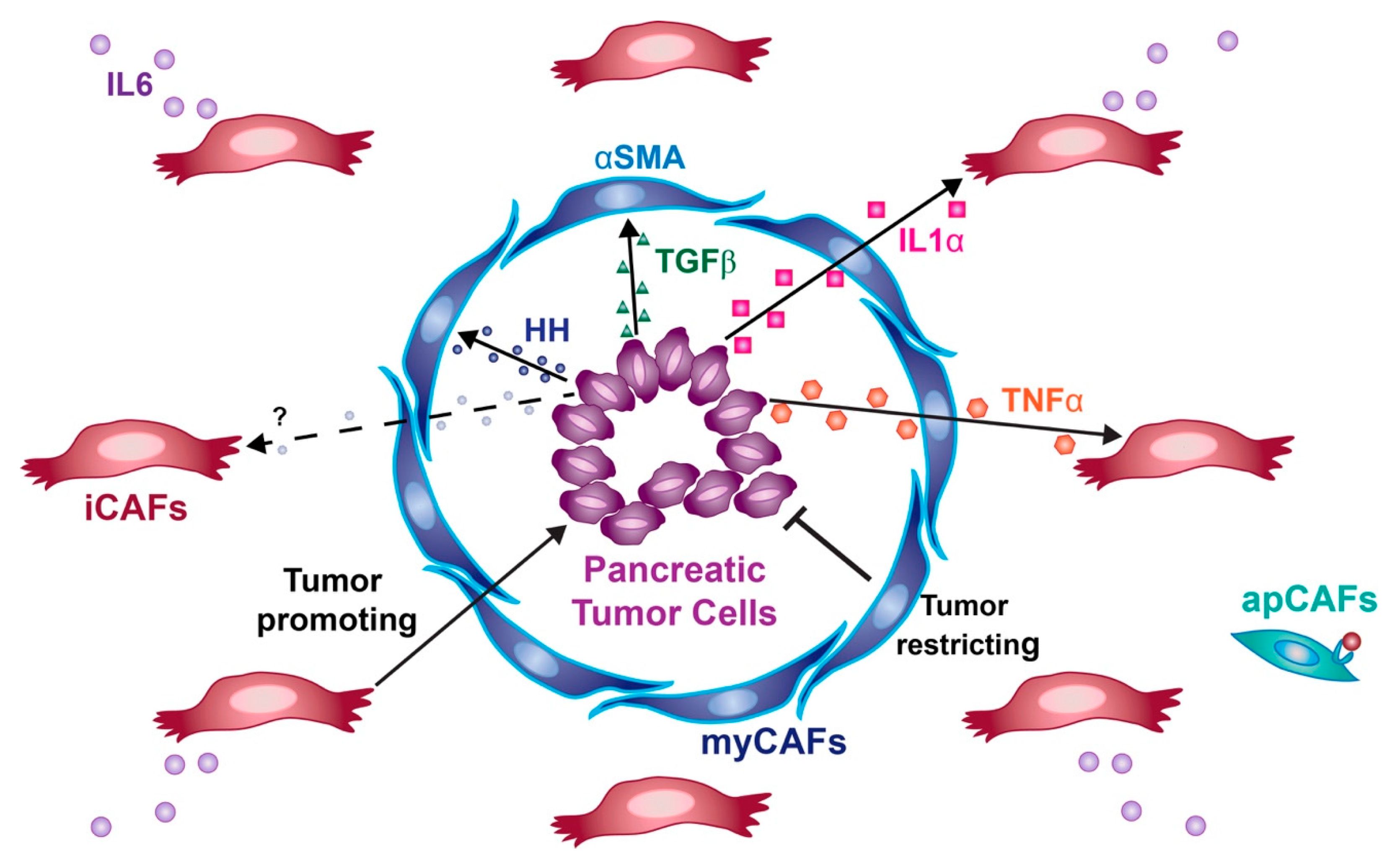
5.1. Positional Heterogeneity
5.2. Interactive Heterogeneity
5.2.1. Hedgehog Signaling
5.2.2. TGF-β Signaling
5.3. Transcriptional Heterogeneity
6. Origin of Cancer-Associated Fibroblasts
7. Challenges in Studying Fibroblast Heterogeneity
8. Targeting Cancer-Associated Fibroblasts in the Clinic
9. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- McAnulty, R.J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J. Biochem. Cell Biol. 2007, 39, 666–671. [Google Scholar] [CrossRef]
- Darby, I.A.; Laverdet, B.; Bonté, F.; Desmoulière, A. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301. [Google Scholar] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 157–188. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Mathew, E.; Zhang, Y.; Holtz, A.M.; Kane, K.T.; Song, J.Y.; Allen, B.L.; di Magliano, M.P. Dosage-dependent regulation of pancreatic cancer growth and angiogenesis by Hedgehog signaling. Cell Rep. 2014, 9, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.-C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091-100. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Van Mansfeld, A.D.M.; Offerhaus, G.J.A.; Van Weering, D.H.J.; Allison, D.C.; Goodman, S.N.; Kensler, T.W.; Bose, K.K.; Cameron, J.L.; Bos, J.L. K-ras oncogene activation in adenocarcinoma of the human pancreas: A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am. J. Pathol. 1993, 143, 545–554. [Google Scholar]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; Depinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Carrière, C.; Young, A.L.; Gunn, J.R.; Longnecker, D.S.; Korc, M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem. Biophys. Res. Commun. 2009, 382, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Schönhuber, N.; Seidler, B.; Schuck, K.; Veltkamp, C.; Schachtler, C.; Zukowska, M.; Eser, S.; Feyerabend, T.B.; Paul, M.C.; Eser, P.; et al. A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat. Med. 2014, 20, 1340–1347. [Google Scholar] [CrossRef]
- Chen, Y.; LeBleu, V.S.; Carstens, J.L.; Sugimoto, H.; Zheng, X.; Malasi, S.; Saur, D.; Kalluri, R. Dual reporter genetic mouse models of pancreatic cancer identify an epithelial-to-mesenchymal transition-independent metastasis program. EMBO Mol. Med. 2018, 10, e9085. [Google Scholar] [CrossRef]
- Wen, H.-J.; Gao, S.; Wang, Y.; Ray, M.; Magnuson, M.A.; Wright, C.V.E.; Di Magliano, M.P.; Frankel, T.L.; Crawford, H.C. Myeloid Cell-Derived HB-EGF Drives Tissue Recovery After Pancreatitis. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 173–192. [Google Scholar] [CrossRef]
- Garcia, P.E.; Adoumie, M.; Kim, E.C.; Zhang, Y.; Scales, M.K.; El-Tawil, Y.S.; Shaikh, A.Z.; Wen, H.-J.; Bednar, F.; Allen, B.L.; et al. Differential Contribution of Pancreatic Fibroblast Subsets to the Pancreatic Cancer Stroma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 581–599. [Google Scholar] [CrossRef]
- Buch, T.; Heppner, F.L.; Tertilt, C.; Heinen, T.J.A.J.; Kremer, M.; Wunderlich, F.T.; Jung, S.; Waisman, A. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat. Methods 2005, 2, 419–426. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, L.E.; Wong, D.M.; Subramaniam, M.; Meyer, N.P.; Gilchrist, C.L.; Knox, S.M.; Tward, A.D.; Ye, C.J.; Sneddon, J.B. Lineage dynamics of murine pancreatic development at single-cell resolution. Nat. Commun. 2018, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.; Veres, A.; Wolock, S.L.; Klein, A.M.; Melton, D.A.; Yanai, I. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 2016, 3, 346–360.e4. [Google Scholar] [CrossRef]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 1102–1123. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef]
- Zorn, A.M.; Wells, J.M. Vertebrate Endoderm Development and Organ Formation. Annu. Rev. Cell Dev. Biol. 2009, 25, 221–251. [Google Scholar] [CrossRef]
- Golosow, N.; Grobstein, C. Epitheliomesenchymal Pancreatic Interaction in Morphogenesis. Dev. Biol. 1962, 233. [Google Scholar] [CrossRef]
- Wessells, N.K.; Cohen, J.H. Early pancreas organogenesis: Morphogenesis, tissue interactions, and mass effects. Dev. Biol. 1967, 15, 237–270. [Google Scholar] [CrossRef]
- Landsman, L.; Nijagal, A.; Whitchurch, T.J.; VanderLaan, R.L.; Zimmer, W.E.; MacKenzie, T.C.; Hebrok, M. Pancreatic mesenchyme regulates epithelial organogenesis throughout development. PLoS Biol. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Hebrok, M.; Kim, S.K.; Melton, D.A. Notochord repression of endodermal sonic hedgehog permits pancreas development. Genes Dev. 1998, 12, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, A.; Itoh, N.; Kato, S.; Thiery, J.P.; Czernichow, P.; Bellusci, S.; Scharfmann, R. Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development 2001, 128, 5109–5117. [Google Scholar] [PubMed]
- Stafford, D.; Prince, V.E. Retinoic acid signaling is required for a critical early step in zebrafish pancreatic development. Curr. Biol. 2002, 12, 1215–1220. [Google Scholar] [CrossRef]
- Martín, M.; Gallego-Llamas, J.; Ribes, V.; Kedinger, M.; Niederreither, K.; Chambon, P.; Dollé, P.; Gradwohl, G. Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Dev. Biol. 2005, 284, 399–411. [Google Scholar] [CrossRef]
- Molotkov, A.; Molotkova, N.; Duester, G. Retinoic acid generated by Raldh2 in mesoderm is required for mouse dorsal endodermal pancreas development. Dev. Dyn. 2005, 232, 950–957. [Google Scholar] [CrossRef]
- Ahnfelt-Rønne, J.; Ravassard, P.; Pardanaud-Glavieux, C.; Scharfmann, R.; Serup, P. Mesenchymal bone morphogenetic protein signaling is required for normal pancreas development. Diabetes 2010, 59, 1948–1956. [Google Scholar] [CrossRef]
- Rossi, J.M.; Dunn, N.R.; Hogan, B.L.M.; Zaret, K.S. Distinct mesodermal signals, including BMPs from the septum, transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev. 2001, 15, 1998–2009. [Google Scholar] [CrossRef]
- Shin, D.; Hyun Shin, C.; Tucker, J.; Ober, E.A.; Rentzsch, F.; Poss, K.D.; Hammerschmidt, M.; Mullins, M.C.; Stanier, D.Y.R. Bmp and Fgf signaling are essential for liver specification in zebrafish. Development 2007, 134, 2041–2050. [Google Scholar] [CrossRef]
- Chung, W.S.; Shin, C.H.; Stainier, D.Y.R. Bmp2 Signaling Regulates the Hepatic versus Pancreatic Fate Decision. Dev. Cell 2008, 15, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Wandzioch, E.; Zaret, K.S. Dynamic signaling network for the specification of embryonic pancreas and liver progenitors. Science 2009, 324, 1707–1710. [Google Scholar] [CrossRef]
- Apelqvist, Å.; Ahlgren, U.; Edlund, H. Sonic hedgehog directs specialised mesoderm differentiation in the intestine and pancreas. Curr. Biol. 1997, 7, 801–804. [Google Scholar] [CrossRef]
- Kawahira, H.; Ma, N.H.; Tzanakakis, E.S.; McMahon, A.P.; Chuang, P.-T.; Hebrok, M. Combined activities of hedgehog signaling inhibitors regulate pancreas development. Development 2003, 130, 4871–4879. [Google Scholar] [CrossRef] [PubMed]
- Hibsher, D.; Epshtein, A.; Oren, N.; Landsman, L. Pancreatic Mesenchyme Regulates Islet Cellular Composition in a Patched/Hedgehog-Dependent Manner. Sci. Rep. 2016, 1–12. [Google Scholar] [CrossRef]
- Larsen, B.M.; Hrycaj, S.M.; Newman, M.; Li, Y.; Wellik, D.M. Mesenchymal Hox6 function is required for mouse pancreatic endocrine cell differentiation. Development 2015, 142, 3859–3868. [Google Scholar] [CrossRef]
- Murtaugh, L.C.; Law, A.C.; Dor, Y.; Melton, D.A. B-Catenin is essential for pancreatic acinar but not islet development. Development 2005, 132, 4663–4674. [Google Scholar] [CrossRef]
- Wells, J.M.; Esni, F.; Boivin, G.P.; Aronow, B.J.; Stuart, W.; Combs, C.; Sklenka, A.; Leach, S.D.; Lowy, A.M. Wnt/β-catenin signaling is required for development of the exocrine pancreas. BMC Dev. Biol. 2007, 7, 1–18. [Google Scholar] [CrossRef]
- Heiser, P.W.; Lau, J.; Taketo, M.M.; Herrera, P.L.; Hebrok, M. Stabilization of β-catenin impacts pancreas growth. Development 2006, 133, 2023–2032. [Google Scholar] [CrossRef]
- Harari, N.; Sakhneny, L.; Khalifa-Malka, L.; Busch, A.; Hertel, K.J.; Hebrok, M.; Landsman, L. Pancreatic pericytes originate from the embryonic pancreatic mesenchyme. Dev. Biol. 2019, 449, 14–20. [Google Scholar] [CrossRef]
- Sasson, A.; Rachi, E.; Sakhneny, L.; Baer, D.; Lisnyansky, M.; Epshtein, A.; Landsman, L. Islet pericytes are required for β-cell maturity. Diabetes 2016, 65, 3008–3014. [Google Scholar] [CrossRef] [PubMed]
- Russ, H.A.; Landsman, L.; Moss, C.L.; Higdon, R.; Greer, R.L.; Kaihara, K.; Salamon, R.; Kolker, E.; Hebrok, M. Dynamic Proteomic Analysis of Pancreatic Mesenchyme Reveals Novel Factors That Enhance Human Embryonic Stem Cell to Pancreatic Cell Differentiation. Stem Cells Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; Von Gise, A.; Ikeda, S.; Chien, K.R.; et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.R.; Moss, M.C. A Morphometric Study of the Endocrine and Exocrine Capillaries of the Pancreas. Q. J. Exp. Physiol. 1985, 70, 347–356. [Google Scholar] [CrossRef]
- Ozerdem, U.; Grako, K.A.; Dahlin-Huppe, K.; Monosov, E.; Stallcup, W.B. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev. Dyn. 2001, 222, 218–227. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef]
- Almaça, J.; Weitz, J.; Rodriguez-Diaz, R.; Pereira, E.; Caicedo, A. The Pericyte of the Pancreatic Islet Regulates Capillary Diameter and Local Blood Flow. Cell Metab. 2018, 27, 630–644.e4. [Google Scholar] [CrossRef]
- Lebleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Seeberger, K.L.; Dufour, J.M.; James Shapiro, A.M.; Lakey, J.R.T.; Rajotte, R.V.; Korbutt, G.S. Expansion of mesenchymal stem cells from human pancreatic ductal epithelium. Lab. Investig. 2006, 86, 141–153. [Google Scholar] [CrossRef]
- Baertschiger, R.M.; Bosco, D.; Morel, P.; Serre-Beinier, V.; Berney, T.; Buhler, L.H.; Gonelle-Gispert, C. Mesenchymal stem cells derived from human exocrine pancreas express transcription factors implicated in beta-cell development. Pancreas 2008, 37, 75–84. [Google Scholar] [CrossRef]
- Mathew, E.; Brannon, A.L.; Del Vecchio, A.; Garcia, P.E.; Penny, M.K.; Kane, K.T.; Vinta, A.; Buckanovich, R.J.; Pasca di Magliano, M. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization. Neoplasia 2016, 18, 142–151. [Google Scholar] [CrossRef]
- Waghray, M.; Yalamanchili, M.; Dziubinski, M.; Zeinali, M.; Erkkinen, M.; Yang, H.; Schradle, K.A.; Urs, S.; Pasca di Magliano, M.; Welling, T.H.; et al. GM-CSF Mediates Mesenchymal–Epithelial Cross-talk in Pancreatic Cancer. Cancer Discov. 2016, 6, 886–899. [Google Scholar] [CrossRef]
- Apte, M.V.; Wilson, J.S. Dangerous liaisons: Pancreatic stellate cells and pancreatic cancer cells. J. Gastroenterol. Hepatol. 2012, 27, 69–74. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; Mccaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef]
- Shek, F.W.T.; Benyon, R.C.; Walker, F.M.; McCrudden, P.R.; Pender, S.L.F.; Williams, E.J.; Johnson, P.A.; Johnson, C.D.; Bateman, A.C.; Fine, D.R.; et al. Expression of transforming growth factor-β1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am. J. Pathol. 2002, 160, 1787–1798. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Investig. 2007, 117, 50–59. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schneider, E.; Groß, H.; Weidenbach, H.; Schmid, R.M.; Adler, G.; Menke, A.; Siech, M.; Beger, H.; Gru, A. Identification, Culture, and Characterization of Pancreatic Stellate Cells in Rats and Humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Identification of markers for quiescent pancreatic stellate cells in the normal human pancreas. Histochem. Cell Biol. 2017. [Google Scholar] [CrossRef]
- Apte, M.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cell: Physiologic role, role in fibrosis and cancer. Curr. Opin. Gastroenterol. 2015, 31, 416–423. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andrén-Sandberg, A.; Domellöf, L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Machicado, J.D.; Yadav, D. Epidemiology of Recurrent Acute and Chronic Pancreatitis: Similarities and Differences. Dig. Dis. Sci. 2017, 62, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Whitcomb, D.C.; Shimosegawa, T.; Esposito, I.; Lerch, M.M.; Gress, T.; Mayerle, J.; Drewes, A.M.; Rebours, V.; Akisik, F.; et al. Chronic pancreatitis. Nat. Rev. Dis. Prim. 2017, 3, 1–18. [Google Scholar] [CrossRef]
- Klöppel, G. Chronic pancreatitis, pseudotumors and other tumor-like lesions. Mod. Pathol. 2007, 20, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Lerch, M.M.; Gorelick, F.S. Models of acute and chronic pancreatitis. Gastroenterology 2013, 144, 1180–1193. [Google Scholar] [CrossRef]
- Geisz, A.; Sahin-Tóth, M. A preclinical model of chronic pancreatitis driven by trypsinogen autoactivation. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Engle, D.D.; Tiriac, H.; Rivera, K.D.; Pommier, A.; Whalen, S.; Oni, T.E.; Alagesan, B.; Lee, E.J.; Yao, M.A.; Lucito, M.S.; et al. The glycan CA19-9 promotes pancreatitis and pancreatic cancer in mice. Science 2019, 364, 1156–1162. [Google Scholar] [CrossRef]
- Gui, F.; Zhang, Y.; Wan, J.; Zhan, X.; Yao, Y.; Li, Y.; Haddock, A.N.; Shi, J.; Guo, J.; Chen, J.; et al. Trypsin activity governs increased susceptibility to pancreatitis in mice expressing human PRSS1R122H. J. Clin. Investig. 2020, 130, 189–202. [Google Scholar] [CrossRef]
- Huang, H.; Swidnicka-Siergiejko, A.K.; Daniluk, J.; Gaiser, S.; Yao, Y.; Peng, L.; Zhang, Y.; Liu, Y.; Dong, M.; Zhan, X.; et al. Transgenic Expression of PRSS1R122H Sensitizes Mice to Pancreatitis. Gastroenterology 2020, 158, 1072–1082.e7. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.S.; Keogh, G.W.; Apte, M.V.; Moran, C.S.; Stewart, N.L.; Crawford, D.H.G.; Pirola, R.C.; McCaughan, G.W.; Ramm, G.A.; Wilson, J.S. Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am. J. Pathol. 1999, 155, 1087–1095. [Google Scholar] [CrossRef]
- Mews, P.; Phillips, P.; Fahmy, R.; Korsten, M.; Pirola, R.; Wilson, J.; Apte, M. Pancreatic stellate cells respond to inflammatory cytokines: Potential role in chronic pancreatitis. Gut 2002, 50, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Phillips, P.A.; Fahmy, R.R.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Naidoo, D.; Wilson, J.S. Does Alcohol Directly Stimulate Pancreatic Fibrogenesis? Studies With Rat Pancreatic Stellate Cells. Gastroenterology 2000, 118, 780–794. [Google Scholar] [CrossRef]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Sendler, M.; Beyer, G.; Mahajan, U.M.; Kauschke, V.; Maertin, S.; Schurmann, C.; Homuth, G.; Völker, U.; Völzke, H.; Halangk, W.; et al. Complement Component 5 Mediates Development of Fibrosis, via Activation of Stellate Cells, in 2 Mouse Models of Chronic Pancreatitis. Gastroenterology 2015, 149, 765–776.e10. [Google Scholar] [CrossRef]
- Mathew, E.; Collins, M.A.; Fernandez-Barrena, M.G.; Holtz, A.M.; Yan, W.; Hogan, J.O.; Tata, Z.; Allen, B.L.; Fernandez-Zapico, M.E.; Di Magliano, M.P. The transcription factor GLI1 modulates the inflammatory response during pancreatic tissue remodeling. J. Biol. Chem. 2014, 289, 27727–27743. [Google Scholar] [CrossRef]
- Lees, C.W.; Zacharias, W.J.; Tremelling, M.; Noble, C.L.; Nimmo, E.R.; Tenesa, A.; Cornelius, J.; Torkvist, L.; Kao, J.; Farrington, S.; et al. Analysis of Germline GLI1 Variation Implicates Hedgehog Signalling in the Regulation of Intestinal Inflammatory Pathways. PLoS Med. 2008, 5, 1761–1775. [Google Scholar] [CrossRef]
- Haeberle, L.; Steiger, K.; Schlitter, A.M.; Safi, S.A.; Knoefel, W.T.; Erkan, M.; Esposito, I. Stromal heterogeneity in pancreatic cancer and chronic pancreatitis. Pancreatology 2018, 18, 536–549. [Google Scholar] [CrossRef]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-M.; Vincent, A.; Kanda, M.; Leclerc, J.; Omura, N.; Borges, M.; Klein, A.P.; Canto, M.I.; Hruban, R.H.; Goggins, M. Genome-Wide Somatic Copy Number Alterations in Low-Grade PanINs and IPMNs from Individuals with a Family History of Pancreatic Cancer. Clin. Cancer Res. 2012, 18, 4303–4312. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.F.; Dubois, C.L.; Morris, J.P.; Pan, F.C.; Akiyama, H.; Wright, C.V.E.; Jensen, K.; et al. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Hendley, A.M.; Lafaro, K.J.; Pruski, M.A.; Jones, N.C.; Alsina, J.; Younes, M.; Maitra, A.; McAllister, F.; Iacobuzio-Donahue, C.A. p53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene 2016, 35, 4282–4288. [Google Scholar] [CrossRef]
- Ferreira, R.M.M.; Sancho, R.; Messal, H.A.; Nye, E.; Spencer-Dene, B.; Stone, R.K.; Stamp, G.; Rosewell, I.; Quaglia, A.; Behrens, A. Duct- and Acinar-Derived Pancreatic Ductal Adenocarcinomas Show Distinct Tumor Progression and Marker Expression. Cell Rep. 2017, 21, 966–978. [Google Scholar] [CrossRef]
- Kopp, J.L.; Dubois, C.L.; Schaeffer, D.F.; Samani, A.; Taghizadeh, F.; Cowan, R.W.; Rhim, A.D.; Stiles, B.L.; Valasek, M.; Sander, M. Loss of Pten and Activation of Kras Synergistically Induce Formation of Intraductal Papillary Mucinous Neoplasia From Pancreatic Ductal Cells in Mice. Gastroenterology 2018, 154, 1509–1523.e5. [Google Scholar] [CrossRef]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.-C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Pasca di Magliano, M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef]
- Sodir, N.M.; Kortlever, R.M.; Barthet, V.J.A.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Swigart, L.B.; Soucek, L.; Arends, M.J.; et al. MYC instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov. 2020, 10, 588–607. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Oon, C.; Kothari, A.; Horton, W.; Link, J.; Sears, R.C.; Sherman, M.H. Acidic fibroblast growth factor underlies microenvironmental regulation of MYC in pancreatic cancer. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2020. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-santana, A.; Bi, G.; Elyada, E.; Almeida, A.S.; Ponz-sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Bernard, V.; Semaan, A.; Huang, J.; Anthony San Lucas, F.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-cell transcriptomics of pancreatic cancer precursors demonstrates epithelial and microenvironmental heterogeneity as an early event in neoplastic progression. Clin. Cancer Res. 2019, 25, 2194–2205. [Google Scholar] [CrossRef]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.B.; et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Wang, L.C.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef]
- Yuzawa, S.; Kano, M.R.; Einama, T.; Nishihara, H. PDGFRβ expression in tumor stroma of pancreatic adenocarcinoma as a reliable prognostic marker. Med. Oncol. 2012, 29, 2824–2830. [Google Scholar] [CrossRef]
- Djurec, M.; Graña, O.; Lee, A.; Troulé, K.; Espinet, E.; Cabras, L.; Navas, C.; Blasco, M.T.; Martín-Díaz, L.; Burdiel, M.; et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc. Natl. Acad. Sci. USA 2018, 115, E1147–E1156. [Google Scholar] [CrossRef]
- Hirayama, K.; Kono, H.; Nakata, Y.; Akazawa, Y.; Wakana, H.; Fukushima, H.; Fujii, H. Expression of podoplanin in stromal fibroblasts plays a pivotal role in the prognosis of patients with pancreatic cancer. Surg. Today 2018, 48, 110–118. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef]
- Carstens, J.L.; Correa de Sampaio, P.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Madden, J.I. Infinity Reports Update from Phase 2 Study of Saridegib Plus Gemcitabine in Patients with Metastatic Pancreatic Cancer. Available online: https://www.businesswire.com/news/home/20120127005146/en/Infinity-Reports-Update-Phase-2-Study-Saridegib (accessed on 10 November 2020).
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- Dessaud, E.; McMahon, A.P.; Briscoe, J. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 2008, 135, 2489–2503. [Google Scholar] [CrossRef]
- Tenzen, T.; Allen, B.L.; Cole, F.; Kang, J.S.; Krauss, R.S.; McMahon, A.P. The Cell Surface Membrane Proteins Cdo and Boc Are Components and Targets of the Hedgehog Signaling Pathway and Feedback Network in Mice. Dev. Cell 2006, 10, 647–656. [Google Scholar] [CrossRef]
- Zhang, W.; Kang, J.S.; Cole, F.; Yi, M.J.; Krauss, R.S. Cdo Functions at Multiple Points in the Sonic Hedgehog Pathway, and Cdo-Deficient Mice Accurately Model Human Holoprosencephaly. Dev. Cell 2006, 10, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Tenzen, T.; McMahon, A.P. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 2007, 21, 1244–1257. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Song, J.Y.; Izzi, L.; Althaus, I.W.; Kang, J.S.; Charron, F.; Krauss, R.S.; McMahon, A.P. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev. Cell 2011, 20, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pitarresi, J.R.; Cuitiño, M.C.; Kladney, R.D.; Woelke, S.A.; Sizemore, G.M.; Nayak, S.G.; Egriboz, O.; Schweickert, P.G.; Yu, L.; et al. Genetic ablation of Smoothened in pancreatic fibroblasts increases acinar—ductal metaplasia. Genes Dev. 2016, 30, 1943–1955. [Google Scholar] [CrossRef]
- Pitarresi, J.R.; Liu, X.; Avendano, A.; Thies, K.A.; Sizemore, G.M.; Hammer, A.M.; Hildreth, B.E.; Wang, D.J.; Steck, S.A.; Donohue, S.; et al. Disruption of stromal hedgehog signaling initiates RNF5-mediated proteasomal degradation of PTEN and accelerates pancreatic tumor growth. Life Sci. Alliance 2018, 1. [Google Scholar] [CrossRef]
- Savage, N.D.L.; de Boer, T.; Walburg, K.V.; Joosten, S.A.; van Meijgaarden, K.; Geluk, A.; Ottenhoff, T.H.M. Human Anti-Inflammatory Macrophages Induce Foxp3 + GITR + CD25 + Regulatory T Cells, Which Suppress via Membrane-Bound TGFβ-1. J. Immunol. 2008, 181, 2220–2226. [Google Scholar] [CrossRef]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.-J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef]
- Truty, M.J.; Urrutia, R. Basics of TGF-β and pancreatic cancer. Pancreatology 2007, 7, 423–435. [Google Scholar] [CrossRef]
- Adrian, K.; Strouch, M.J.; Zeng, Q.; Barron, M.R.; Cheon, E.C.; Honasoge, A.; Xu, Y.; Phukan, S.; Sadim, M.; Bentrem, D.J.; et al. Tgfbr1 haploinsufficiency inhibits the development of murine mutant Kras-induced pancreatic precancer. Cancer Res. 2009, 69, 9169–9174. [Google Scholar] [CrossRef]
- Principe, D.R.; DeCant, B.; Mascariñas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.; et al. TGFβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.E.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-β signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Cheng, K.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 3130–3146. [Google Scholar] [CrossRef] [PubMed]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A. KrasG12D and Smad4/Dpc4 Haploinsufficiency Cooperate to Induce Mucinous Cystic Neoplasms and Invasive Adenocarcinoma of the Pancreas. Cancer Cell 2007, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Vickers, S.M.; Adsay, N.V.; Jhala, N.C.; Kim, H.G.; Schoeb, T.R.; Grizzle, W.E.; Klug, C.A. Inactivation of Smad4 accelerates KrasG12D-mediated pancreatic neoplasia. Cancer Res. 2007, 67, 8121–8130. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Tumour microenvironment - TGFΒ: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF- β: Duality of Function Between Tumor Prevention and Carcinogenesis. J. Natl. Cancer Inst. 2014, 1–16. [Google Scholar] [CrossRef]
- Principe, D.R.; Park, A.; Dorman, M.J.; Kumar, S.; Viswakarma, N.; Rubin, J.; Torres, C.; McKinney, R.; Munshi, H.G.; Grippo, P.J.; et al. TGFb blockade augments PD-1 inhibition to promote T-cell–mediated regression of pancreatic cancer. Mol. Cancer Ther. 2019, 18, 613–620. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef]
- Rodriguez-Aznar, E.; Wiesmüller, L.; Sainz, B.; Hermann, P.C. EMT and stemness—Key players in pancreatic cancer stem cells. Cancers 2019, 11, 1136. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, R.T.; Veronese, N.; Nottegar, A.; Malleo, G.; Smith, L.; Demurtas, J.; Cheng, L.; Wood, L.D.; Silvestris, N.; Salvia, R.; et al. Prognostic role of high-grade tumor budding in pancreatic ductal adenocarcinoma: A systematic review and meta-analysis with a focus on epithelial to mesenchymal transition. Cancers 2019, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Huang, Y.-H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.A.; Freitas, J.P.; Mazher Hussain, S.; Glazer, E.S. TGF-β Inhibitors in Metastatic Pancreatic Ductal Adenocarcinoma. J. Gastrointest. Cancer 2019, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Dardare, J.; Witz, A.; Merlin, J.L.; Gilson, P.; Harlé, A. SMAD4 and the TGFΒ pathway in patients with pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 3534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Crawford, H.C.; Pasca di Magliano, M. Epithelial-Stromal Interactions in Pancreatic Cancer. Annu. Rev. Physiol. 2019, 81, 211–233. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.-P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Lin, S.-L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Lin, S.-C.J.; Aronow, B.J.; Tallquist, M.D. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef]
- El Agha, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Szibor, M.; Kosanovic, D.; Schwind, F.; Schermuly, R.T.; et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 2017, 20, 261–273.e3. [Google Scholar] [CrossRef]
- Xie, T.; Liang, J.; Liu, N.; Huan, C.; Zhang, Y.; Liu, W.; Kumar, M.; Xiao, R.; D’Armiento, J.; Metzger, D.; et al. Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J. Clin. Investig. 2016, 126, 3063–3079. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, C.J.; Colvin, E.K.; Pinese, M.; Chang, D.K.; Morey, A.L.; Musgrove, E.A.; Pajic, M.; Apte, M.; Henshall, S.M.; Sutherland, R.L.; et al. Recruitment and Activation of Pancreatic Stellate Cells from the Bone Marrow in Pancreatic Cancer: A Model of Tumor-Host Interaction. PLoS ONE 2011, 6, e26088. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed]
- Shindo, K.; Aishima, S.; Ohuchida, K.; Fujiwara, K.; Fujino, M.; Mizuuchi, Y.; Hattori, M.; Mizumoto, K.; Tanaka, M.; Oda, Y. Podoplanin expression in cancer-associated fibroblasts enhances tumor progression of invasive ductal carcinoma of the pancreas. Mol. Cancer 2013, 12, 168. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]
- Vennin, C.; Mélénec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 3637. [Google Scholar] [CrossRef]
- Farran, B.; Nagaraju, G.P. The dynamic interactions between the stroma, pancreatic stellate cells and pancreatic tumor development: Novel therapeutic targets. Cytokine Growth Factor Rev. 2019, 48, 11–23. [Google Scholar] [CrossRef]
- Ko, A.H.; Loconte, N.; Tempero, M.A.; Walker, E.J.; Kelley, R.K.; Lewis, S.; Chang, W.; Kantoff, E.; Vannier, M.W.; Daniel, V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Büchler, P.; Reber, H.A.; Büchler, M.W.; Friess, H.; Hines, O.J.; Russell, R.C.G.; Poon, R. VEGF-RII influences the prognosis of pancreatic cancer. Ann. Surg. 2002, 236, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Baba, H.; Fukuda, T.; Takashima, M.; Sugimachi, K. High expression of vascular endothelial growth factor is associated with liver metastasis and a poor prognosis for patients with ductal pancreatic adenocarcinoma. Cancer 2000, 88, 2239–2245. [Google Scholar] [CrossRef]
- Kindler, H.L.; Ioka, T.; Richel, D.J.; Bennouna, J.; Létourneau, R.; Okusaka, T.; Funakoshi, A.; Furuse, J.; Park, Y.S.; Ohkawa, S.; et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: A double-blind randomised phase 3 study. Lancet. Oncol. 2011, 12, 256–262. [Google Scholar] [CrossRef]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef]
- Mantoni, T.S.; Lunardi, S.; Al-Assar, O.; Masamune, A.; Brunner, T.B. Pancreatic stellate cells radioprotect pancreatic cancer cells through β1-integrin signaling. Cancer Res. 2011, 71, 3453–3458. [Google Scholar] [CrossRef]
- Chan, T.S.; Hsu, C.C.; Pai, V.C.; Liao, W.Y.; Huang, S.S.; Tan, K.T.; Yen, C.J.; Hsu, S.C.; Chen, W.Y.; Shan, Y.S.; et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J. Exp. Med. 2016, 213, 2967–2988. [Google Scholar] [CrossRef]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef]
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141. [Google Scholar] [CrossRef]
- Muranen, T.; Iwanicki, M.P.; Curry, N.L.; Hwang, J.; DuBois, C.D.; Coloff, J.L.; Hitchcock, D.S.; Clish, C.B.; Brugge, J.S.; Kalaany, N.Y. Starved epithelial cells uptake extracellular matrix for survival. Nat. Commun. 2017, 8, 13989. [Google Scholar] [CrossRef]
- Francescone, R.; Barbosa Vendramini-Costa, D.; Franco-Barraza, J.; Wagner, J.; Muir, A.; Lau, A.N.; Gabitova, L.; Pazina, T.; Gupta, S.; Luong, T.; et al. Netrin G1 promotes pancreatic tumorigenesis through cancer associated fibroblast driven nutritional support and immunosuppression. Cancer Discov. 2020. [Google Scholar] [CrossRef] [PubMed]
- Toole, B.P.; Slomiany, M.G. Hyaluronan: A constitutive regulator of chemoresistance and malignancy in cancer cells. Semin. Cancer Biol. 2008, 18, 244–250. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Doherty, G.J.; Tempero, M.; Corrie, P.G. HALO-109–301: A Phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Futur. Oncol. 2018, 14, 13–22. [Google Scholar] [CrossRef]
- Kildani, A. Halozyme Announces HALO-301 Phase 3 Study Fails To Meet Primary Endpoint. Available online: https://www.halozyme.com/investors/news-releases/news-release-details/2019/Halozyme-Announces-HALO-301-Phase-3-Study-Fails-To-Meet-Primary-Endpoint/default.aspx (accessed on 10 November 2020).
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Froeling, F.E.M.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wntβ-catenin signaling to slow tumor progression. Gastroenterology 2011, 141, 1486–1497.e14. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Gorchs, L.; Ahmed, S.; Mayer, C.; Knauf, A.; Fernández Moro, C.; Svensson, M.; Heuchel, R.; Rangelova, E.; Bergman, P.; Kaipe, H. The vitamin D analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hilmi, M.; Bartholin, L.; Neuzillet, C. Immune therapies in pancreatic ductal adenocarcinoma: Where are we now? World J. Gastroenterol. 2018, 24, 2137–2151. [Google Scholar] [CrossRef]
- Lazarus, J.; Maj, T.; Smith, J.J.; Perusina Lanfranca, M.; Rao, A.; D’Angelica, M.I.; Delrosario, L.; Girgis, A.; Schukow, C.; Shia, J.; et al. Spatial and phenotypic immune profiling of metastatic colon cancer. JCI Insight 2018, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fassler, D.J.; Abousamra, S.; Gupta, R.; Chen, C.; Zhao, M.; Paredes, D.; Batool, S.A.; Knudsen, B.S.; Escobar-Hoyos, L.; Shroyer, K.R.; et al. Deep learning-based image analysis methods for brightfield-acquired multiplex immunohistochemistry images. Diagn. Pathol. 2020, 15, 116. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-lopez, J.; Haas, W.; Aryee, M.J.; Ting, D.T. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer Article Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 1–16. [Google Scholar] [CrossRef]


{kind=link}
| Fibroblast Population | Defining Features | Known Function in PDA | Key References |
|---|---|---|---|
| myCAFs | αSMAhigh TGF-β dependent Tumor-adjacent | Tumor-restricting | [15,36,37,38,111,112,113] |
| iCAFs | IL6+ IL1/Jak-stat dependent Distant from tumor | Tumor-promoting | [36,37,38,111,112,113] |
| apCAFs | MHC Class II+ Have also been described as mesothelial cells | Antigen presentation to T cells | [37,38] |
| FAP+ Fibroblasts | Fap+ | Immunosuppressive | [5,6,114,115] |
| Gli1 Lineage | Derived from Gli1+ fibroblasts in healthy pancreas | Not fully understood Partially contributes to myCAFs | [28] |
| MSCs | CD45−;CD44+;CD49a+;CD73+; CD90+ | Tumor-promoting | [72,73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, P.E.; Scales, M.K.; Allen, B.L.; Pasca di Magliano, M. Pancreatic Fibroblast Heterogeneity: From Development to Cancer. Cells 2020, 9, 2464. https://doi.org/10.3390/cells9112464
Garcia PE, Scales MK, Allen BL, Pasca di Magliano M. Pancreatic Fibroblast Heterogeneity: From Development to Cancer. Cells. 2020; 9(11):2464. https://doi.org/10.3390/cells9112464
Chicago/Turabian StyleGarcia, Paloma E., Michael K. Scales, Benjamin L. Allen, and Marina Pasca di Magliano. 2020. "Pancreatic Fibroblast Heterogeneity: From Development to Cancer" Cells 9, no. 11: 2464. https://doi.org/10.3390/cells9112464
APA StyleGarcia, P. E., Scales, M. K., Allen, B. L., & Pasca di Magliano, M. (2020). Pancreatic Fibroblast Heterogeneity: From Development to Cancer. Cells, 9(11), 2464. https://doi.org/10.3390/cells9112464